
Enhanced Palmitate-Induced Interleukin-8 Formation in Human Macrophages by Insulin or Prostaglandin E2

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cultivation of Human Macrophage Cell Line THP-1
2.2. Real-Time RT-PCR Analysis
2.3. Determination of IL-8 and PGE2
2.4. Statistical Analysis
3. Results
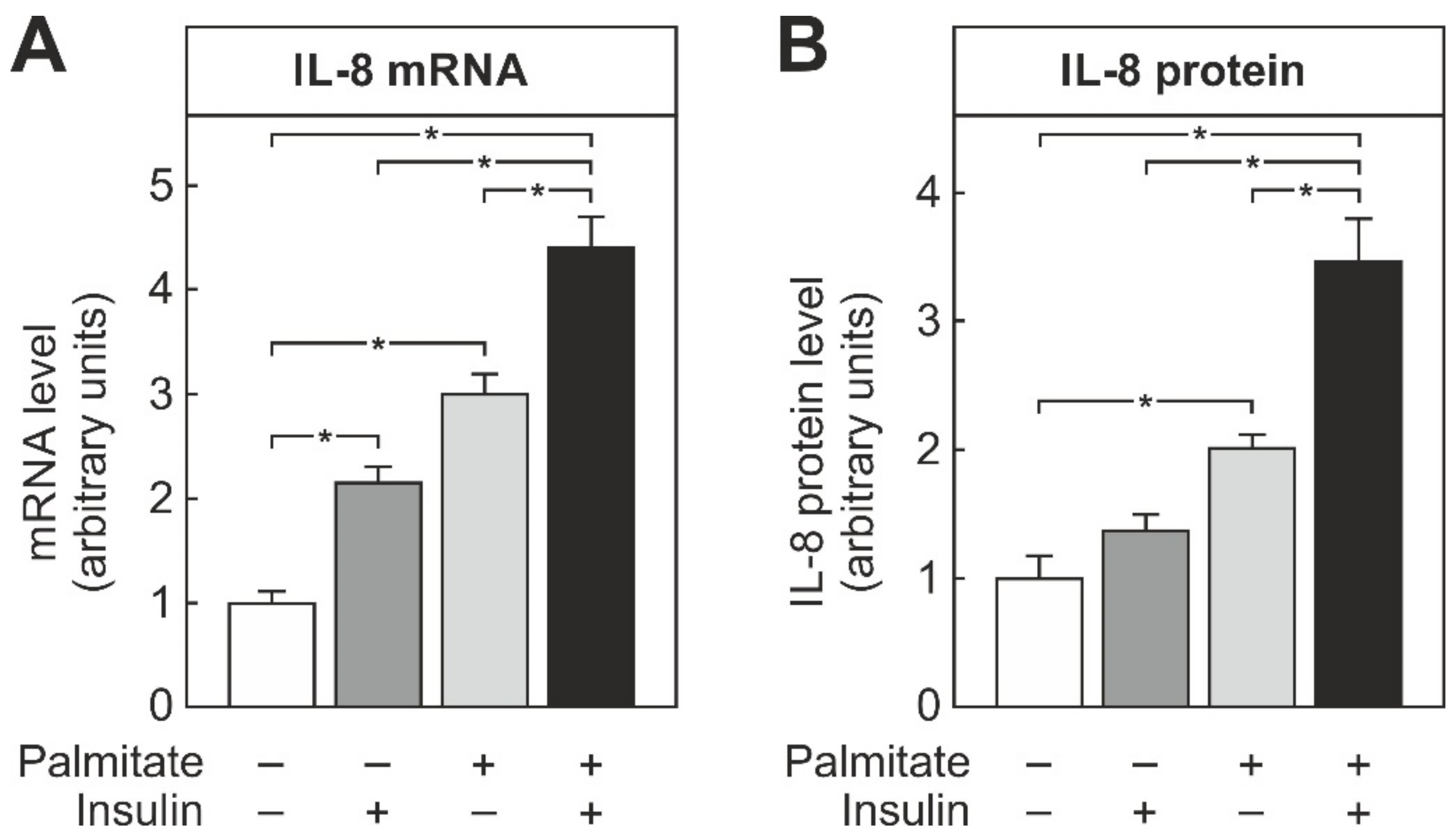
3.1. Insulin-Enhanced Palmitate-Dependent Induction of IL-8 in THP-1 Macrophages
3.2. Palmitate- and Insulin-Dependent Induction of PGE2 Synthesis in THP-1 Macrophages
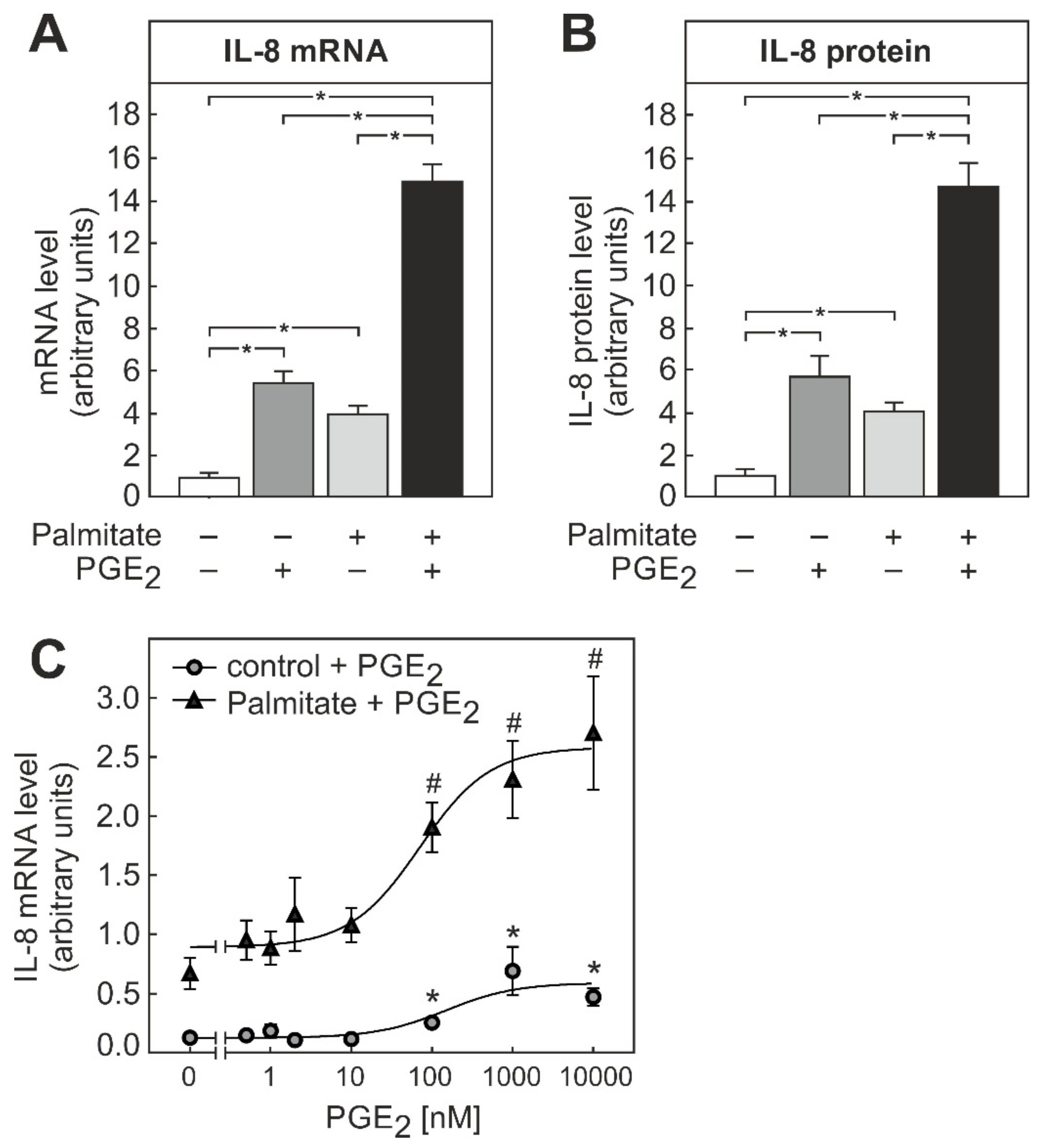
3.3. PGE2-Dependent Modulation of IL-8 Formation in THP-1 Macrophages
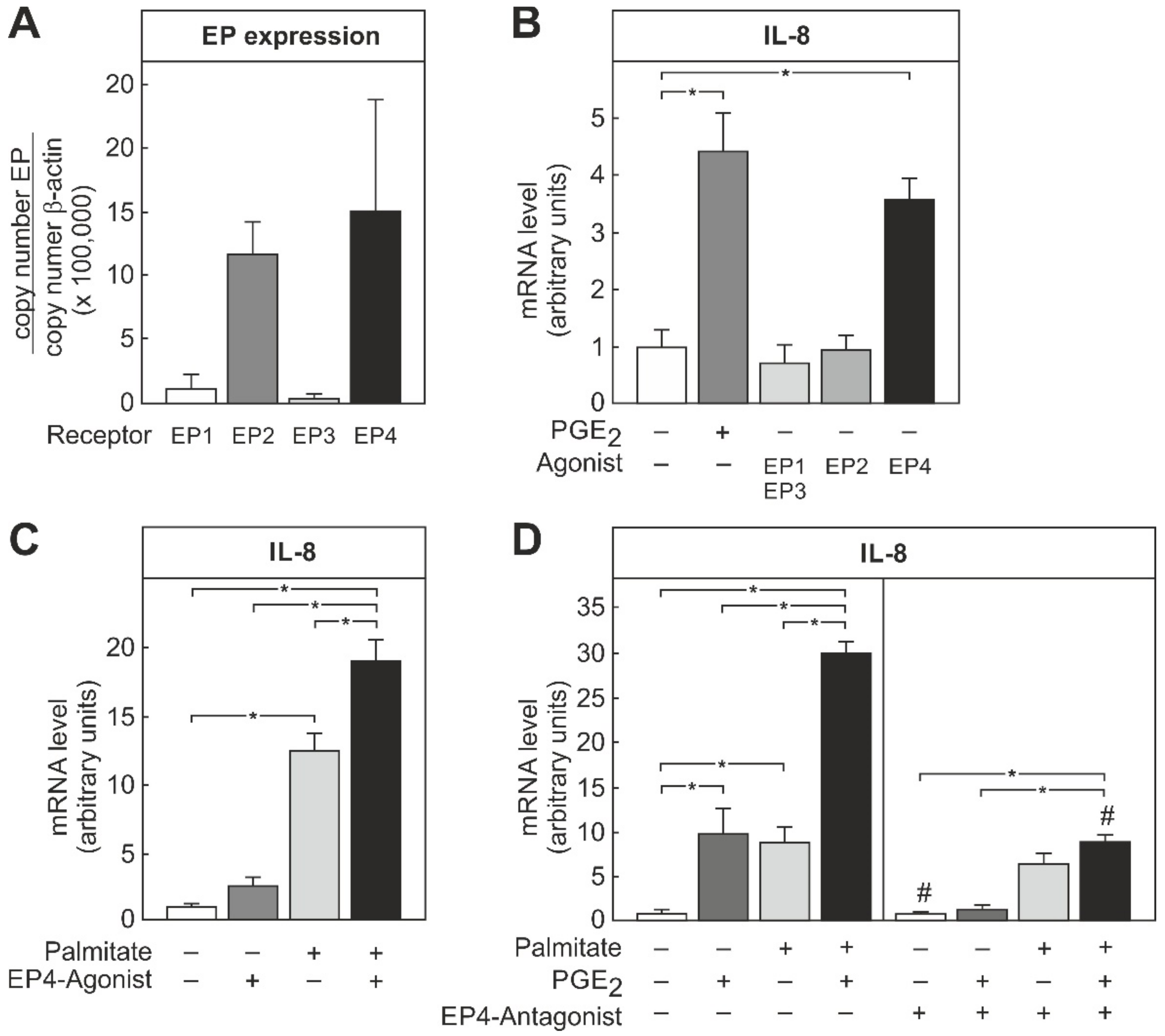
3.4. Involvement of EP4 Receptor on THP-1 Macrophages in PGE2-Dependent Modulation of IL-8 mRNA Induction
4. Discussion
4.1. Interaction of Different Factors to Enhance Inflammatory Response
4.2. Possible Role of PGE2 in an Autocrine or Paracrine Feed-Forward Loop
4.3. EP-Receptor Specificity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wellen, K.E.; Hotamisligil, G.S. Obesity-induced inflammatory changes in adipose tissue. J. Clin. Investig. 2003, 112, 1785–1788. [Google Scholar] [CrossRef] [PubMed]
- Moreira, A.P.B.; Texeira, T.F.S.; Ferreira, A.B.; Peluzio, M.d.C.G.; Alfenas, R.d.C.G. Influence of a high-fat diet on gut microbiota, intestinal permeability and metabolic endotoxaemia. Br. J. Nutr. 2012, 108, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Rohr, M.W.; Narasimhulu, C.A.; Rudeski-Rohr, T.A.; Parthasarathy, S. Negative Effects of a High-Fat Diet on Intestinal Permeability: A Review. Adv. Nutr. 2020, 11, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Sonnenburg, J.L.; Bäckhed, F. Diet-microbiota interactions as moderators of human metabolism. Nature 2016, 535, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, G.I.; Langley, K.G.; Berglund, N.A.; Kammoun, H.L.; Reibe, S.; Estevez, E.; Weir, J.; Mellett, N.A.; Pernes, G.; Conway, J.R.W.; et al. Evidence that TLR4 Is Not a Receptor for Saturated Fatty Acids but Mediates Lipid-Induced Inflammation by Reprogramming Macrophage Metabolism. Cell Metab. 2018, 27, 1096–1110.e5. [Google Scholar] [CrossRef]
- Pal, D.; Dasgupta, S.; Kundu, R.; Maitra, S.; Das, G.; Mukhopadhyay, S.; Ray, S.; Majumdar, S.S.; Bhattacharya, S. Fetuin-A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat. Med. 2012, 18, 1279–1285. [Google Scholar] [CrossRef]
- Ni, Y.; Zhao, L.; Yu, H.; Ma, X.; Bao, Y.; Rajani, C.; Loo, L.W.M.; Shvetsov, Y.B.; Yu, H.; Chen, T.; et al. Circulating Unsaturated Fatty Acids Delineate the Metabolic Status of Obese Individuals. EBioMedicine 2015, 2, 1513–1522. [Google Scholar] [CrossRef]
- De Almeida, I.T.; Cortez-Pinto, H.; Fidalgo, G.; Rodrigues, D.; Camilo, M.E. Plasma total and free fatty acids composition in human non-alcoholic steatohepatitis. Clin. Nutr. 2002, 21, 219–223. [Google Scholar] [CrossRef]
- Jensen, M.D.; Haymond, M.W.; Rizza, R.A.; Cryer, P.E.; Miles, J.M. Influence of body fat distribution on free fatty acid metabolism in obesity. J. Clin. Investig. 1989, 83, 1168–1173. [Google Scholar] [CrossRef]
- Klauder, J.; Henkel, J.; Vahrenbrink, M.; Wohlenberg, A.-S.; Camargo, R.G.; Püschel, G.P. Direct and indirect modulation of LPS-induced cytokine production by insulin in human macrophages. Cytokine 2020, 136, 155241. [Google Scholar] [CrossRef]
- Manowsky, J.; Camargo, R.G.; Kipp, A.P.; Henkel, J.; Püschel, G.P. Insulin-induced cytokine production in macrophages causes insulin resistance in hepatocytes. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E938–E946. [Google Scholar] [CrossRef] [PubMed]
