
The Challenges and Strategies of Antisense Oligonucleotide Drug Delivery

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. ASO Mechanisms of Action and FDA-Approved Drugs
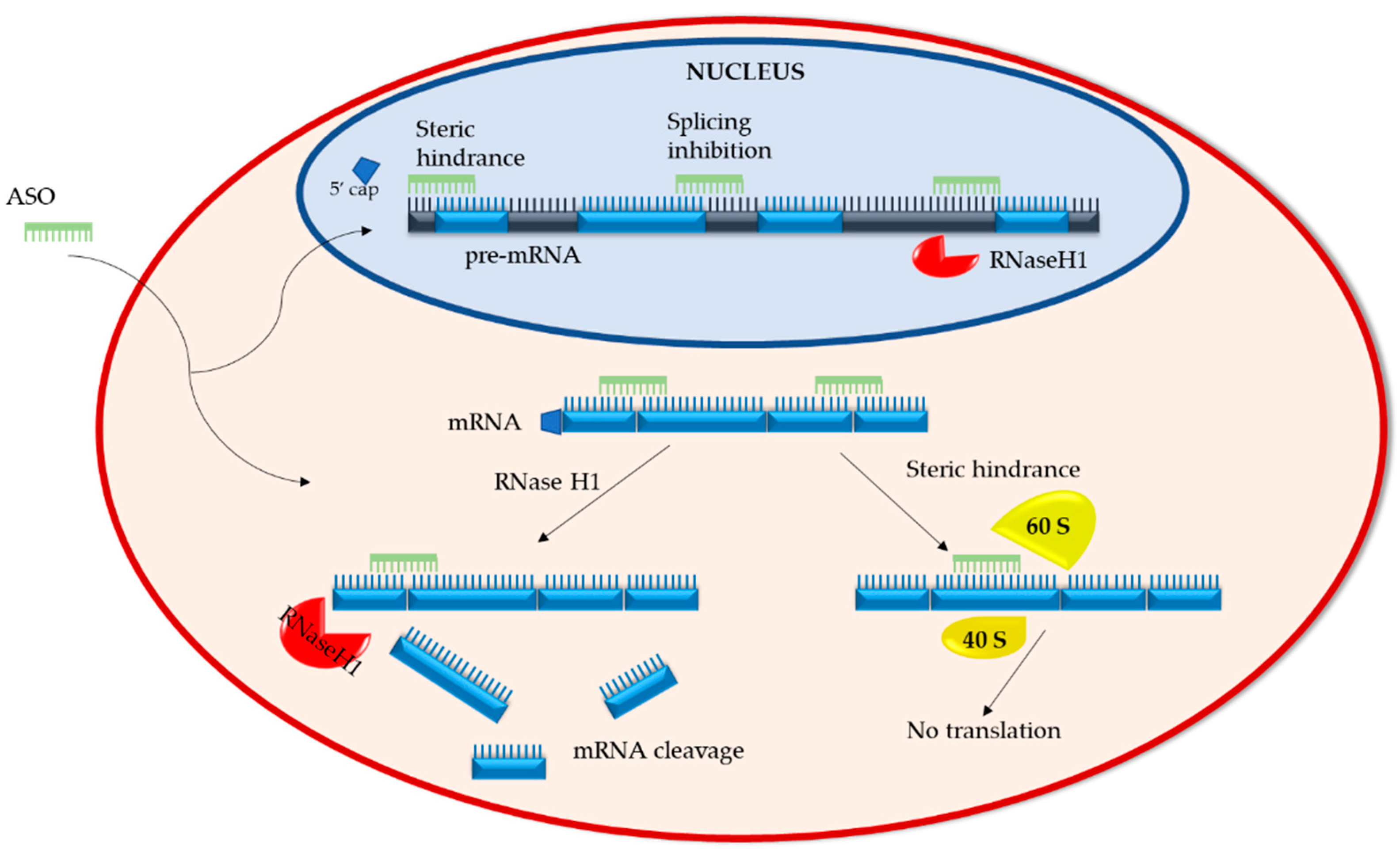
2.1. Mechanisms of Action
2.1.1. RNA Degradation
2.1.2. RNA Steric Hindrance
2.2. FDA-Approved ASO Drugs
2.2.1. RNaseH1-Dependent ASOs
2.2.2. Splice-Altering ASOs
3. Strategies to Optimize ASO Drug Delivery
3.1. Chemical Modifications of ASOs
3.2. Bioconjugation and Targeting
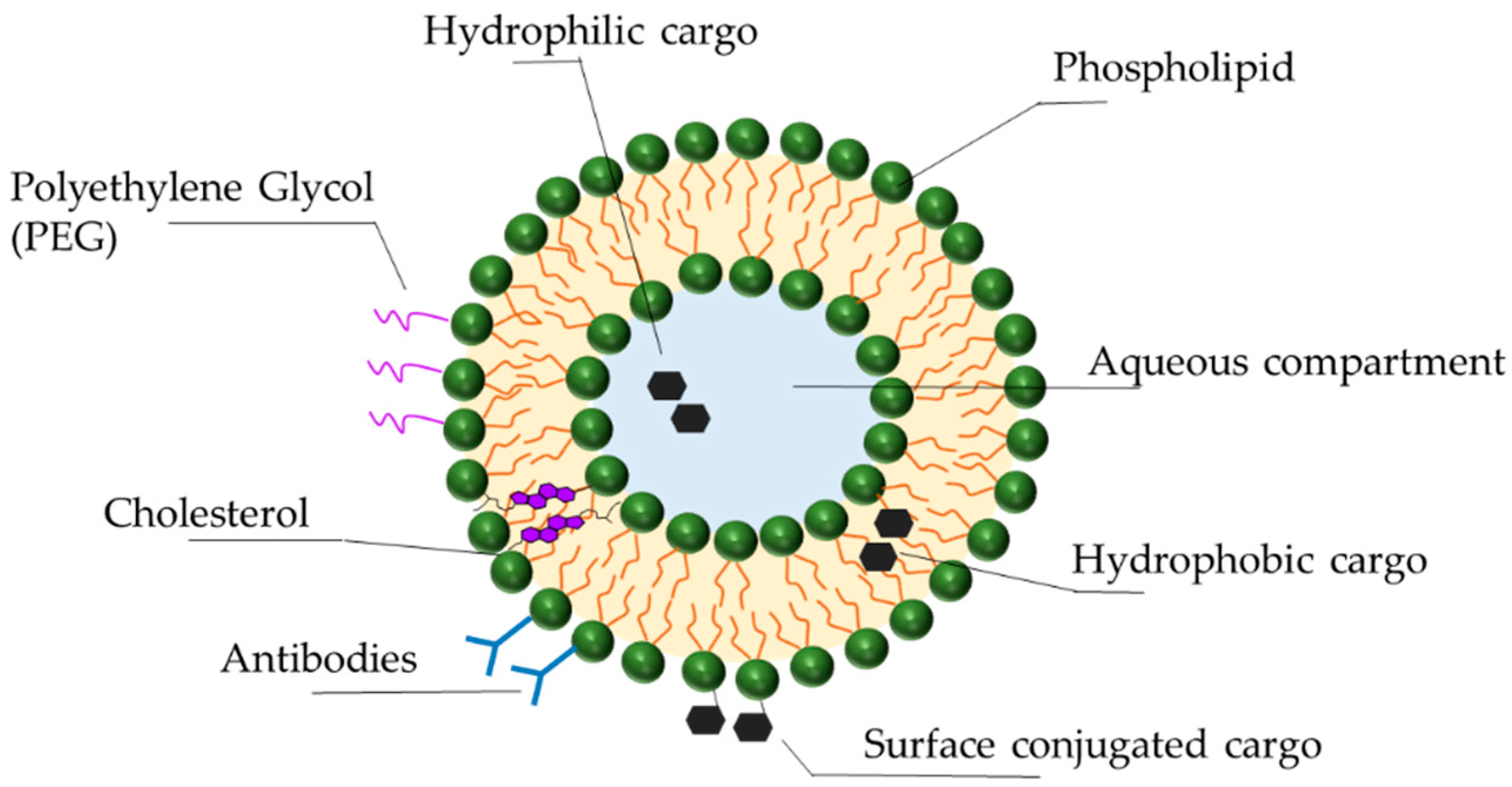
3.3. Delivery Vehicles
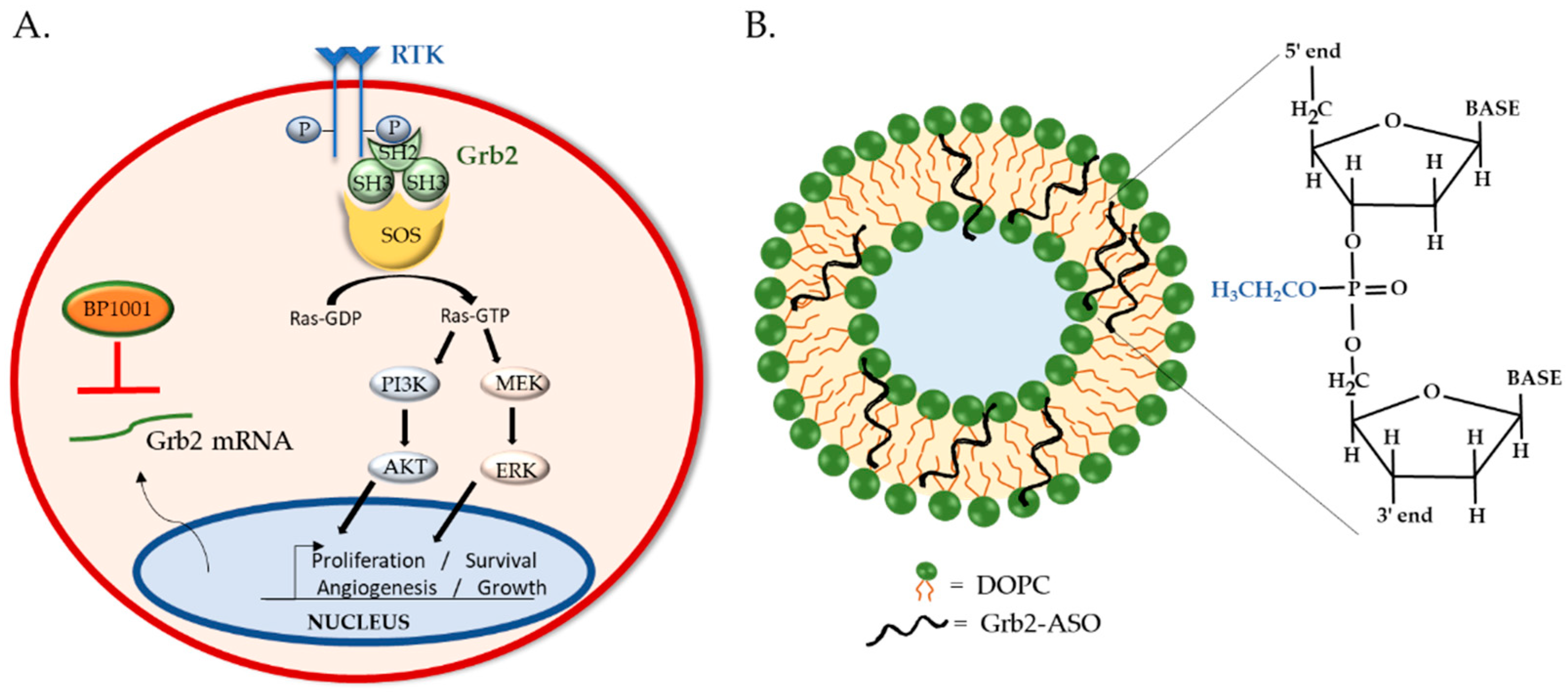
4. BP1001: Liposomal P-Ethoxy-Grb2 ASO
4.1. BP1001 Pre-Clinical Studies
4.2. BP1001 Clinical Trials
5. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rodriguez-Esteban, R.; Jiang, X. Differential gene expression in disease: A comparison between high-throughput studies and the literature. BMC Med. Genom. 2017, 10, 59. [Google Scholar] [CrossRef]
- Bajan, S.; Hutvagner, G. RNA-Based Therapeutics: From Antisense Oligonucleotides to miRNAs. Cells 2020, 9, 137. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, M.L.; Zamecnik, P.C. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Panagiotopoulos, A.-L.; Karguth, N.; Pavlou, M.; Böhm, S.; Gasparoni, G.; Walter, J.; Graf, A.; Blum, H.; Biel, M.; Riedmayr, L.M.; et al. Antisense Oligonucleotide- and CRISPR-Cas9-Mediated Rescue of mRNA Splicing for a Deep Intronic CLRN1 Mutation. Mol. Ther. Nucleic Acids 2020, 21, 1050–1061. [Google Scholar] [CrossRef] [PubMed]
- Takakura, K.; Kawamura, A.; Torisu, Y.; Koido, S.; Yahagi, N.; Saruta, M. The Clinical Potential of Oligonucleotide Therapeutics against Pancreatic Cancer. Int. J. Mol. Sci. 2019, 20, 3331. [Google Scholar] [CrossRef] [PubMed]
- Dias, N.; Stein, C.A. Antisense oligonucleotides: Basic concepts and mechanisms. Mol. Cancer Ther. 2002, 1, 347–355. [Google Scholar]
- Crooke, S.T.; Baker, B.F.; Crooke, R.M.; Liang, X.-H. Antisense technology: An overview and prospectus. Nat. Rev. Drug Discov. 2021, 1–27. [Google Scholar] [CrossRef]
- Sajid, M.I.; Moazzam, M.; Kato, S.; Cho, K.Y.; Tiwari, R.K. Overcoming Barriers for siRNA Therapeutics: From Bench to Bedside. Pharmaceuticals 2020, 13, 294. [Google Scholar] [CrossRef]
- Yu, D.; Pendergraff, H.; Liu, J.; Kordasiewicz, H.B.; Cleveland, D.W.; Swayze, E.E.; Lima, W.F.; Crooke, S.T.; Prakash, T.P.; Corey, D.R. Single-Stranded RNAs Use RNAi to Potently and Allele-Selectively Inhibit Mutant Huntingtin Expression. Cell 2012, 150, 895–908. [Google Scholar] [CrossRef]
- Nishina, K.; Piao, W.; Yoshida-Tanaka, K.; Sujino, Y.; Nishina, T.; Yamamoto, T.; Nitta, K.; Yoshioka, K.; Kuwahara, H.; Yasuhara, H.; et al. DNA/RNA heteroduplex oligonucleotide for highly efficient gene silencing. Nat. Commun. 2015, 6, 7969. [Google Scholar] [CrossRef]
- Sciabola, S.; Xi, H.; Cruz, D.; Cao, Q.; Lawrence, C.; Zhang, T.; Rotstein, S.; Hughes, J.D.; Caffrey, D.R.; Stanton, R.V. PFRED: A computational platform for siRNA and antisense oligonucleotides design. PLoS ONE 2021, 16, e0238753. [Google Scholar] [CrossRef]
- Kim, J.; Hu, C.; El Achkar, C.M.; Black, L.E.; Douville, J.; Larson, A.; Pendergast, M.K.; Goldkind, S.F.; Lee, E.A.; Kuniholm, A.; et al. Patient-Customized Oligonucleotide Therapy for a Rare Genetic Disease. N. Engl. J. Med. 2019, 381, 1644–1652. [Google Scholar] [CrossRef]
- Martin, H.C.; Wani, S.; Steptoe, A.L.; Krishnan, K.; Nones, K.; Nourbakhsh, E.; Vlassov, A.; Grimmond, S.M.; Cloonan, N. Imperfect centered miRNA binding sites are common and can mediate repression of target mRNAs. Genome Biol. 2014, 15, R51. [Google Scholar] [CrossRef]
- Chakraborty, C.; Sharma, A.R.; Sharma, G.; Lee, S.-S. Therapeutic advances of miRNAs: A preclinical and clinical update. J. Adv. Res. 2021, 28, 127–138. [Google Scholar] [CrossRef]
- Detassis, S.; Grasso, M.; Del Vescovo, V.; Denti, M.A. microRNAs Make the Call in Cancer Personalized Medicine. Front. Cell Dev. Biol. 2017, 5, 86. [Google Scholar] [CrossRef] [PubMed]
- Pakunlu, R.I.; Wang, Y.; Saad, M.; Khandare, J.J.; Starovoytov, V.; Minko, T. In vitro and in vivo intracellular liposomal delivery of antisense oligonucleotides and anticancer drug. J. Control. Release 2006, 114, 153–162. [Google Scholar] [CrossRef]
