
Bromodomain and Extraterminal Protein Inhibitor, Apabetalone (RVX-208), Reduces ACE2 Expression and Attenuates SARS-Cov-2 Infection In Vitro

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemical Compounds
2.2. Cell Culture
2.3. Real-Time PCR
2.4. Immunoblot Analysis
2.5. Flow Cytometry
2.6. SARS-CoV-2 Spike Protein Binding
2.7. MTS Proliferation Assay
2.8. SARS-CoV-2 Infection of Calu-3 cells
2.9. Statistical Analysis
3. Results
3.1. Apabetalone Downregulates ACE2 Gene Expression in Multiple Cell Types
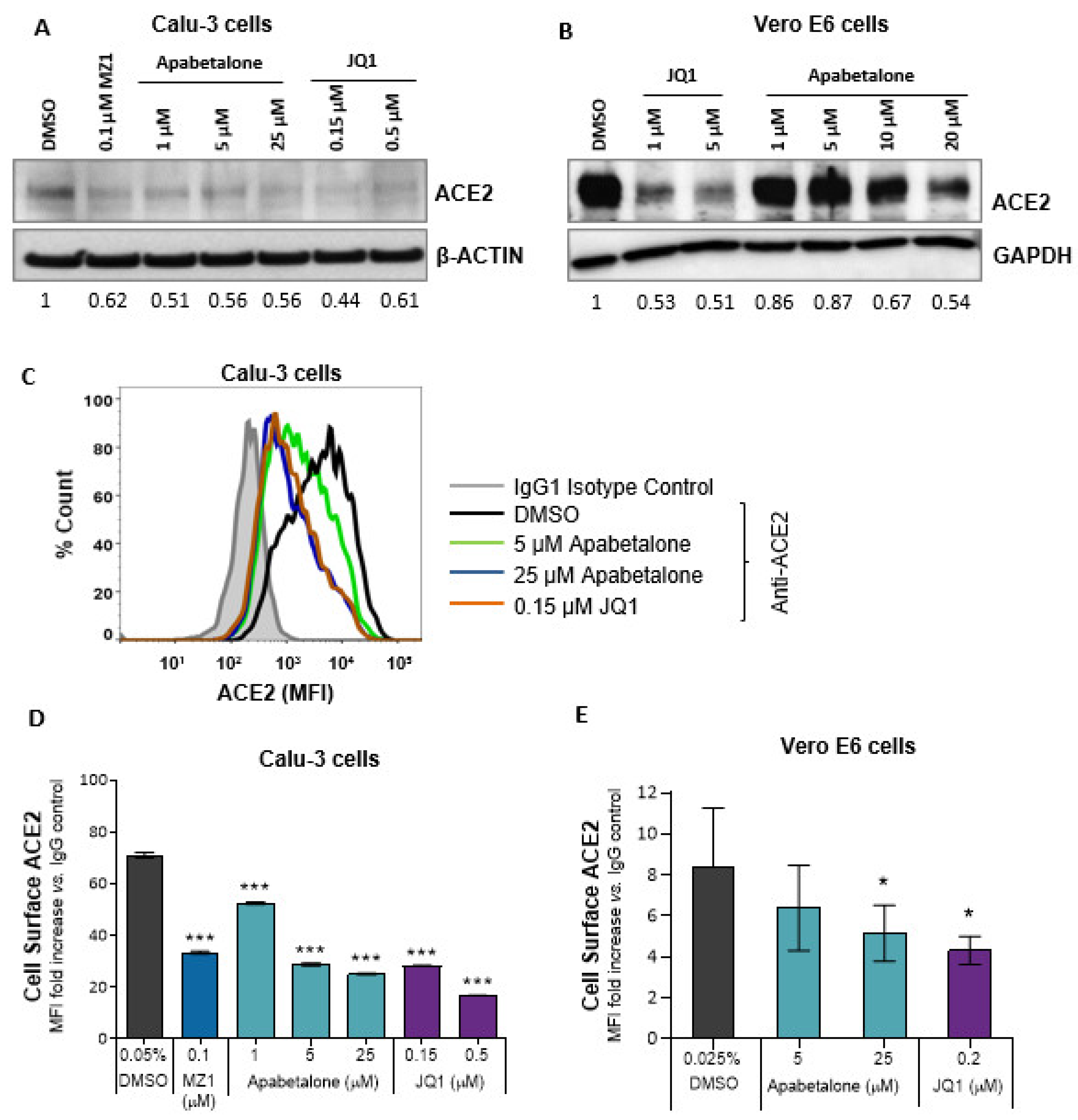
3.2. Apabetalone Reduces ACE2 Protein Levels in Calu-3 and Vero E6
3.3. Apabetalone Downregulates DPP4 (CD26) Expression
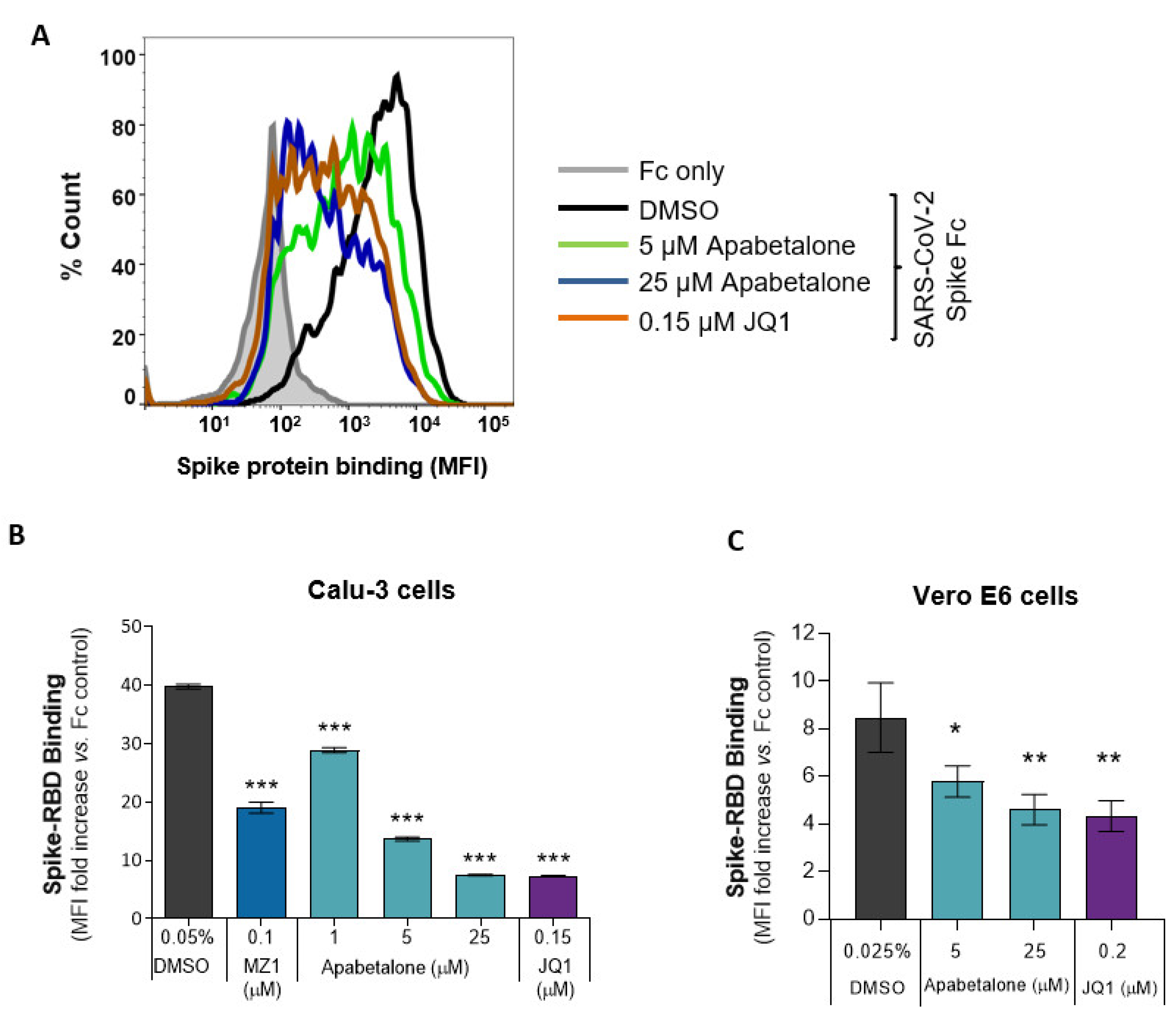
3.4. Apabetalone Attenuates SARS-CoV-2 Spike Protein Binding
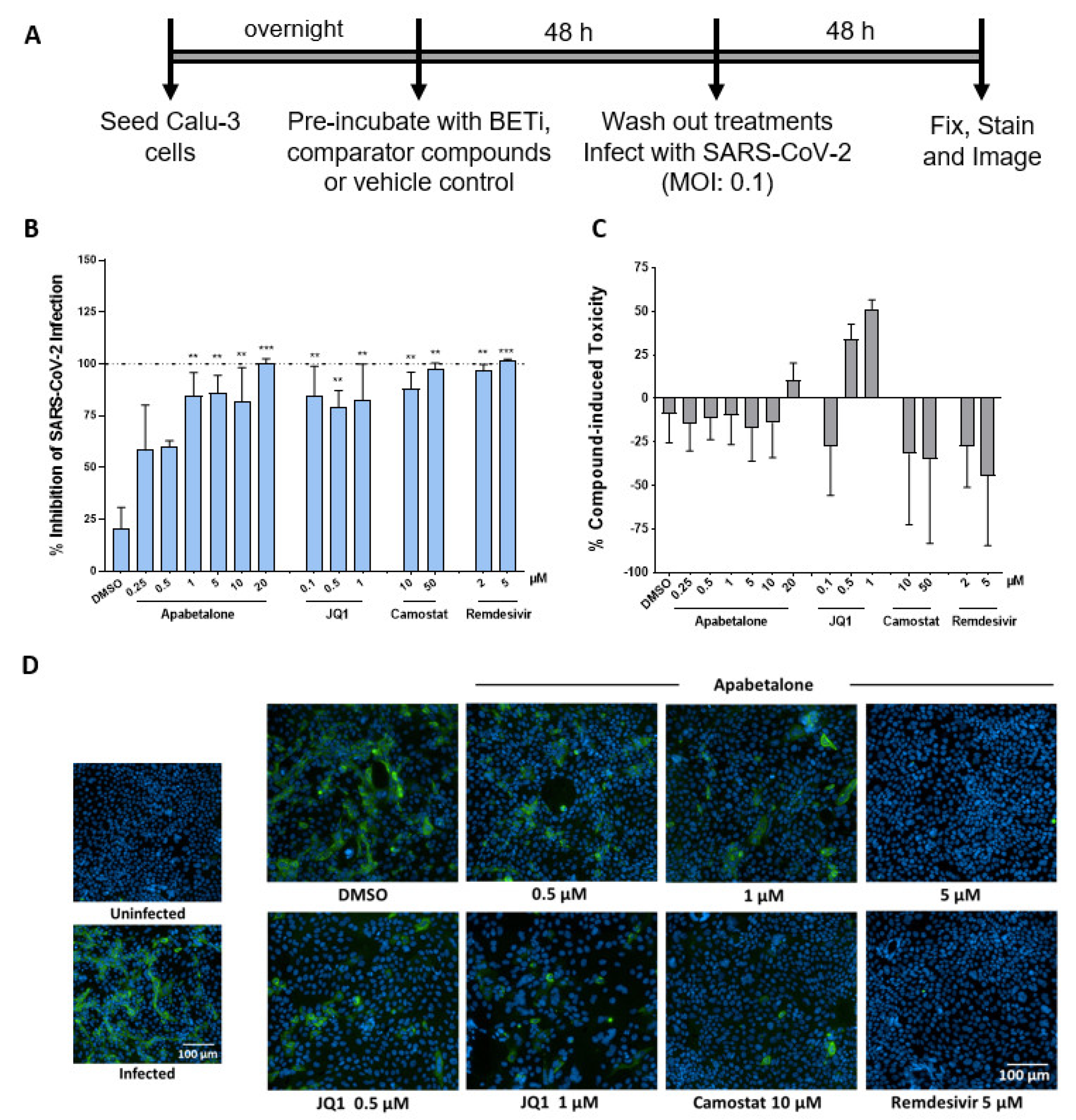
3.5. Apabetalone Abrogates SARS-CoV-2 Infection
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Soy, M.; Keser, G.; Atagündüz, P.; Tabak, F.; Atagündüz, I.; Kayhan, S. Cytokine storm in COVID-19: Pathogenesis and overview of anti-inflammatory agents used in treatment. Clin. Rheumatol. 2020, 39, 2085–2094. [Google Scholar] [CrossRef]
- The World Health Organization: Statement on the second meeting of the International Health Regulations (2005) Emergency Committee regarding the outbreak of novel coronavirus (2019-nCoV). Available online: https://www.who.int/news/item/30-01-2020-statement-on-the-second-meeting-of-the-international-health-regulations-(2005)-emergency-committee-regarding-the-outbreak-of-novel-coronavirus-(2019-ncov) (accessed on 17 April 2021).
- Stawicki, S.P.; Jeanmonod, R.; Miller, A.C.; Paladino, L.; Gaieski, D.F.; Yaffee, A.Q.; De Wulf, A.; Grover, J.; Papadimos, T.J.; Bloem, C.; et al. The 2019–2020 novel coronavirus (severe acute respiratory syndrome coronavirus 2) pandemic: A joint american college of academic international medicine-world academic council of emergency medicine multidisciplinary COVID-19 working group consensus paper. J. Glob. Infect. Dis. 2020, 12, 47–93. [Google Scholar] [CrossRef]