- Mukaida, N.; Harada, A.; Matsushima, K. Interleukin-8 (IL-8) and monocyte chemotactic and activating factor (MCAF/MCP-1), chemokines essentially involved in inflammatory and immune reactions. Cytokine Growth Factor Rev. 1998, 9, 9–23. [Google Scholar] [CrossRef]
- Kobashi, C.; Asamizu, S.; Ishiki, M.; Iwata, M.; Usui, I.; Yamazaki, K.; Tobe, K.; Kobayashi, M.; Urakaze, M. Inhibitory effect of IL-8 on insulin action in human adipocytes via MAP kinase pathway. J. Inflamm. (Lond.) 2009, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.-S.; Park, H.-S.; Kawada, T.; Kim, J.-H.; Lim, D.; Hubbard, N.E.; Kwon, B.-S.; Erickson, K.L.; Yu, R. Circulating levels of MCP-1 and IL-8 are elevated in human obese subjects and associated with obesity-related parameters. Int. J. Obes. (Lond.) 2006, 30, 1347–1355. [Google Scholar] [CrossRef] [PubMed]
- Schell, M.; Chudoba, C.; Leboucher, A.; Alfine, E.; Flore, T.; Ritter, K.; Weiper, K.; Wernitz, A.; Henkel, J.; Kleinridders, A. Interplay of Dietary Fatty Acids and Cholesterol Impacts Brain Mitochondria and Insulin Action. Nutrients 2020, 12, 1518. [Google Scholar] [CrossRef]
- Henkel, J.; Alfine, E.; Saín, J.; Jöhrens, K.; Weber, D.; Castro, J.P.; König, J.; Stuhlmann, C.; Vahrenbrink, M.; Jonas, W.; et al. Soybean Oil-Derived Poly-Unsaturated Fatty Acids Enhance Liver Damage in NAFLD Induced by Dietary Cholesterol. Nutrients 2018, 10, 1326. [Google Scholar] [CrossRef]
- Caspar-Bauguil, S.; Kolditz, C.-I.; Lefort, C.; Vila, I.; Mouisel, E.; Beuzelin, D.; Tavernier, G.; Marques, M.-A.; Zakaroff-Girard, A.; Pecher, C.; et al. Fatty acids from fat cell lipolysis do not activate an inflammatory response but are stored as triacylglycerols in adipose tissue macrophages. Diabetologia 2015, 58, 2627–2636. [Google Scholar] [CrossRef]