- Zylberberg, C.; Matosevic, S. Pharmaceutical liposomal drug delivery: A review of new delivery systems and a look at the regulatory landscape. Drug Deliv. 2016, 23, 3319–3329. [Google Scholar] [CrossRef]
- Kouhara, H.; Hadari, Y.; Spivak-Kroizman, T.; Schilling, J.; Bar-Sagi, D.; Lax, I.; Schlessinger, J. A Lipid-Anchored Grb2-Binding Protein That Links FGF-Receptor Activation to the Ras/MAPK Signaling Pathway. Cell 1997, 89, 693–702. [Google Scholar] [CrossRef]
- Lowenstein, E.; Daly, R.; Batzer, A.; Li, W.; Margolis, B.; Lammers, R.; Ullrich, A.; Skolnik, E.; Bar-Sagi, D.; Schlessinger, J. The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell 1992, 70, 431–442. [Google Scholar] [CrossRef]
- Liang, X.-H.; Sun, H.; Nichols, J.G.; Crooke, S.T. RNase H1-Dependent Antisense Oligonucleotides Are Robustly Active in Directing RNA Cleavage in Both the Cytoplasm and the Nucleus. Mol. Ther. 2017, 25, 2075–2092. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, V.K.; Jones, C.I.; Newbury, S.F.; Green, P.J. XRN 5′→3′ exoribonucleases: Structure, mechanisms and functions. Biochim. et Biophys. Acta (BBA) Bioenerg. 2013, 1829, 590–603. [Google Scholar] [CrossRef]
- Lima, W.F.; De Hoyos, C.L.; Liang, X.-H.; Crooke, S.T. RNA cleavage products generated by antisense oligonucleotides and siRNAs are processed by the RNA surveillance machinery. Nucleic Acids Res. 2016, 44, 3351–3363. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Lima, W.F.; Zhang, H.; Fan, A.; Sun, H.; Crooke, S.T. Determination of the Role of the Human RNase H1 in the Pharmacology of DNA-like Antisense Drugs. J. Biol. Chem. 2004, 279, 17181–17189. [Google Scholar] [CrossRef] [PubMed]
- Vickers, T.A.; Crooke, S.T. The rates of the major steps in the molecular mechanism of RNase H1-dependent antisense oligonucleotide induced degradation of RNA. Nucleic Acids Res. 2015, 43, 8955–8963. [Google Scholar] [CrossRef]
- Liang, X.-H.; Nichols, J.G.; De Hoyos, C.L.; Crooke, S.T. Some ASOs that bind in the coding region of mRNAs and induce RNase H1 cleavage can cause increases in the pre-mRNAs that may blunt total activity. Nucleic Acids Res. 2020, 48, 9840–9858. [Google Scholar] [CrossRef]
- Yoo, B.H. 2’-O-methyl-modified phosphorothioate antisense oligonucleotides have reduced non-specific effects in vitro. Nucleic Acids Res. 2004, 32, 2008–2016. [Google Scholar] [CrossRef]
- Baker, B.F.; Lot, S.S.; Condon, T.P.; Cheng-Flournoy, S.; Lesnik, E.A.; Sasmor, H.M.; Bennett, C.F. 2′-O-(2-Methoxy)ethyl-modified Anti-intercellular Adhesion Molecule 1 (ICAM-1) Oligonucleotides Selectively Increase the ICAM-1 mRNA Level and Inhibit Formation of the ICAM-1 Translation Initiation Complex in Human Umbilical Vein Endothelial Cells. J. Biol. Chem. 1997, 272, 11994–12000. [Google Scholar] [CrossRef] [PubMed]
- Dominski, Z.; Kole, R. Restoration of correct splicing in thalassemic pre-mRNA by antisense oligonucleotides. Proc. Natl. Acad. Sci. USA 1993, 90, 8673–8677. [Google Scholar] [CrossRef]
- Liang, X.-H.; Shen, W.; Sun, H.; Migawa, M.T.; Vickers, T.A.; Crooke, S.T. Translation efficiency of mRNAs is increased by antisense oligonucleotides targeting upstream open reading frames. Nat. Biotechnol. 2016, 34, 875–880. [Google Scholar] [CrossRef]
- Somers, J.; Pöyry, T.; Willis, A.E. A perspective on mammalian upstream open reading frame function. Int. J. Biochem. Cell Biol. 2013, 45, 1690–1700. [Google Scholar] [CrossRef]
- Mercatante, D.R.; Mohler, J.L.; Kole, R. Cellular Response to an Antisense-mediated Shift of Bcl-x Pre-mRNA Splicing and Antineoplastic Agents. J. Biol. Chem. 2002, 277, 49374–49382. [Google Scholar] [CrossRef]
- Wu, L.; Mao, C.; Ming, X. Modulation of Bcl-x Alternative Splicing Induces Apoptosis of Human Hepatic Stellate Cells. BioMed Res. Int. 2016, 2016, 7478650. [Google Scholar] [CrossRef] [PubMed]
- Azad, R.F.; Driver, V.B.; Tanaka, K.; Crooke, R.M.; Anderson, K.P. Antiviral activity of a phosphorothioate oligonucleotide complementary to RNA of the human cytomegalovirus major immediate-early region. Antimicrob. Agents Chemother. 1993, 37, 1945–1954. [Google Scholar] [CrossRef]