- Dong, Y.; Mo, X.; Hu, Y.; Qi, X.; Jiang, F.; Jiang, Z.; Tong, S. Epidemiology of COVID-19 among children in China. Pediatrics 2020, 145, e20200702. [Google Scholar] [CrossRef]
- Barker-Davies, R.M.; O’Sullivan, O.; Senaratne, K.P.P.; Baker, P.; Cranley, M.; Dharm-Datta, S.; Ellis, H.; Goodall, D.; Gough, M.; Lewis, S.; et al. The Stanford Hall consensus statement for post-COVID-19 rehabilitation. Br. J. Sports Med. 2020, 54, 949–959. [Google Scholar] [CrossRef]
- Del Rio, C.; Collins, L.F.; Malani, P. Long-term health consequences of COVID. J. Am. Med. Assoc. 2020, 324, 1723. [Google Scholar] [CrossRef]
- Greenhalgh, T.; Knight, M.; A’Court, C.; Buxton, M.; Husain, L. Management of post-acute covid-19 in primary care. Brit. Med. J. 2020, 370, m3026. [Google Scholar] [CrossRef]
- Ragab, D.; Eldin, H.S.; Taeimah, M.; Khattab, R.; Salem, R. The COVID-19 cytokine storm; What we know so far. Front. Immunol. 2020, 11, 1446. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.Y.; Uspal, W.E.; Wei, T. Airborne transmission of COVID-19: Aerosol dispersion, lung deposition, and virus-receptor interactions. ACS Nano 2020, 14, 16502–16524. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed]
- V’Kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV. Nat. Rev. Genet. 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Li, H.; Liu, L.; Zhang, D.; Xu, J.; Dai, H.; Tang, N.; Su, X.; Cao, B. SARS-CoV-2 and viral sepsis: Observations and hypotheses. Lancet 2020, 395, 1517–1520. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, S.; Liu, H.; Li, W.; Lin, F.; Jiang, L.; Li, X.; Xu, P.; Zhang, L.; Zhao, L.; et al. SARS-CoV-2 infection of the liver directly contributes to hepatic impairment in patients with COVID. J. Hepatol. 2020, 73, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.I.; Feng, G.; Liu, W.; Targher, G.; Byrne, C.D.; Zheng, M. Extrapulmonary complications of COVID-19: A multisystem disease? J. Med. Virol. 2021, 93, 323–335. [Google Scholar] [CrossRef]
