
Pharmacological and Therapeutic Approaches in the Treatment of Epilepsy

 ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Role of Genes, Genetics and Inheritance
3. Epigenetics Involved in Epilepsy
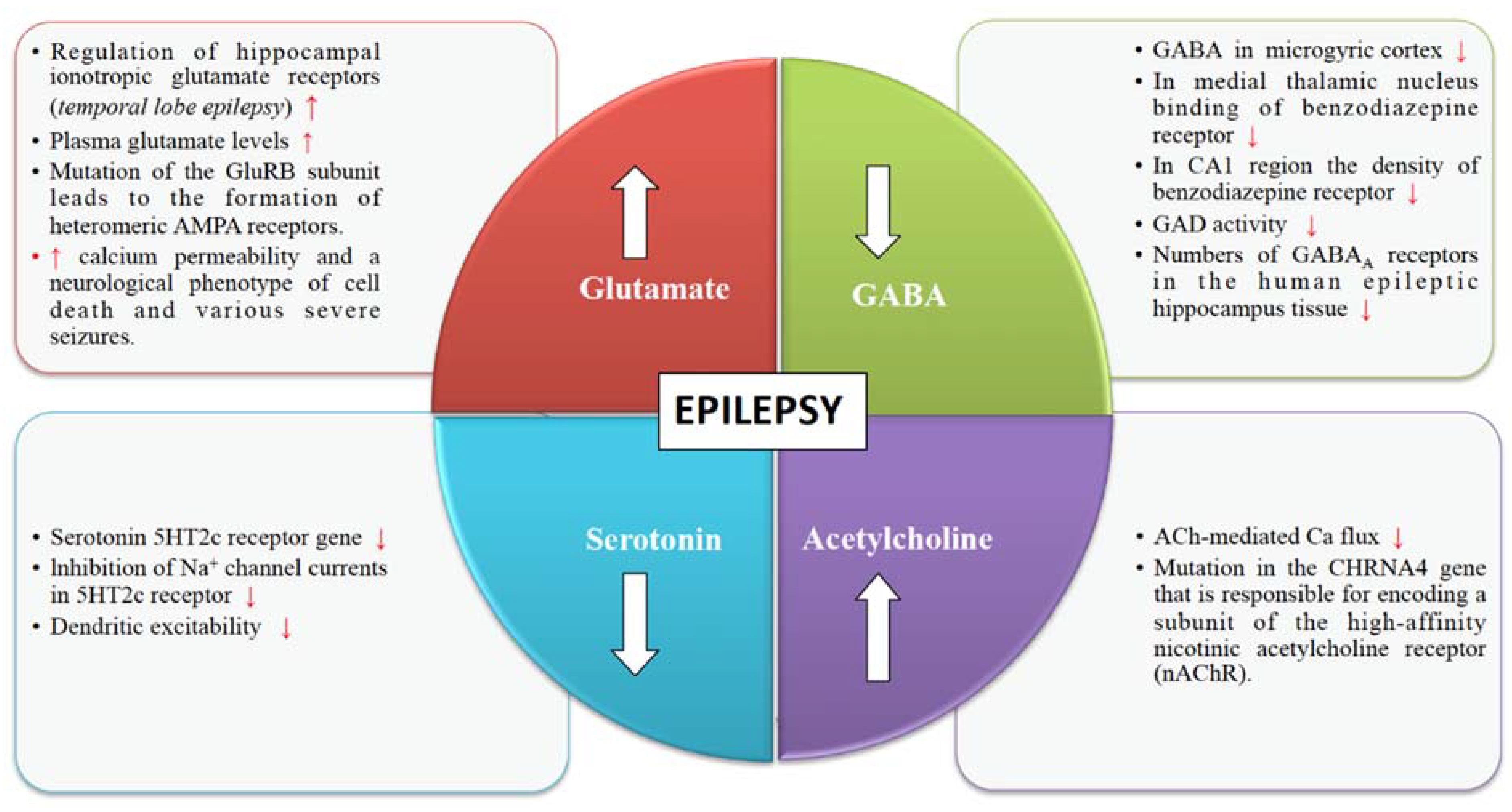
4. Neurotransmitter Release Machinery in Epilepsy
4.1. Glutamate Receptors
4.2. GABA Receptors
4.3. Cholinergic Receptors
4.4. Serotonin Receptors
5. Drug Resistant Epilepsy
5.1. Alterations in the Drug Targets
5.2. The Inability of the Drugs to Reach Their Targets
5.3. Real Targets Missed by the Drugs
6. Non-Conventional Therapeutic Strategies
6.1. Ruling Out Pseudo-Resistance
6.2. Combination Therapy
6.3. Non-Drug Treatment
7. Modern Approaches for Treatment
7.1. MTOR Pathway
7.2. Inflammatory Pathways
7.3. Breakdown of Blood-Brain Barrier
8. Conclusions

{kind=link}
| S. No. | Treatment Approaches | Interventions Used | Action Mechanism | Main Uses | References |
|---|---|---|---|---|---|
| A. | PHARMACEUTICAL APPROACHES | Gabapentin | Ca2+ blockage | Used for generalised and focal seizures. | [74,75] |
| (Anti-epileptic drugs) | Carbamazepine | Na+ channel blockage | Decrease nerve impulses that are responsible for causing seizures. | [74] | |
| Lamotrigine | Na+ channel blockage | Used as a first- line drug for generalized and focal seizures. | [60,75] | ||
| Tiagabine | GABA potentiation | Used for partial seizures in adjunctive therapy. | [74] | ||
| Zonisamide | Na+ channel blockage | Used for generalized and focal seizures. | [74,75] | ||
| Vigabatrin | GABA potentiation | Used for infantile spasms and for focal onset of seizures. | [74] | ||
| Perampanel | Glutamate (AMPA) antagonist | Used for partial seizures with focal onset. | [74,75] | ||
| B. | THERAPEUTIC APPROACHES | Progressive muscle relaxation | Tense a group of muscles while breathing in and relaxes them while breathing out. | Improves sleep and overall well-being. Enhances control over epilepsy by the patients. | [76] |
| Yoga | Release tension in key joints through combination of body postures. | Decrease in automatic dysfunction during onset of seizures. | [76,77] | ||
| Cognitive behavioural therapy | Restructuring of maladaptive thought patterns. | Improvement in anxiety and depression and enhanced psychosocial functioning. | [76,78] | ||
| Vagus nerve stimulation | Used to generate impulse through electric current in vagus nerve. | Used as an adjunctive therapy for partial onset of seizures. | [65] | ||
| C. | NATURAL APPROACHES | Ketogenic diet | Neurotransmitter modulation in brain by ketone bodies. | Successful in reducing seizures and enhancing motor function. | [27,64,79] |
| Vitamin D3 | Increase Ca2+ uptake and decrease neuronal excitability. | Produces anti-convulsant effect and prevent seizures. | [80,81] | ||
| Herbal treatments | Found to be involved in potentiation of GABAergic activity in brain. | Herbal medications control epileptic seizures and reduce side effects and increase cognitive effects of AEDs. | [82,83] |