- Detrick, B.; Nagineni, C.N.; Grillone, L.R.; Anderson, K.P.; Henry, S.P.; Hooks, J.J. Inhibition of human cytomegalovirus replication in a human retinal epithelial cell model by antisense oligonucleotides. Investig. Ophthalmol. Vis. Sci. 2001, 42, 163–169. [Google Scholar]
- Perry, C.M.; Balfour, J.A.B. Fomivirsen. Drugs 1999, 57, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Ray, M.; Logan, R.; Sterne, J.A.C.; Hernández-Díaz, S.; Robins, J.M.; Sabin, C.; Bansi, L.; Van Sighem, A.; De Wolf, F.; Costagliola, D.; et al. The effect of combined antiretroviral therapy on the overall mortality of HIV-infected individuals. AIDS 2010, 24, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Geary, R.S.; Baker, B.F.; Crooke, S.T. Clinical and Preclinical Pharmacokinetics and Pharmacodynamics of Mipomersen (Kynamro®): A Second-Generation Antisense Oligonucleotide Inhibitor of Apolipoprotein B. Clin. Pharmacokinet. 2015, 54, 133–146. [Google Scholar] [CrossRef]
- Raal, F.J.; Santos, R.D.; Blom, D.J.; Marais, A.D.; Charng, M.-J.; Cromwell, W.C.; Lachmann, R.H.; Gaudet, D.; Tan, J.L.; Chasan-Taber, S.; et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: A randomised, double-blind, placebo-controlled trial. Lancet 2010, 375, 998–1006. [Google Scholar] [CrossRef]
- Fogacci, F.; Ferri, N.; Toth, P.P.; Ruscica, M.; Corsini, A.; Cicero, A.F.G. Efficacy and Safety of Mipomersen: A Systematic Review and Meta-Analysis of Randomized Clinical Trials. Drugs 2019, 79, 751–766. [Google Scholar] [CrossRef]
- Keam, S.J. Inotersen: First Global Approval. Drugs 2018, 78, 1371–1376. [Google Scholar] [CrossRef]
- Mathew, V.; Wang, A.K. Inotersen: New promise for the treatment of hereditary transthyretin amyloidosis. Drug Des. Dev. Ther. 2019, 13, 1515–1525. [Google Scholar] [CrossRef]
- Sikora, J.L.; Logue, M.W.; Chan, G.G.; Spencer, B.H.; Prokaeva, T.B.; Baldwin, C.T.; Seldin, D.C.; Connors, L.H. Genetic variation of the transthyretin gene in wild-type transthyretin amyloidosis (ATTRwt). Qual. Life Res. 2015, 134, 111–121. [Google Scholar] [CrossRef]
- Connors, L.H.; Lim, A.; Prokaeva, T.; Roskens, V.A.; Costello, C.E. Tabulation of human transthyretin (TTR) variants, 2003. Amyloid 2003, 10, 160–184. [Google Scholar] [CrossRef]
- Benson, M.D.; Waddington-Cruz, M.; Berk, J.L.; Polydefkis, M.; Dyck, P.J.; Wang, A.K.; Planté-Bordeneuve, V.; Barroso, F.A.; Merlini, G.; Obici, L.; et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 22–31. [Google Scholar] [CrossRef]
- Neil, E.E.; Bisaccia, E.K. Nusinersen: A Novel Antisense Oligonucleotide for the Treatment of Spinal Muscular Atrophy. J. Pediatr. Pharmacol. Ther. 2019, 24, 194–203. [Google Scholar] [CrossRef]
- Wirth, B. An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA). Hum. Mutat. 2000, 15, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Wurster, C.D.; Ludolph, A.C. Nusinersen for spinal muscular atrophy. Ther. Adv. Neurol. Disord. 2018, 11, 10–12. [Google Scholar] [CrossRef] [PubMed]
- Jodelka, F.M.; Ebert, A.D.; Duelli, D.M.; Hastings, M.L. A feedback loop regulates splicing of the spinal muscular atrophy-modifying gene, SMN2. Hum. Mol. Genet. 2010, 19, 4906–4917. [Google Scholar] [CrossRef]
- Mercuri, E.; Darras, B.T.; Chiriboga, C.A.; Day, J.W.; Campbell, C.; Connolly, A.M.; Iannaccone, S.T.; Kirschner, J.; Kuntz, N.L.; Saito, K.; et al. Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2018, 378, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Syed, Y.Y. Eteplirsen: First Global Approval. Drugs 2016, 76, 1699–1704. [Google Scholar] [CrossRef] [PubMed]
- Amantana, A.; Iversen, P.L. Pharmacokinetics and biodistribution of phosphorodiamidate morpholino antisense oligomers. Curr. Opin. Pharmacol. 2005, 5, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.R.Q.; Maruyama, R.; Yokota, T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des. Dev. Ther. 2017, 11, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Kinali, M.; Arechavala-Gomeza, V.; Feng, L.; Cirak, S.; Hunt, D.; Adkin, C.; Guglieri, M.; Ashton, E.; Abbs, S.; Nihoyannopoulos, P.; et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: A single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009, 8, 918–928. [Google Scholar] [CrossRef]