- Alsoussi, W.B.; Turner, J.S.; Case, J.B.; Zhao, H.; Schmitz, A.J.; Zhou, J.Q.; Chen, R.E.; Lei, T.; Rizk, A.A.; McIntire, K.M.; et al. A potently neutralizing antibody protects mice against SARS-CoV-2 infection. J. Immunol. 2020, 205, 915–922. [Google Scholar] [CrossRef]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkrüys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Del Pozo, C.H.; Prosper, F.; et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE. Cell 2020, 181, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Tortorici, M.A.; Beltramello, M.; Lempp, F.A.; Pinto, D.; Dang, H.V.; Rosen, L.E.; McCallum, M.; Bowen, J.; Minola, A.; Jaconi, S.; et al. Ultrapotent human antibodies protect against SARS-CoV-2 challenge via multiple mechanisms. Science 2020, 370, 950–957. [Google Scholar] [CrossRef]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Marc, G.P.; Moreira, E.D.; Zerbini, C.; et al. Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef] [PubMed]
- Walensky, R.P.; Walke, H.T.; Fauci, A.S. SARS-CoV-2 variants of concern in the United States—Challenges and opportunities. J. Am. Med. Assoc. 2021, 325, 1037. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Wang, X.-M.; Mannan, R.; Pitchiaya, S.; Zhang, Y.; Wotring, J.W.; Xiao, L.; Robinson, D.R.; Wu, Y.-M.; Tien, J.C.-Y.; et al. Targeting transcriptional regulation of SARS-CoV-2 entry factors ACE2 and TMPRSS. Proc. Natl. Acad. Sci. USA 2021, 118, e2021450118. [Google Scholar] [CrossRef]
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.-W.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of inflammation by a synthetic histone mimic. Nat. Cell Biol. 2010, 468, 1119–1123. [Google Scholar] [CrossRef] [PubMed]
- Gilan, O.; Rioja, I.; Knezevic, K.; Bell, M.J.; Yeung, M.M.; Harker, N.R.; Lam, E.Y.N.; Chung, C.-W.; Bamborough, P.; Petretich, M.; et al. Selective targeting of BD1 and BD2 of the BET proteins in cancer and immunoinflammation. Science 2020, 368, 387–394. [Google Scholar] [CrossRef]