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Scharfman, H.E. The neurobiology of epilepsy. Curr. Neurol. Neurosci. Rep. 2007, 7, 348–354. [Google Scholar] [CrossRef]
- Kobylarek, D.; Iwanowski, P.; Lewandowska, Z.; Limphaibool, N.; Szafranek, S.; Labrzycka, A.; Kozubski, W. Advances in the Potential Biomarkers of Epilepsy. Front. Neurol. 2019, 10, 685. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.S.; Acevedo, C.; Arzimanoglou, A.; Bogacz, A.; Cross, J.H.; Elger, C.E.; Engel, J.; Forsgren, L.; French, J.A.; Glynn, M.; et al. ILAE Official Report: A practical clinical definition of epilepsy. Epilepsia 2014, 55, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.; Sillanpää, M. Evidence-based review on the natural history of the epilepsies. Curr. Opin. Neurol. 2012, 25, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Berg, A.T.; Berkovic, S.F.; Brodie, M.J.; Buchhalter, J.; Cross, J.H.; Boas, W.V.E.; Engel, J.; French, J.; Glauser, T.A.; Mathern, G.W.; et al. Revised terminology and concepts for organization of seizures and epilepsies: Report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia 2010, 51, 676–685. [Google Scholar] [CrossRef]
- Löscher, W.; Klitgaard, H.; Twyman, R.E.; Schmidt, D. New avenues for anti-epileptic drug discovery and development. Nat. Rev. Drug Discov. 2013, 12, 757–776. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D. Is antiepileptogenesis a realistic goal in clinical trials? Concerns and new horizons. Epileptic Disord 2012, 14, 105–113. [Google Scholar] [CrossRef]
- Brodie, M.J.; Barry, S.J.E.; Bamagous, G.A.; Norrie, J.D.; Kwan, P. Patterns of treatment response in newly diagnosed epilepsy. Neurology 2012, 78, 1548–1554. [Google Scholar] [CrossRef]
- Mishra, P.; Mittal, A.K.; Rajput, S.K.; Sinha, J.K. Cognition and memory impairment attenuation via reduction of oxidative stress in acute and chronic mice models of epilepsy using antiepileptogenic Nux vomica. J. Ethnopharmacol. 2021, 267, 113509. [Google Scholar] [CrossRef]
- Wang, J.; Lin, Z.-J.; Liu, L.; Xu, H.-Q.; Shi, Y.-W.; Yi, Y.-H.; He, N.; Liao, W.-P. Epilepsy-associated genes. Seizure 2017, 44, 11–20. [Google Scholar] [CrossRef]
- Martins-Ferreira, R.; Chaves, J.; Carvalho, C.; Bettencourt, A.; Chorão, R.; Freitas, J.; Samões, R.; Boleixa, D.; Lopes, J.; Ramalheira, J.; et al. Circulating microRNAs as potential biomarkers for genetic generalized epilepsies: A three microRNA panel. Eur. J. Neurol. 2020, 27, 660–666. [Google Scholar] [CrossRef] [PubMed]
- Caramaschi, D.; Hatcher, C.; Mulder, R.H.; Felix, J.F.; Cecil, C.A.M.; Relton, C.L.; Walton, E. Epigenome-wide association study of seizures in childhood and adolescence. Clin. Epigenet. 2020, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Grønskov, K.; Brøndum-Nielsen, K.; Dedic, A.; Hjalgrim, H. A nonsense mutation in FMR1 causing fragile X syndrome. Eur. J. Hum. Genet. 2011, 19, 489–491. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Albert, J.; Lipinski, M.; Lopez-Cascales, M.T.; Rowley, M.J.; Martin-Gonzalez, A.M.; Del Blanco, B.; Corces, V.G.; Barco, A. Immediate and deferred epigenomic signatures of in vivo neuronal activation in mouse hippocampus. Nat. Neurosci. 2019, 22, 1718–1730. [Google Scholar] [CrossRef] [PubMed]