- Heo, Y.A. Golodirsen: First Approval. Drugs 2020, 80, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.E.; Schnell, F.J.; Akana, C.; El-Husayni, S.H.; Desjardins, C.A.; Morgan, J.; Charleston, J.S.; Sardone, V.; Domingos, J.; Dickson, G.; et al. Increased dystrophin production with golodirsen in patients with Duchenne muscular dystrophy. Neurology 2020, 94, e2270–e2282. [Google Scholar] [CrossRef]
- Dhillon, S. Viltolarsen: First Approval. Drugs 2020, 80, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Roshmi, R.; Yokota, T. Viltolarsen for the treatment of Duchenne muscular dystrophy. Drugs Today 2019, 55, 627–639. [Google Scholar] [CrossRef]
- Komaki, H.; Takeshima, Y.; Matsumura, T.; Ozasa, S.; Funato, M.; Takeshita, E.; Iwata, Y.; Yajima, H.; Egawa, Y.; Toramoto, T.; et al. Viltolarsen in Japanese Duchenne muscular dystrophy patients: A phase 1/2 study. Ann. Clin. Transl. Neurol. 2020, 7, 2393–2408. [Google Scholar] [CrossRef]
- Clemens, P.R.; Rao, V.K.; Connolly, A.M.; Harper, A.D.; Mah, J.K.; Smith, E.C.; McDonald, C.M.; Zaidman, C.M.; Morgenroth, L.P.; Osaki, H.; et al. Safety, Tolerability, and Efficacy of Viltolarsen in Boys With Duchenne Muscular Dystrophy Amenable to Exon 53 Skipping. JAMA Neurol. 2020, 77, 982. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-targeted-treatment-rare-duchenne-muscular-dystrophy-mutation-0 (accessed on 28 March 2021).
- Rodrigues, M.; Yokota, T. An Overview of Recent Advances and Clinical Applications of Exon Skipping and Splice Modulation for Muscular Dystrophy and Various Genetic Diseases. Methods Mol. Biol. 2018, 1828, 31–55. [Google Scholar] [CrossRef]
- Clinical Trials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02500381 (accessed on 28 March 2021).
- Juliano, R.L. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016, 44, 6518–6548. [Google Scholar] [CrossRef]
- Linnane, E.; Davey, P.; Zhang, P.; Puri, S.; Edbrooke, M.; Chiarparin, E.; Revenko, A.S.; MacLeod, A.R.; Norman, J.C.; Ross, S.J. Differential uptake, kinetics and mechanisms of intracellular trafficking of next-generation antisense oligonucleotides across human cancer cell lines. Nucleic Acids Res. 2019, 47, 4375–4392. [Google Scholar] [CrossRef] [PubMed]
- Thomas, O.S.; Weber, W. Overcoming Physiological Barriers to Nanoparticle Delivery—Are We There Yet? Front. Bioeng. Biotechnol. 2019, 7, 415. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T.; Vickers, T.A.; Liang, X.-H. Phosphorothioate modified oligonucleotide–protein interactions. Nucleic Acids Res. 2020, 48, 5235–5253. [Google Scholar] [CrossRef] [PubMed]
- Gaus, H.J.; Gupta, R.; Chappell, A.E.; Østergaard, M.E.; Swayze, E.E.; Seth, P.P. Characterization of the interactions of chemically-modified therapeutic nucleic acids with plasma proteins using a fluorescence polarization assay. Nucleic Acids Res. 2018, 47, 1110–1122. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.-H.; Sun, H.; Shen, W.; Crooke, S.T. Identification and characterization of intracellular proteins that bind oligonucleotides with phosphorothioate linkages. Nucleic Acids Res. 2015, 43, 2927–2945. [Google Scholar] [CrossRef]
- Miller, C.M.; Wan, W.B.; Seth, P.P.; Harris, E.N. Endosomal Escape of Antisense Oligonucleotides Internalized by Stabilin Receptors Is Regulated by Rab5C and EEA1 During Endosomal Maturation. Nucleic Acid Ther. 2018, 28, 86–96. [Google Scholar] [CrossRef]
- Majlessi, M.; Nelson, N.C.; Becker, M.M. Advantages of 2’-O-methyl oligoribonucleotide probes for detecting RNA targets. Nucleic Acids Res. 1998, 26, 2224–2229. [Google Scholar] [CrossRef]
- Shen, W.; De Hoyos, C.L.; Sun, H.; Vickers, T.A.; Liang, X.-H.; Crooke, S.T. Acute hepatotoxicity of 2′ fluoro-modified 5–10–5 gapmer phosphorothioate oligonucleotides in mice correlates with intracellular protein binding and the loss of DBHS proteins. Nucleic Acids Res. 2018, 46, 2204–2217. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Liang, X.-H.; Sun, H.; Crooke, S.T. 2’-Fluoro-modified phosphorothioate oligonucleotide can cause rapid degradation of P54nrb and PSF. Nucleic Acids Res. 2015, 43, 4569–4578. [Google Scholar] [CrossRef]