- Nicholls, S.J.; The BETonMACE Investigators; Schwartz, G.G.; Buhr, K.A.; Ginsberg, H.N.; Johansson, J.O.; Kalantar-Zadeh, K.; Kulikowski, E.; Toth, P.P.; Wong, N.; et al. Apabetalone and hospitalization for heart failure in patients following an acute coronary syndrome: A prespecified analysis of the BETonMACE study. Cardiovasc. Diabetol. 2021, 20, 1–9. [Google Scholar] [CrossRef]
- Ray, K.K.; Nicholls, S.J.; Buhr, K.A.; Ginsberg, H.N.; Johansson, J.O.; Kalantar-Zadeh, K.; Kulikowski, E.; Toth, P.P.; Wong, N.; Sweeney, M.; et al. Effect of Apabetalone added to standard therapy on major adverse cardiovascular events in patients with recent acute coronary syndrome and type 2 diabetes. J. Am. Med. Assoc. 2020, 323, 1565–1573. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Z.; Yang, L.; Lian, X.; Xie, Y.; Li, S.; Xin, S.; Cao, P.; Lu, J. The MERS-CoV receptor DPP4 as a candidate binding target of the SARS-CoV-2 Spike. iScience 2020, 23, 101160. [Google Scholar] [CrossRef]
- Solerte, S.B.; Di Sabatino, A.; Galli, M.; Fiorina, P. Dipeptidyl peptidase-4 (DPP4) inhibition in COVID-19. Acta Diabetol. 2020, 57, 779–783. [Google Scholar] [CrossRef]
- Gilham, D.; Wasiak, S.; Tsujikawa, L.M.; Halliday, C.; Norek, K.; Patel, R.G.; Kulikowski, E.; Johansson, J.; Sweeney, M.; Wong, N.C. RVX-208, a BET-inhibitor for treating atherosclerotic cardiovascular disease, raises ApoA-I/HDL and represses pathways that contribute to cardiovascular disease. Atherosclerosis 2016, 247, 48–57. [Google Scholar] [CrossRef]
- Gilham, D.; Tsujikawa, L.M.; Sarsons, C.D.; Halliday, C.; Wasiak, S.; Stotz, S.C.; Jahagirdar, R.; Sweeney, M.; Johansson, J.O.; Wong, N.C.; et al. Apabetalone downregulates factors and pathways associated with vascular calcification. Atherosclerosis 2019, 280, 75–84. [Google Scholar] [CrossRef]