- Namgaladze, D.; Lips, S.; Leiker, T.J.; Murphy, R.C.; Ekroos, K.; Ferreiros, N.; Geisslinger, G.; Brüne, B. Inhibition of macrophage fatty acid β-oxidation exacerbates palmitate-induced inflammatory and endoplasmic reticulum stress responses. Diabetologia 2014, 57, 1067–1077. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-E.; Kim, T.H.; Yi, S.-A.; Hwang, Y.C.; Hwang, W.S.; Choe, S.J.; Han, S.J.; Kim, H.J.; Kim, D.J.; Kang, Y.; et al. Capsaicin attenuates palmitate-induced expression of macrophage inflammatory protein 1 and interleukin 8 by increasing palmitate oxidation and reducing c-Jun activation in THP-1 (human acute monocytic leukemia cell) cells. Nutr. Res. 2011, 31, 468–478. [Google Scholar] [CrossRef] [PubMed]
- Håversen, L.; Danielsson, K.N.; Fogelstrand, L.; Wiklund, O. Induction of proinflammatory cytokines by long-chain saturated fatty acids in human macrophages. Atherosclerosis 2009, 202, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Hasan, A.; Akhter, N.; Al-Roub, A.; Thomas, R.; Kochumon, S.; Wilson, A.; Koshy, M.; Al-Ozairi, E.; Al-Mulla, F.; Ahmad, R. TNF-α in Combination with Palmitate Enhances IL-8 Production via The MyD88- Independent TLR4 Signaling Pathway: Potential Relevance to Metabolic Inflammation. Int. J. Mol. Sci. 2019, 20, 4112. [Google Scholar] [CrossRef] [PubMed]
- Cuschieri, J.; Bulger, E.; Grinsell, R.; Garcia, I.; Maier, R.V. Insulin regulates macrophage activation through activin A. Shock 2008, 29, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Verratti, V.; Brunetti, L.; Ferrante, C.; Orlando, G.; Recinella, L.; Chiavaroli, A.; Leone, S.; Wang, R.; Berardinelli, F. Physiological and pathological levels of prostaglandin E2 in renal parenchyma and neoplastic renal tissue. Prostaglandins Other Lipid Mediat. 2019, 141, 11–13. [Google Scholar] [CrossRef]