- Khvorova, A.; Watts, A.K.J.K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 2017, 35, 238–248. [Google Scholar] [CrossRef]
- Østergaard, M.E.; Southwell, A.L.; Kordasiewicz, H.; Watt, A.T.; Skotte, N.H.; Doty, C.N.; Vaid, K.; Villanueva, E.B.; Swayze, E.E.; Bennett, C.F.; et al. Rational design of antisense oligonucleotides targeting single nucleotide polymorphisms for potent and allele selective suppression of mutant Huntingtin in the CNS. Nucleic Acids Res. 2013, 41, 9634–9650. [Google Scholar] [CrossRef]
- Østergaard, M.E.; Nichols, J.; Dwight, T.A.; Lima, W.; Jung, M.E.; Swayze, E.E.; Seth, P.P. Fluorinated Nucleotide Modifications Modulate Allele Selectivity of SNP-Targeting Antisense Oligonucleotides. Mol. Ther. Nucleic Acids 2017, 7, 20–30. [Google Scholar] [CrossRef]
- Freier, S.M. The ups and downs of nucleic acid duplex stability: Structure-stability studies on chemically-modified DNA:RNA duplexes. Nucleic Acids Res. 1997, 25, 4429–4443. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, S.; Milam, V.T. Modified Nucleic Acids: Expanding the Capabilities of Functional Oligonucleotides. Molecules 2020, 25, 4659. [Google Scholar] [CrossRef]
- McCampbell, A.; Cole, T.; Wegener, A.J.; Tomassy, G.S.; Setnicka, A.; Farley, B.J.; Schoch, K.M.; Hoye, M.L.; Shabsovich, M.; Sun, L.; et al. Antisense oligonucleotides extend survival and reverse decrement in muscle response in ALS models. J. Clin. Investig. 2018, 128, 3558–3567. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.; Cudkowicz, M.; Shaw, P.J.; Andersen, P.M.; Atassi, N.; Bucelli, R.C.; Genge, A.; Glass, J.; Ladha, S.; Ludolph, A.L.; et al. Phase 1–2 Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med. 2020, 383, 109–119. [Google Scholar] [CrossRef]
- Pallan, P.S.; Allerson, C.R.; Berdeja, A.; Seth, P.P.; Swayze, E.E.; Prakash, T.P.; Egli, M. Structure and nuclease resistance of 2′,4′-constrained 2′-O-methoxyethyl (cMOE) and 2′-O-ethyl (cEt) modified DNAs. Chem. Commun. 2012, 48, 8195–8197. [Google Scholar] [CrossRef] [PubMed]
- Frieden, M.; Christensen, S.M.; Mikkelsen, N.D.; Rosenbohm, C.; Thrue, C.A.; Westergaard, M.; Hansen, H.F.; Ørum, H.; Koch, T. Expanding the design horizon of antisense oligonucleotides with alpha-L-LNA. Nucleic Acids Res. 2003, 31, 6365–6372. [Google Scholar] [CrossRef] [PubMed]
- Yılmaz-Eliş, A.S.; Aartsma-Rus, A.; Hoen, P.A.T.; Safdar, H.; Breukel, C.; Van Vlijmen, B.J.; Van Deutekom, J.; De Kimpe, S.; Van Ommen, G.-J.; Verbeek, J.S. Inhibition of IL-1 Signaling by Antisense Oligonucleotide-mediated Exon Skipping of IL-1 Receptor Accessory Protein (IL-1RAcP). Mol. Ther. Nucleic Acids 2013, 2, e66. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, D.; Omlin, A.; Pezaro, C.; Lorente, D.; Ferraldeschi, R.; Mukherji, D.; Crespo, M.; Figueiredo, I.; Miranda, S.; Riisnaes, R.; et al. First-in-human Phase I study of EZN-4176, a locked nucleic acid antisense oligonucleotide to exon 4 of the androgen receptor mRNA in patients with castration-resistant prostate cancer. Br. J. Cancer 2013, 109, 2579–2586. [Google Scholar] [CrossRef] [PubMed]
- Sewing, S.; Boess, F.; Moisan, A.; Bertinetti-Lapatki, C.; Minz, T.; Hedtjaern, M.; Tessier, Y.; Schuler, F.; Singer, T.; Roth, A.B. Establishment of a Predictive In Vitro Assay for Assessment of the Hepatotoxic Potential of Oligonucleotide Drugs. PLoS ONE 2016, 11, e0159431. [Google Scholar] [CrossRef] [PubMed]
- Tari, A.M.; Stephens, C.; Rosenblum, M.; Lopez-Berestein, G. Pharmacokinetics, Tissue Distribution, and Safety of P-Ethoxy Oligonucleotides Incorporated in Liposomes. J. Liposome Res. 1998, 8, 251–264. [Google Scholar] [CrossRef]
- Volpi, S.; Cancelli, U.; Neri, M.; Corradini, R. Multifunctional Delivery Systems for Peptide Nucleic Acids. Pharmaceuticals 2020, 14, 14. [Google Scholar] [CrossRef]
- Shen, X.; Corey, D.R. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res. 2018, 46, 1584–1600. [Google Scholar] [CrossRef] [PubMed]