- Tsujikawa, L.M.; Fu, L.; Das, S.; Halliday, C.; Rakai, B.D.; Stotz, S.C.; Sarsons, C.D.; Gilham, D.; Daze, E.; Wasiak, S.; et al. Apabetalone (RVX-208) reduces vascular inflammation in vitro and in CVD patients by a BET-dependent epigenetic mechanism. Clin. Epigenetics 2019, 11, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Hamming, N.V.; Timens, W.; Bulthuis, M.L.C.; Lely, A.T.; Navis, G.J.; Van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef]
- Hikmet, F.; Méar, L.; Edvinsson, Å.; Micke, P.; Uhlén, M.; Lindskog, C. The protein expression profile of ACE2 in human tissues. Mol. Syst. Biol. 2020, 16, e9610. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, M.C.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Zengerle, M.; Chan, K.-H.; Ciulli, A. Selective small molecule induced degradation of the BET bromodomain protein BRD. ACS Chem. Biol. 2015, 10, 1770–1777. [Google Scholar] [CrossRef]
- Kerscher, B.; Barlow, J.L.; Batika, M.; Rana, B.M.; Jolin, H.E.; Gogoi, M.; Bartholomew, M.A.; Jhamb, D.; Pandey, A.; Tough, D.F.; et al. BET bromodomain inhibitor IBET151 impedes human ILC2 activation and prevents experimental allergic lung inflammation. Front. Immunol. 2019, 10, 678. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Kawase, M.; Shirato, K.; Van Der Hoek, L.; Taguchi, F.; Matsuyama, S. simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome Coronavirus entry. J. Virol. 2012, 86, 6537–6545. [Google Scholar] [CrossRef]
- Fajgenbaum, D.C.; June, C.H. Cytokine storm. N. Engl. J. Med. 2020, 383, 2255–2273. [Google Scholar] [CrossRef] [PubMed]
- Jahagirdar, R.; Zhang, H.; Azhar, S.; Tobin, J.; Attwell, S.; Yu, R.; Wu, J.; McLure, K.G.; Hansen, H.C.; Wagner, G.S.; et al. A novel BET bromodomain inhibitor, RVX-208, shows reduction of atherosclerosis in hyperlipidemic ApoE deficient mice. Atherosclerosis 2014, 236, 91–100. [Google Scholar] [CrossRef]
- Wasiak, S.; Dzobo, K.E.; Rakai, B.D.; Kaiser, Y.; Versloot, M.; Bahjat, M.; Stotz, S.C.; Fu, L.; Sweeney, M.; Johansson, J.O.; et al. BET protein inhibitor apabetalone (RVX-208) suppresses pro-inflammatory hyper-activation of monocytes from patients with cardiovascular disease and type 2 diabetes. Clin. Epigenetics 2020, 12, 1–19. [Google Scholar] [CrossRef]