- Henkel, J.; Coleman, C.D.; Schraplau, A.; Jöhrens, K.; Weiss, T.S.; Jonas, W.; Schürmann, A.; Püschel, G.P. Augmented liver inflammation in a microsomal prostaglandin E synthase 1 (mPGES-1)-deficient diet-induced mouse NASH model. Sci. Rep. 2018, 8, 16127. [Google Scholar] [CrossRef]
- Henkel, J.; Neuschäfer-Rube, F.; Pathe-Neuschäfer-Rube, A.; Püschel, G.P. Aggravation by prostaglandin E2 of interleukin-6-dependent insulin resistance in hepatocytes. Hepatology 2009, 50, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.W.; Burke, P.A.; Drotar, M.E.; Chavali, S.R.; Forse, R.A. Effects of prostaglandin E2, cholera toxin and 8-bromo-cyclic AMP on lipopolysaccharide-induced gene expression of cytokines in human macrophages. Immunology 1995, 84, 446–452. [Google Scholar] [PubMed]
- Neuschäfer-Rube, F.; Pathe-Neuschäfer-Rube, A.; Hippenstiel, S.; Püschel, G.P. PGE2 enhanced TNFα-mediated IL-8 induction in monocytic cell lines and PBMC. Cytokine 2019, 113, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; García-Cardena, G.; Sukhova, G.K.; Comander, J.; Gimbrone, M.A.; Libby, P. Prostaglandin E2 suppresses chemokine production in human macrophages through the EP4 receptor. J. Biol. Chem. 2002, 277, 44147–44154. [Google Scholar] [CrossRef]
- Standiford, T.J.; Kunkel, S.L.; Rolfe, M.W.; Evanoff, H.L.; Allen, R.M.; Strieter, R.M. Regulation of human alveolar macrophage- and blood monocyte-derived interleukin-8 by prostaglandin E2 and dexamethasone. Am. J. Respir. Cell Mol. Biol. 1992, 6, 75–81. [Google Scholar] [CrossRef]
- Saleh, L.S.; Vanderheyden, C.; Frederickson, A.; Bryant, S.J. Prostaglandin E2 and Its Receptor EP2 Modulate Macrophage Activation and Fusion in Vitro. ACS Biomater. Sci. Eng. 2020, 6, 2668–2681. [Google Scholar] [CrossRef]