- Lundin, K.E.; Gissberg, O.; Smith, C.E. Oligonucleotide Therapies: The Past and the Present. Hum. Gene Ther. 2015, 26, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Copolovici, D.M.; Langel, K.; Eriste, E.; Langel, Ü. Cell-Penetrating Peptides: Design, Synthesis, and Applications. ACS Nano 2014, 8, 1972–1994. [Google Scholar] [CrossRef]
- McClorey, G.; Banerjee, S. Cell-Penetrating Peptides to Enhance Delivery of Oligonucleotide-Based Therapeutics. Biomedicines 2018, 6, 51. [Google Scholar] [CrossRef]
- Betts, C.; Saleh, A.F.; Arzumanov, A.A.; Hammond, S.M.; Godfrey, C.; Coursindel, T.; Gait, M.J.; Wood, M.J. Pip6-PMO, A New Generation of Peptide-oligonucleotide Conjugates with Improved Cardiac Exon Skipping Activity for DMD Treatment. Mol. Ther. Nucleic Acids 2012, 1, e38. [Google Scholar] [CrossRef]
- Klein, A.F.; Varela, M.A.; Arandel, L.; Holland, A.; Naouar, N.; Arzumanov, A.; Seoane, D.; Revillod, L.; Bassez, G.; Ferry, A.; et al. Peptide-conjugated oligonucleotides evoke long-lasting myotonic dystrophy correction in patient-derived cells and mice. J. Clin. Investig. 2019, 129, 4739–4744. [Google Scholar] [CrossRef]
- Arnold, A.E.; Malek-Adamian, E.; Le, P.U.; Meng, A.; Martínez-Montero, S.; Petrecca, K.; Damha, M.J.; Shoichet, M.S. Antibody-Antisense Oligonucleotide Conjugate Downregulates a Key Gene in Glioblastoma Stem Cells. Mol. Ther. Nucleic Acids 2018, 11, 518–527. [Google Scholar] [CrossRef]
- Satake, N.; Duong, C.; Yoshida, S.; Oestergaard, M.; Chen, C.; Peralta, R.; Guo, S.; Seth, P.P.; Li, Y.; Beckett, L.; et al. Novel Targeted Therapy for Precursor B-Cell Acute Lymphoblastic Leukemia: Anti-CD22 Antibody-MXD3 Antisense Oligonucleotide Conjugate. Mol. Med. 2016, 22, 632–642. [Google Scholar] [CrossRef]
- Ni, S.; Zhuo, Z.; Pan, Y.; Yu, Y.; Li, F.; Liu, J.; Wang, L.; Wu, X.; Li, D.; Wan, Y.; et al. Recent Progress in Aptamer Discoveries and Modifications for Therapeutic Applications. ACS Appl. Mater. Interfaces 2021, 13, 9500–9519. [Google Scholar] [CrossRef] [PubMed]
- Prakash, T.P.; Graham, M.J.; Yu, J.; Carty, R.; Low, A.; Chappell, A.; Schmidt, K.; Zhao, C.; Aghajan, M.; Murray, H.F.; et al. Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N-acetyl galactosamine improves potency 10-fold in mice. Nucleic Acids Res. 2014, 42, 8796–8807. [Google Scholar] [CrossRef] [PubMed]
- Viney, N.J.; van Capelleveen, J.C.; Geary, R.S.; Xia, S.; Tami, J.A.; Yu, R.Z.; Marcovina, S.M.; Hughes, S.G.; Graham, M.J.; Crooke, R.M.; et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): Two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet 2016, 388, 2239–2253. [Google Scholar] [CrossRef]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef] [PubMed]
- Arrighetti, N.; Corbo, C.; Evangelopoulos, M.; Pastò, A.; Zuco, V.; Tasciotti, E. Exosome-like Nanovectors for Drug Delivery in Cancer. Curr. Med. Chem. 2019, 26, 6132–6148. [Google Scholar] [CrossRef]
- Chi, Q.; Yang, Z.; Xu, K.; Wang, C.; Liang, H. DNA Nanostructure as an Efficient Drug Delivery Platform for Immunotherapy. Front. Pharmacol. 2020, 10, 1–17. [Google Scholar] [CrossRef]
- Ashizawa, A.T.; Cortes, J. Liposomal delivery of nucleic acid-based anticancer therapeutics: BP-100-1.01. Expert Opin. Drug Deliv. 2014, 12, 1107–1120. [Google Scholar] [CrossRef]
- Daraee, H.; Etemadi, A.; Kouhi, M.; Alimirzalu, S.; Akbarzadeh, A. Application of liposomes in medicine and drug delivery. Artif. Cells Nanomed. Biotechnol. 2016, 44, 381–391. [Google Scholar] [CrossRef]
- Bozzuto, G.; Molinari, A. Liposomes as nanomedical devices. Int. J. Nanomed. 2015, 10, 975–999. [Google Scholar] [CrossRef] [PubMed]
- Maclachlan, I. Liposomal formulations for nucleic acid delivery. In Antisense Drug Technology; Crooke, S.T., Ed.; CRC Press: Boca Raton, FL, USA, 2007; pp. 237–270. [Google Scholar]
- Abu Lila, A.S.; Kiwada, H.; Ishida, T. The accelerated blood clearance (ABC) phenomenon: Clinical challenge and approaches to manage. J. Control. Release 2013, 172, 38–47. [Google Scholar] [CrossRef]
- Zelphati, O.; Uyechi, L.S.; Barron, L.G.; Szoka, F.C. Effect of serum components on the physico-chemical properties of cationic lipid/oligonucleotide complexes and on their interactions with cells. Biochim. et Biophys. Acta (BBA) Lipids Lipid Metab. 1998, 1390, 119–133. [Google Scholar] [CrossRef]