- Wasiak, S.; Gilham, D.; Daze, E.; Tsujikawa, L.M.; Halliday, C.; Stotz, S.C.; Rakai, B.D.; Fu, L.; Jahagirdar, R.; Sweeney, M.; et al. Epigenetic modulation by apabetalone counters cytokine-driven acute phase response in vitro, in mice and in patients with cardiovascular disease. Cardiovasc. Ther. 2020, 2020, 1–12. [Google Scholar] [CrossRef]
- Wasiak, S.; Gilham, D.; Tsujikawa, L.M.; Halliday, C.; Calosing, C.; Jahagirdar, R.; Johansson, J.; Sweeney, M.; Wong, N.C.; Kulikowski, E. Downregulation of the complement cascade in vitro, in mice and in patients with cardiovascular disease by the BET protein inhibitor apabetalone (RVX-208). J. Cardiovasc. Transl. Res. 2017, 10, 337–347. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.-P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Müller, S.; Pawson, T.; et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef] [PubMed]
- Gacias, M.; Gerona-Navarro, G.; Plotnikov, A.N.; Zhang, G.; Zeng, L.; Kaur, J.; Moy, G.; Rusinova, E.; Rodriguez, Y.; Matikainen, B.; et al. Selective chemical modulation of gene transcription favors oligodendrocyte lineage progression. Chem. Biol. 2014, 21, 841–854. [Google Scholar] [CrossRef] [PubMed]
- Picaud, S.; Wells, C.; Felletar, I.; Brotherton, D.; Martin, S.; Savitsky, P.; Diez-Dacal, B.; Philpott, M.; Bountra, C.; Lingard, H.; et al. RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proc. Natl. Acad. Sci. USA 2013, 110, 19754–19759. [Google Scholar] [CrossRef] [PubMed]
- McLure, K.G.; Gesner, E.M.; Tsujikawa, L.; Kharenko, O.A.; Attwell, S.; Campeau, E.; Wasiak, S.; Stein, A.; White, A.; Fontano, E.; et al. RVX-208, an inducer of ApoA-I in humans, is a bet bromodomain antagonist. PLoS ONE 2013, 8, e83190. [Google Scholar] [CrossRef] [PubMed]