- Pederick, D.T.; Richards, K.L.; Piltz, S.G.; Kumar, R.; Mincheva-Tasheva, S.; Mandelstam, S.A.; Dale, R.C.; Scheffer, I.E.; Gecz, J.; Petrou, S.; et al. Abnormal Cell Sorting Underlies the Unique X-Linked Inheritance of PCDH19 Epilepsy. Neuron 2018, 97, 59–66.e5. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, J.L.; Kostadinov, D.; Chen, W.V.; Maniatis, T.; Sanes, J.R. Protocadherins mediate dendritic self-avoidance in the mammalian nervous system. Nat. Cell Biol. 2012, 488, 517–521. [Google Scholar] [CrossRef]
- Toth, M. Epigenetic Neuropharmacology: Drugs Affecting the Epigenome in the Brain. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 181–201. [Google Scholar] [CrossRef]
- Eboison, D. The Biochemistry and Epigenetics of Epilepsy: Focus on Adenosine and Glycine. Front. Mol. Neurosci. 2016, 9, 26. [Google Scholar] [CrossRef]
- Kobow, K.; Blümcke, I. The emerging role of DNA methylation in epileptogenesis. Epilepsia 2012, 53, 11–20. [Google Scholar] [CrossRef]
- Symonds, J.D. CHD2 epilepsy: Epigenetics and the quest for precision medicine. Dev. Med. Child Neurol. 2019, 62, 549–550. [Google Scholar] [CrossRef]
- Feng, J.; Zhou, Y.; Campbell, S.L.; Le, T.; Li, E.; Sweatt, J.D.; Silva, A.J.; Fan, G. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat. Neurosci. 2010, 13, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Henshall, D.C.; Kobow, K. Epigenetics and Epilepsy. Cold Spring Harb. Perspect. Med. 2015, 5, a022731. [Google Scholar] [CrossRef] [PubMed]
- De Nijs, L.; Choe, K.; Steinbusch, H.; Schijns, O.E.M.G.; Dings, J.; Hove, D.L.A.V.D.; Rutten, B.P.F.; Hoogland, G. DNA methyltransferase isoforms expression in the temporal lobe of epilepsy patients with a history of febrile seizures. Clin. Epigenetics 2019, 11, 118. [Google Scholar] [CrossRef] [PubMed]
- Boison, D.; Rho, J.M. Epigenetics and epilepsy prevention: The therapeutic potential of adenosine and metabolic therapies. Neuropharmacol. 2020, 167, 107741. [Google Scholar] [CrossRef] [PubMed]
- Younus, I.; Reddy, D.S. Epigenetic interventions for epileptogenesis: A new frontier for curing epilepsy. Pharmacol. Ther. 2017, 177, 108–122. [Google Scholar] [CrossRef]
- Boison, D. Adenosine Kinase: Exploitation for Therapeutic Gain. Pharmacol. Rev. 2013, 65, 906–943. [Google Scholar] [CrossRef]
- Longo, R.; Peri, C.; Cricrì, D.; Coppi, L.; Caruso, D.; Mitro, N.; De Fabiani, E.; Crestani, M. Ketogenic Diet: A New Light Shining on Old but Gold Biochemistry. Nutrients 2019, 11, 2497. [Google Scholar] [CrossRef]
- Jago, S.C.S.; Lubin, F.D. Epigenetic Therapeutic Intervention for a Rare Epilepsy Disorder. Epilepsy Curr. 2020, 20, 111–112. [Google Scholar] [CrossRef]
- Chapman, A.G. Glutamate and Epilepsy. J. Nutr. 2000, 130, 1043S–1045S. [Google Scholar] [CrossRef]