- Nag, O.K.; Awasthi, V. Surface Engineering of Liposomes for Stealth Behavior. Pharmaceutics 2013, 5, 542–569. [Google Scholar] [CrossRef]
- Gabizon, A.A. Pegylated Liposomal Doxorubicin: Metamorphosis of an Old Drug into a New Form of Chemotherapy. Cancer Investig. 2001, 19, 424–436. [Google Scholar] [CrossRef]
- Schöttler, S.; Becker, G.; Winzen, S.; Steinbach, T.; Mohr, K.; Landfester, K.; Mailänder, S.S.V.; Wurm, F. Protein adsorption is required for stealth effect of poly(ethylene glycol)- and poly(phosphoester)-coated nanocarriers. Nat. Nanotechnol. 2016, 11, 372–377. [Google Scholar] [CrossRef]
- Thi, T.T.H.; Pilkington, E.H.; Nguyen, D.H.; Lee, J.S.; Park, K.D.; Truong, N.P. The Importance of Poly(ethylene glycol) Alternatives for Overcoming PEG Immunogenicity in Drug Delivery and Bioconjugation. Polymers 2020, 12, 298. [Google Scholar] [CrossRef]
- Romberg, B.; Oussoren, C.; Snel, C.J.; Carstens, M.G.; Hennink, W.E.; Storm, G. Pharmacokinetics of poly(hydroxyethyl-l-asparagine)-coated liposomes is superior over that of PEG-coated liposomes at low lipid dose and upon repeated administration. Biochim. et Biophys. Acta (BBA) Biomembr. 2007, 1768, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Barenholz, Y. (Chezy) Doxil®—The first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef]
- Estephan, Z.G.; Schlenoff, P.S.; Schlenoff, J.B. Zwitteration As an Alternative to PEGylation. Langmuir 2011, 27, 6794–6800. [Google Scholar] [CrossRef]
- Gallová, J.; Uhríková, D.; Islamov, A.; Kuklin, A.; Balgavý, P. Effect of cholesterol on the bilayer thickness in unilamellar extruded DLPC and DOPC liposomes: SANS contrast variation study. Gen. Physiol. Biophys. 2004, 23, 113–128. [Google Scholar]
- Magarkar, A.; Dhawan, V.; Kallinteri, P.; Viitala, T.; Elmowafy, M.; Róg, T.; Bunker, A. Cholesterol level affects surface charge of lipid membranes in saline solution. Sci. Rep. 2014, 4, 5005. [Google Scholar] [CrossRef]
- Briuglia, M.-L.; Rotella, C.; McFarlane, A.; Lamprou, D.A. Influence of cholesterol on liposome stability and on in vitro drug release. Drug Deliv. Transl. Res. 2015, 5, 231–242. [Google Scholar] [CrossRef]
- Sicard, G.; Paris, C.; Giacometti, S.; Rodallec, A.; Ciccolini, J.; Rocchi, P.; Fanciullino, R. Enhanced Antisense Oligonucleotide Delivery Using Cationic Liposomes Grafted with Trastuzumab: A Proof-of-Concept Study in Prostate Cancer. Pharmaceutics 2020, 12, 1166. [Google Scholar] [CrossRef]
- Lee, Y.; Thompson, D. Stimuli-responsive liposomes for drug delivery. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017, 9, e1450. [Google Scholar] [CrossRef]
- Li, J.; Wang, X.; Zhang, T.; Wang, C.; Huang, Z.; Luo, X.; Deng, Y. A review on phospholipids and their main applications in drug delivery systems. Asian J. Pharm. Sci. 2015, 10, 81–98. [Google Scholar] [CrossRef]
- Paliwal, S.R.; Paliwal, R.; Vyas, S.P. A review of mechanistic insight and application of pH-sensitive liposomes in drug delivery. Drug Deliv. 2014, 22, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Xiang, B.; Jia, X.-L.; Qi, J.-L.; Yang, L.-P.; Sun, W.-H.; Yan, X.; Yang, S.-K.; Cao, D.-Y.; Du, Q.; Qi, X.-R. Enhancing siRNA-based cancer therapy using a new pH-responsive activatable cell-penetrating peptide-modified liposomal system. Int. J. Nanomed. 2017, 12, 2385–2405. [Google Scholar] [CrossRef]
- Tagami, T.; Ozeki, T. Recent Trends in Clinical Trials Related to Carrier-Based Drugs. J. Pharm. Sci. 2017, 106, 2219–2226. [Google Scholar] [CrossRef]
- Cheng, P.F.; Dummer, R.; Levesque, M.P. Data mining The Cancer Genome Atlas in the era of precision cancer medicine. Swiss Med. Wkly. 2015, 145, w14183. [Google Scholar] [CrossRef]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. Review The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Współczesna Onkologia 2015, 1A, 68–77. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Program. Available online: https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga (accessed on 15 February 2021).