- Kulikowski, E.; Halliday, C.; Johansson, J.; Sweeney, M.; Lebioda, K.; Wong, N.; Haarhaus, M.; Brandenburg, V.; Beddhu, S.; Tonelli, M.; et al. Apabetalone mediated epigenetic modulation is associated with favorable kidney function and alkaline phosphatase profile in patients with chronic kidney disease. Kidney Blood Press. Res. 2018, 43, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Haarhaus, M.; Gilham, D.; Kulikowski, E.; Magnusson, P.; Kalantar-Zadeh, K. Pharmacologic epigenetic modulators of alkaline phosphatase in chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2020, 29, 4–15. [Google Scholar] [CrossRef]
- Dyer, O. Covid-19: Remdesivir has little or no impact on survival, WHO trial shows. Br. Med. J. 2020, 371, m4057. [Google Scholar] [CrossRef]
- Mills, R.J.; Humphrey, S.J.; Fortuna, P.R.; Lor, M.; Foster, S.R.; Quaife-Ryan, G.A.; Johnston, R.L.; Dumenil, T.; Bishop, C.; Ruraraju, R.; et al. BET inhibition blocks inflammation-induced cardiac dysfunction and SARS-CoV-2 infection. Cell 2021, 184, 1–16. [Google Scholar] [CrossRef]
- Bian, J.; Li, Z. Angiotensin-converting enzyme 2 (ACE2): SARS-CoV-2 receptor and RAS modulator. Acta Pharm. Sin. B 2021, 11, 1–12. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Gordon, A.; Johansson, J.; Wolski, K.; Ballantyne, C.M.; Kastelein, J.J.; Taylor, A.; Borgman, M.; Nissen, S.E. Efficacy and safety of a novel oral inducer of apolipoprotein a-i synthesis in statin-treated patients with stable coronary artery disease. J. Am. Coll. Cardiol. 2011, 57, 1111–1119. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, S.J.; Puri, R.; Wolski, K.; Ballantyne, C.M.; Barter, P.J.; Brewer, H.B.; Kastelein, J.J.P.; Hu, B.; Uno, K.; Kataoka, Y.; et al. Effect of the BET protein inhibitor, RVX-208, on progression of coronary atherosclerosis: Results of the phase 2b, randomized, double-blind, multicenter, ASSURE trial. Am. J. Cardiovasc. Drugs 2015, 16, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, S.J.; Ray, K.K.; Johansson, J.O.; Gordon, A.; Sweeney, M.; Halliday, C.; Kulikowski, E.; Wong, N.; Kim, S.W.; Schwartz, G.G. Selective BET protein inhibition with apabetalone and cardiovascular events: A pooled analysis of trials in patients with coronary artery disease. Am. J. Cardiovasc. Drugs 2018, 18, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Thornberry, N.A.; Gallwitz, B. Mechanism of action of inhibitors of dipeptidyl-peptidase-4 (DPP-4). Best Pract. Res. Clin. Endocrinol. 