- Salpietro, V.; SYNAPS Study Group; Dixon, C.L.; Guo, H.; Bello, O.D.; Vandrovcova, J.; Efthymiou, S.; Maroofian, R.; Heimer, G.; Burglen, L.; et al. AMPA receptor GluA2 subunit defects are a cause of neurodevelopmental disorders. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Kumar, S.S.; Bacci, A.; Kharazia, V.; Huguenard, J.R. A Developmental Switch of AMPA Receptor Subunits in Neocortical Pyramidal Neurons. J. Neurosci. 2002, 22, 3005–3015. [Google Scholar] [CrossRef] [PubMed]
- Meldrum, B.S.; Akbar, M.T.; Chapman, A.G. Glutamate receptors and transporters in genetic and acquired models of epilepsy. Epilepsy Res. 1999, 36, 189–204. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Chen, Z. Double-edged GABAergic synaptic transmission in seizures: The importance of chloride plasticity. Brain Res. 2018, 1701, 126–136. [Google Scholar] [CrossRef]
- Treiman, D.M. GABAergic Mechanisms in Epilepsy. Epilepsia 2001, 42, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.W.; De Lanerolle, N.C.; Kim, J.H.; Sundaresan, S.; Spencer, D.D.; Mattson, R.H.; Zoghbi, S.S.; Baldwin, R.M.; Hoffer, P.B.; Seibyl, J.P.; et al. “Central” and “peripheral” benzodiazepine receptors: Opposite changes in human epileptogenic tissue. Neurology 1992, 42, 811. [Google Scholar] [CrossRef]
- Grunewald, R.A.; Thompson, P.J.; Corcoran, R.; Corden, Z.; Jackson, G.D.; Duncan, J.S. Effects of vigabatrin on partial seizures and cognitive function. J. Neurol. Neurosurg. Psychiatry 1994, 57, 1057–1063. [Google Scholar] [CrossRef]
- Dani, J.A.; Bertrand, D. Nicotinic Acetylcholine Receptors and Nicotinic Cholinergic Mechanisms of the Central Nervous System. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 699–729. [Google Scholar] [CrossRef]
- Raggenbass, M.; Bertrand, D. Nicotinic receptors in circuit excitability and epilepsy. J. Neurobiol. 2002, 53, 580–589. [Google Scholar] [CrossRef]
- Ghasemi, M.; Hadipour-Niktarash, A. Pathologic role of neuronal nicotinic acetylcholine receptors in epileptic disorders: Implication for pharmacological interventions. Rev. Neurosci. 2015, 26, 199–223. [Google Scholar] [CrossRef]
- Bertrand, D.; Picard, F.; Le Hellard, S.; Weiland, S.; Favre, I.; Phillips, H.; Berkovic, S.F.; Malafosse, A.; Mulley, J. How Mutations in the nAChRs Can Cause ADNFLE Epilepsy. Epilepsia 2002, 43, 112–122. [Google Scholar] [CrossRef]
- Hayashi, M.; Nakajima, K.; Miyata, R.; Tanuma, N.; Kodama, T. Lesions of Acetylcholine Neurons in Refractory Epilepsy. ISRN Neurol. 2012, 2012, 1–6. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bourin, M. Mechanisms of Action of Anxiolytics. In Psychiatry and Neuroscience Update; Springer Science and Business Media LLC: New York, NY, USA, 2021; pp. 195–211. [Google Scholar]
- Maia, G.H.; Brazete, C.S.; Soares, J.I.; Luz, L.L.; Lukoyanov, N.V. Serotonin depletion increases seizure susceptibility and worsens neuropathological outcomes in kainate model of epilepsy. Brain Res. Bull. 2017, 134, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Singh, T.; Goel, R.K. Managing epilepsy-associated depression: Serotonin enhancers or serotonin producers? Epilepsy Behav. 2017, 66, 93–99. [Google Scholar] [CrossRef]