- Vallerie, S.N.; Kramer, F.; Barnhart, S.; Kanter, J.E.; Breyer, R.M.; Andreasson, K.I.; Bornfeldt, K.E. Myeloid Cell Prostaglandin E2 Receptor EP4 Modulates Cytokine Production but Not Atherogenesis in a Mouse Model of Type 1 Diabetes. PLoS ONE 2016, 11, e0158316. [Google Scholar] [CrossRef] [PubMed]
- Fennekohl, A.; Sugimoto, Y.; Segi, E.; Maruyama, T.; Ichikawa, A.; Püschel, G.P. Contribution of the two Gs-coupled PGE2-receptors EP2-receptor and EP4-receptor to the inhibition by PGE2 of the LPS-induced TNFalpha-formation in Kupffer cells from EP2-or EP4-receptor-deficient mice. Pivotal role for the EP4-receptor in wild type Kupffer cells. J. Hepatol. 2002, 36, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Makabe, T.; Koga, K.; Nagabukuro, H.; Asada, M.; Satake, E.; Taguchi, A.; Takeuchi, A.; Miyashita, M.; Harada, M.; Hirata, T.; et al. Use of selective PGE2 receptor antagonists on human endometriotic stromal cells and peritoneal macrophages. Mol. Hum. Reprod. 2021, 27. [Google Scholar] [CrossRef] [PubMed]
- McCoy, J.M.; Wicks, J.R.; Audoly, L.P. The role of prostaglandin E2 receptors in the pathogenesis of rheumatoid arthritis. J. Clin. Investig. 2002, 110, 651–658. [Google Scholar] [CrossRef]
- Neuschäfer-Rube, F.; Hermosilla, R.; Rehwald, M.; Rönnstrand, L.; Schülein, R.; Wernstedt, C.; Püschel, G.P. Identification of a Ser/Thr cluster in the C-terminal domain of the human prostaglandin receptor EP4 that is essential for agonist-induced beta-arrestin1 recruitment but differs from the apparent principal phosphorylation site. Biochem. J. 2004, 379, 573–585. [Google Scholar] [CrossRef] [PubMed]
- Katsuyama, M.; Ikegami, R.; Karahashi, H.; Amano, F.; Sugimoto, Y.; Ichikawa, A. Characterization of the LPS-stimulated expression of EP2 and EP4 prostaglandin E receptors in mouse macrophage-like cell line, J774.1. Biochem. Biophys. Res. Commun. 1998, 251, 727–731. [Google Scholar] [CrossRef]