2009, 23, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Klemann, C.; Wagner, L.; Stephan, M.; Von Hörsten, S. Cut to the chase: A review of CD26/dipeptidyl peptidase-4’s (DPP4) entanglement in the immune system. Clin. Exp. Immunol. 2016, 185, 1–21. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gilham, D.; Smith, A.L.; Fu, L.; Moore, D.Y.; Muralidharan, A.; Reid, S.P.M.; Stotz, S.C.; Johansson, J.O.; Sweeney, M.; Wong, N.C.W.; et al. Bromodomain and Extraterminal Protein Inhibitor, Apabetalone (RVX-208), Reduces ACE2 Expression and Attenuates SARS-Cov-2 Infection In Vitro. Biomedicines 2021, 9, 437. https://doi.org/10.3390/biomedicines9040437
Gilham D, Smith AL, Fu L, Moore DY, Muralidharan A, Reid SPM, Stotz SC, Johansson JO, Sweeney M, Wong NCW, et al. Bromodomain and Extraterminal Protein Inhibitor, Apabetalone (RVX-208), Reduces ACE2 Expression and Attenuates SARS-Cov-2 Infection In Vitro. Biomedicines. 2021; 9(4):437. https://doi.org/10.3390/biomedicines9040437
Chicago/Turabian StyleGilham, Dean, Audrey L. Smith, Li Fu, Dalia Y. Moore, Abenaya Muralidharan, St. Patrick M. Reid, Stephanie C. Stotz, Jan O. Johansson, Michael Sweeney, Norman C. W. Wong, and et al. 2021. "Bromodomain and Extraterminal Protein Inhibitor, Apabetalone (RVX-208), Reduces ACE2 Expression and Attenuates SARS-Cov-2 Infection In Vitro" Biomedicines 9, no. 4: 437. https://doi.org/10.3390/biomedicines9040437
APA StyleGilham, D., Smith, A. L., Fu, L., Moore, D. Y., Muralidharan, A., Reid, S. P. M., Stotz, S. C., Johansson, J. O., Sweeney, M., Wong, N. C. W., Kulikowski, E., & El-Gamal, D. (2021). Bromodomain and Extraterminal Protein Inhibitor, Apabetalone (RVX-208), Reduces ACE2 Expression and Attenuates SARS-Cov-2 Infection In Vitro. Biomedicines, 9(4), 437. https://doi.org/10.3390/biomedicines9040437