- Krishnakumar, A.; Abraham, P.M.; Paul, J.; Paulose, C. Down-regulation of cerebellar 5-HT2C receptors in pilocarpine-induced epilepsy in rats: Therapeutic role of Bacopa monnieri extract. J. Neurol. Sci. 2009, 284, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Bagdy, G.; Kecskeméti, V.; Riba, P.; Jakus, R. Serotonin and epilepsy. J. Neurochem. 2007, 100, 857–873. [Google Scholar] [CrossRef] [PubMed]
- Kwan, P.; Schachter, S.C.; Brodie, M.J. Drug-Resistant Epilepsy. N. Engl. J. Med. 2011, 365, 919–926. [Google Scholar] [CrossRef]
- Mohanraj, R.; Norrie, J.; Stephen, L.J.; Kelly, K.; Hitiris, N.; Brodie, M.J. Mortality in adults with newly diagnosed and chronic epilepsy: A retrospective comparative study. Lancet Neurol. 2006, 5, 481–487. [Google Scholar] [CrossRef]
- Lawn, N.D.; Bamlet, W.R.; Radhakrishnan, K.; O’Brien, P.C.; So, E.L. Injuries due to seizures in persons with epilepsy: A population-based study. Neurology 2004, 63, 1565–1570. [Google Scholar] [CrossRef]
- McCagh, J.; Fisk, J.E.; Baker, G.A. Epilepsy, psychosocial and cognitive functioning. Epilepsy Res. 2009, 86, 1–14. [Google Scholar] [CrossRef]
- Loup, F.; Picard, F.; Yonekawa, Y.; Wieser, H.-G.; Fritschy, J.-M. Selective changes in GABAA receptor subtypes in white matter neurons of patients with focal epilepsy. Brain 2009, 132, 2449–2463. [Google Scholar] [CrossRef]
- Remy, S.; Beck, H. Molecular and cellular mechanisms of pharmacoresistance in epilepsy. Brain 2005, 129, 18–35. [Google Scholar] [CrossRef]
- Tishler, D.M.; Weinberg, K.I.; Hinton, D.R.; Barbaro, N.; Annett, G.M.; Raffel, C. MDR1 Gene Expression in Brain of Patients with Medically Intractable Epilepsy. Epilepsia 1995, 36, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.; Irani, S.R.; Lang, B. The growing recognition of immunotherapy-responsive seizure disorders with autoantibodies to specific neuronal proteins. Curr. Opin. Neurol. 2010, 23, 144–150. [Google Scholar] [CrossRef]
- McKnight, K.; Jiang, Y.; Hart, Y.; Cavey, A.; Wroe, S.; Blank, M.; Shoenfeld, Y.; Vincent, A.; Palace, J.; Lang, B. Serum antibodies in epilepsy and seizure-associated disorders. Neurology 2005, 65, 1730–1736. [Google Scholar] [CrossRef]
- Elger, C.E.; Schmidt, D. Modern management of epilepsy: A practical approach. Epilepsy Behav. 2008, 12, 501–539. [Google Scholar] [CrossRef]
- Smith, D.; DeFalla, B.; Chadwick, D. The misdiagnosis of epilepsy and the management of refractory epilepsy in a specialist clinic. Qjm: Int. J. Med. 1999, 92, 15–23. [Google Scholar] [CrossRef]
- Faught, E.; Duh, M.S.; Weiner, J.R.; Guerin, A.; Cunnington, M.C. Nonadherence to anti-epileptic drugs and increased mortality: Findings from the RANSOM Study. Neurology 2008, 71, 1572–1578. [Google Scholar] [CrossRef]
- Mishra, P.; Sinha, J.K.; Rajput, S.K. Efficacy of Cicuta virosa medicinal preparations against pentylenetetrazole-induced seizures. Epilepsy Behav. 2021, 115, 107653. [Google Scholar] [CrossRef] [PubMed]
- Brodie, M.; Yuen, A. Lamotrigine substitution study: Evidence for synergism with sodium valproate? Epilepsy Res. 1997, 26, 423–432. [Google Scholar] [CrossRef]