- Samuchiwal, S.K.; Balestrieri, B.; Raff, H.; Boyce, J.A. Endogenous prostaglandin E2 amplifies IL-33 production by macrophages through an E prostanoid (EP)2/EP4-cAMP-EPAC-dependent pathway. J. Biol. Chem. 2017, 292, 8195–8206. [Google Scholar] [CrossRef] [PubMed]
- Zasłona, Z.; Pålsson-McDermott, E.M.; Menon, D.; Haneklaus, M.; Flis, E.; Prendeville, H.; Corcoran, S.E.; Peters-Golden, M.; O’Neill, L.A.J. The Induction of Pro-IL-1β by Lipopolysaccharide Requires Endogenous Prostaglandin E2 Production. J. Immunol. 2017, 198, 3558–3564. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Henkel, J.; Klauder, J.; Statz, M.; Wohlenberg, A.-S.; Kuipers, S.; Vahrenbrink, M.; Püschel, G.P. Enhanced Palmitate-Induced Interleukin-8 Formation in Human Macrophages by Insulin or Prostaglandin E2. Biomedicines 2021, 9, 449. https://doi.org/10.3390/biomedicines9050449
Henkel J, Klauder J, Statz M, Wohlenberg A-S, Kuipers S, Vahrenbrink M, Püschel GP. Enhanced Palmitate-Induced Interleukin-8 Formation in Human Macrophages by Insulin or Prostaglandin E2. Biomedicines. 2021; 9(5):449. https://doi.org/10.3390/biomedicines9050449
Chicago/Turabian StyleHenkel, Janin, Julia Klauder, Meike Statz, Anne-Sophie Wohlenberg, Sonja Kuipers, Madita Vahrenbrink, and Gerhard Paul Püschel. 2021. "Enhanced Palmitate-Induced Interleukin-8 Formation in Human Macrophages by Insulin or Prostaglandin E2" Biomedicines 9, no. 5: 449. https://doi.org/10.3390/biomedicines9050449
APA StyleHenkel, J., Klauder, J., Statz, M., Wohlenberg, A.-S., Kuipers, S., Vahrenbrink, M., & Püschel, G. P. (2021). Enhanced Palmitate-Induced Interleukin-8 Formation in Human Macrophages by Insulin or Prostaglandin E2. Biomedicines, 9(5), 449. https://doi.org/10.3390/biomedicines9050449