- Spencer, S.; Huh, L. Outcomes of epilepsy surgery in adults and children. Lancet Neurol. 2008, 7, 525–537. [Google Scholar] [CrossRef]
- Ghosh, S.; Sinha, J.K.; Muralikrishna, B.; Putcha, U.K.; Raghunath, M. Chronic transgenerational vitamin B12 deficiency of severe and moderate magnitudes modulates adiposity-probable underlying mechanisms. BioFactors 2017, 43, 400–414. [Google Scholar] [CrossRef]
- Ghosh, S.; Sinha, J.K.; Khandelwal, N.; Chakravarty, S.; Kumar, A.; Raghunath, M. Increased stress and altered expression of histone modifying enzymes in brain are associated with aberrant behaviour in vitamin B12 deficient female mice. Nutr. Neurosci. 2020, 23, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Neal, E.G.; Chaffe, H.; Schwartz, R.H.; Lawson, M.S.; Edwards, N.; Fitzsimmons, G.; Whitney, A.; Cross, J.H. The ketogenic diet for the treatment of childhood epilepsy: A randomised controlled trial. Lancet Neurol. 2008, 7, 500–506. [Google Scholar] [CrossRef]
- Milby, A.H.; Halpern, C.H.; Baltuch, G.H. Vagus nerve stimulation in the treatment of refractory epilepsy. J. Neurosurg. 2009, 6, 228–237. [Google Scholar] [CrossRef]
- Ghosh, S.; Durgvanshi, S.; Agarwal, S.; Raghunath, M.; Sinha, J.K. Current Status of Drug Targets and Emerging Therapeutic Strategies in the Management of Alzheimer’s Disease. Curr. Neuropharmacol. 2020, 18, 883–903. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A. Before Epilepsy Unfolds: Finding the epileptogenesis switch. Nat. Med. 2012, 18, 1626–1627. [Google Scholar] [CrossRef] [PubMed]
- Ryther, R.C.C.; Wong, M. Mammalian Target of Rapamycin (mTOR) Inhibition: Potential for Antiseizure, Antiepileptogenic, and Epileptostatic Therapy. Curr. Neurol. Neurosci. Rep. 2012, 12, 410–418. [Google Scholar] [CrossRef]
- Vezzani, A.; Friedman, A.; Dingledine, R.J. The role of inflammation in epileptogenesis. Neuropharmacol. 2013, 69, 16–24. [Google Scholar] [CrossRef]
- Frigerio, F.; Frasca, A.; Weissberg, I.; Parrella, S.; Friedman, A.; Vezzani, A.; Noe’, F.M. Long-lasting pro-ictogenic effects induced in vivo by rat brain exposure to serum albumin in the absence of concomitant pathology. Epilepsia 2012, 53, 1887–1897. [Google Scholar] [CrossRef]
- Cacheaux, L.P.; Ivens, S.; David, Y.; Lakhter, A.J.; Bar-Klein, G.; Shapira, M.Y.; Heinemann, U.; Friedman, A.; Kaufer, D. Transcriptome Profiling Reveals TGF- Signaling Involvement in Epileptogenesis. J. Neurosci. 2009, 29, 8927–8935. [Google Scholar] [CrossRef]
- Grone, B.P.; Baraban, S.C. Animal models in epilepsy research: Legacies and new directions. Nat. Neurosci. 2015, 18, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Sinha, J.K.; Raghunath, M. Epigenomic maintenance through dietary intervention can facilitate DNA repair process to slow down the progress of premature aging. IUBMB Life 2016, 68, 717–721. [Google Scholar] [CrossRef]
- Ventola, C.L. Epilepsy Management: Newer Agents, Unmet Needs, and Future Treatment Strategies. P T Peer-Rev. J. Formul. Manag. 2014, 39, 776–792. [Google Scholar]
- Schmidt, D.; Schachter, S.C. Drug treatment of epilepsy in adults. BMJ 2014, 348, g254. [Google Scholar] [CrossRef] [PubMed]
- Haut, S.R.; Gursky, J.M.; Privitera, M. Behavioral interventions in epilepsy. Curr. Opin. Neurol. 2019, 32, 227–236. [Google Scholar] [CrossRef]
- Panebianco, M.; Sridharan, K.; Ramaratnam, S. Yoga for epilepsy. Cochrane Database Syst. Rev. 2017, 2017, CD001524. [Google Scholar] [CrossRef]
- Ramaratnam, S.; A Baker, G.; Goldstein, L.H. Psychological treatments for epilepsy. Cochrane Database Syst. Rev. 2016, 2, CD002029. [Google Scholar] [CrossRef]
- Çubukçu, D.; Güzel, O.; Arslan, N. Effect of Ketogenic Diet on Motor Functions and Daily Living Activities of Children With Multidrug-Resistant Epilepsy: A Prospective Study. J. Child Neurol. 2018, 33, 718–723. [Google Scholar] [CrossRef] [PubMed]
- Pendo, K.; DeGiorgio, C.M. Vitamin D3 for the Treatment of Epilepsy: Basic Mechanisms, Animal Models, and Clinical Trials. Front. Neurol. 2016, 7, 218. [Google Scholar] [CrossRef]
- Holló, A.; Clemens, Z.; Kamondi, A.; Lakatos, P.; Szűcs, A. Correction of vitamin D deficiency improves seizure control in epilepsy: A pilot study. Epilepsy Behav. 2012, 24, 131–133. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Ge, T.; Pan, Z.; Leng, Y.; Lv, J.; Li, B. The effects of herbal medicine on epilepsy. Oncotarget 2017, 8, 48385–48397. [Google Scholar] [CrossRef] [PubMed]
- Sucher, N.J.; Carles, M.C. A pharmacological basis of herbal medicines for epilepsy. Epilepsy Behav. 2015, 52, 308–318. [Google Scholar] [CrossRef] [PubMed]

| S.No. | Epilepsy Genes | Functional Category | Pattern of Inheritance | Type Of Syndrome | S.No. |
|---|---|---|---|---|---|
| 1. | ALDH7A1 | Enzyme | Autosomal recessive | Pyridoxine dependent epilepsy. | Neonatal period |
| 2. | KCNQ2 | Potassium channel | Autosomal dominant | Benign familial neonatal seizures. | |
| 3. | GABRA1 | Receptor of GABA A | Autosomal dominant | Early infantile epileptic encephalopathy. | Infancy and early childhood |
| 4. | SCN8A, SCN2A | Sodium channel | Autosomal dominant | Benign familial neonatal seizures. Early infantile epileptic encephalopathy. | |
| 5. | CHD2 | Enzyme | Autosomal dominant | Childhood onset epileptic encephalopathy. | |
| 6. | STX1B | Transport across membrane | Autosomal dominant | Generalized epilepsy. | |
| 7. | TBC1D24 | Modulator of enzyme | Autosomal recessive | Familial infantile myoclinic epilepsy. Early infantile epileptic encephalopathy. | |
| 8. | NECAP1 | Not classified | Autosomal recessive | Early infantile epileptic encephalopathy. | |
| 9. | UBA5, GNAO1 | Enzyme | Not known | Early infantile epileptic encephalopathy. | |
| HCN1 | HCN channel | ||||
| 10. | GPR98 | Receptor | Autosomal dominant | Familial febrile seizures. | |
| 11. | KCNMA1 | Potassium channel | Autosomal dominant | Generalized epilepsy with paroxysmal dyskinesia. | |
| 12. | STRGAL3, WWOX | Enzyme | Autosomal recessive | Early infantile epileptic encephalopathy. | |
| 13. | PRRT2 | Not classified | Autosomal dominant | Benign familial infantile seizures. | |
| 14. | SLC6A1 | Transporter | Autosomal dominant | Myoclinic atonic epilepsy. | |
| 15. | ARHGEF9 | Modulator of enzyme | X linked recessive | Early infantile epileptic encephalopathy. | |
| 16. | SCN9A | Sodium channel | Autosomal dominant | Dravet Syndrome. Familial febrile seizures. | |
| 17. | CDKL5 | Enzyme | X linked dominant | Early infantile epileptic encephalopathy. | |
| 18. | GRIN2A | NMDA receptor | Autosomal dominant | Focal epilepsy with speech disorder. | |
| 19. | STRGAL5 | Enzyme | Autosomal recessive | Amish infantile epilepsy. | |
| 20. | CACNA1H | Calcium channel | Not known | Childhood absence epilepsy.Idiopathic generalized epilepsy. | |
| 21. | ALG13 | Enzyme | X linked | Early infantile epileptic encephalopathy. | |
| 22. | CPA6 | Enzyme | Autosomal dominant | Familial temporal lobe epilepsy. | Juvenile phase and later |
| LGI1 | Not classified | ||||
| 23. | CACNB4 | Calcium channel | Autosomal dominant | Juvenile myoclinic epilepsy. Idiopathic generalized epilepsy. | |
| 24. | EFHC1 | Signalling molecule | Autosomal dominant | Juvenile absence epilepsy. Juvenile myoclinic epilepsy. | |
| 25. | CLCN2 | Chloride channel | Autosomal dominant | Juvenile generalized epilepsy. Juvenile absence epilepsy. Juvenile myoclinic epilepsy. | |
| 26. | ADRA2B | Receptor | Autosomal dominant | Familial adult myoclinic epilepsy. | |
| 27. | GABRD | Receptor of GABA A | Autosomal dominant | Generalized epilepsy with febrile seizures. Juvenile myoclinic epilepsy. | |
| 28. | CASR | Receptor | Not known | Idiopathic generalized epilepsy. | |
| 29. | DEPDC5 | Not classified | Autosomal dominant | Familial focal epilepsy. | Unspecified |
| 30. | CHRNB2 | Acetylcholine receptor | Unknown | Nocturnal frontal lobe epilepsy. | |
| 31. | KCNC1 | Potassium channel | Autosomal dominant | Progressive myoclinic epilepsy. | |
| 32. | GOSR2 | Transport across membrane | Autosomal recessive | Progressive myoclinic epilepsy. | |
| CERS1 | Enzyme | ||||
| LMNB2 | Protein for cytoskeleton | ||||
| KCTD7 | Not classified |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghosh, S.; Sinha, J.K.; Khan, T.; Devaraju, K.S.; Singh, P.; Vaibhav, K.; Gaur, P. Pharmacological and Therapeutic Approaches in the Treatment of Epilepsy. Biomedicines 2021, 9, 470. https://doi.org/10.3390/biomedicines9050470
Ghosh S, Sinha JK, Khan T, Devaraju KS, Singh P, Vaibhav K, Gaur P. Pharmacological and Therapeutic Approaches in the Treatment of Epilepsy. Biomedicines. 2021; 9(5):470. https://doi.org/10.3390/biomedicines9050470
Chicago/Turabian StyleGhosh, Shampa, Jitendra Kumar Sinha, Tarab Khan, Kuramkote Shivanna Devaraju, Prabhakar Singh, Kumar Vaibhav, and Pankaj Gaur. 2021. "Pharmacological and Therapeutic Approaches in the Treatment of Epilepsy" Biomedicines 9, no. 5: 470. https://doi.org/10.3390/biomedicines9050470
APA StyleGhosh, S., Sinha, J. K., Khan, T., Devaraju, K. S., Singh, P., Vaibhav, K., & Gaur, P. (2021). Pharmacological and Therapeutic Approaches in the Treatment of Epilepsy. Biomedicines, 9(5), 470. https://doi.org/10.3390/biomedicines9050470