- Dharmawardana, P.G.; Peruzzi, B.; Giubellino, A.; Burke, T.R.; Bottaro, D.P. Molecular targeting of growth factor receptor-bound 2 (Grb2) as an anti-cancer strategy. Anti-Cancer Drugs 2006, 17, 13–20. [Google Scholar] [CrossRef]
- Cheng, A.M.; Saxton, T.M.; Sakai, R.; Kulkarni, S.; Mbamalu, G.; Vogel, W.; Tortorice, C.G.; Cardiff, R.D.; Cross, J.C.; Muller, W.J.; et al. Mammalian Grb2 Regulates Multiple Steps in Embryonic Development and Malignant Transformation. Cell 1998, 95, 793–803. [Google Scholar] [CrossRef]
- Wang, F. Oncogenic Role of Grb2 in Breast Cancer and Grb2 Antagonists as Therapeutic Drugs. Cancer Ther. Oncol. Int. J. 2017, 3, 555618. [Google Scholar] [CrossRef][Green Version]
- Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Kempf, C.R.; Long, J.; Laidler, P.; Mijatovic, S.; Maksimovic-Ivanic, D.; Stivala, F.; Mazzarino, M.C.; et al. Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging 2011, 3, 192–222. [Google Scholar] [CrossRef] [PubMed]
- Nørøxe, D.S.; Yde, C.W.; Østrup, O.; Michaelsen, S.R.; Schmidt, A.Y.; Kinalis, S.; Torp, M.H.; Skjøth-Rasmussen, J.; Brennum, J.; Hamerlik, P.; et al. Genomic profiling of newly diagnosed glioblastoma patients and its potential for clinical utility—A prospective, translational study. Mol. Oncol. 2020, 14, 2727–2743. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, J.; Gao, F.; Fan, S.; Dai, L.; Zhang, J. KPNA2-Associated Immune Analyses Highlight the Dysregulation and Prognostic Effects of GRB2, NRAS, and Their RNA-Binding Proteins in Hepatocellular Carcinoma. Front. Genet. 2020, 11, 593273. [Google Scholar] [CrossRef] [PubMed]
- Li, L.-Y.; Li, E.-M.; Wu, Z.-Y.; Cao, H.-H.; Shen, J.-H.; Xu, X.-E.; Chen, B.; Wu, J.-Y.; Xu, L.-Y. Overexpression of GRB2 is correlated with lymph node metastasis and poor prognosis in esophageal squamous cell carcinoma. Int. J. Clin. Exp. Pathol. 2014, 7, 3132–3140. [Google Scholar]
- Lewis, T.R.; Smith, J.; Griffin, K.; Aguiar, S.; Rueb, K.F.; Holmberg-Douglas, N.; Sampson, E.M.; Tomasetti, S.; Rodriguez, S.; Stachura, D.L.; et al. NHD2-15, a novel antagonist of Growth Factor Receptor-Bound Protein-2 (GRB2), inhibits leukemic proliferation. PLoS ONE 2020, 15, e0236839. [Google Scholar] [CrossRef] [PubMed]
- Ijaz, M.; Wang, F.; Shahbaz, M.; Jiang, W.; Fathy, A.H.; Nesa, E.U. The Role of Grb2 in Cancer and Peptides as Grb2 Antagonists. Protein Pept. Lett. 2018, 24, 1. [Google Scholar] [CrossRef]
- Sarker, D.; Plummer, R.; Meyer, T.; Sodergren, M.H.; Basu, B.; Chee, C.E.; Huang, K.-W.; Palmer, D.H.; Ma, Y.T.; Evans, T.J.; et al. MTL-CEBPA, a Small Activating RNA Therapeutic Upregulating C/EBP-α, in Patients with Advanced Liver Cancer: A First-in-Human, Multicenter, Open-Label, Phase I Trial. Clin. Cancer Res. 2020, 26, 3936–3946. [Google Scholar] [CrossRef] [PubMed]
- Schultheis, B.; Strumberg, D.; Kuhlmann, J.; Wolf, M.; Link, K.; Seufferlein, T.; Kaufmann, J.; Feist, M.; Gebhardt, F.; Khan, M.; et al. Safety, Efficacy and Pharcacokinetics of Targeted Therapy with The Liposomal RNA Interference Therapeutic Atu027 Combined with Gemcitabine in Patients with Pancreatic Adenocarcinoma. A Randomized Phase Ib/IIa Study. Cancers 2020, 12, 3130. [Google Scholar] [CrossRef] [PubMed]
- Miller, P.S.; Barrett, J.C.; Ts’O, P.O.P. Alkyl phosphotriesters of dinucleotides and oligonucleotides. 4. Synthesis of oligodeoxyribonucleotide ethyl phosphotriesters and their specific complex formation with transfer ribonucleic acid. Biochemistry 1974, 13, 4887–4896. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhang, L.; Zhu, W.; Guo, R.; Sun, H.; Chen, X.; Deng, N. Barriers and Strategies of Cationic Liposomes for Cancer Gene Therapy. Mol. Ther. Methods Clin. Dev. 2020, 18, 751–764. [Google Scholar] [CrossRef] [PubMed]
- Arlinghaus, R.; Lopez-Berestein, G.; Belmont, J.; Rosenblum, M.; Sun, T.; Stephens, C.; Monaco, G.; Gutiérrez-Puente, Y.; Tari, A.M. Liposome-incorporated Grb2 antisense oligodeoxynucleotide increases the survival of mice bearing bcr-abl-positive leukemia xenografts. Int. J. Oncol. 2007, 31, 1243–1250. [Google Scholar] [CrossRef][Green Version]
- Tari, A.M.; Hung, M.-C.; Li, K.; Lopez-Berestein, G. Growth inhibition of breast cancer cells by Grb2 downregulation is correlated with inactivation of mitogen-activated protein kinase in EGFR, but not in ErbB2, cells. Oncogene 1999, 18, 1325–1332. [Google Scholar] [CrossRef][Green Version]
- Lara, O.D.; Bayraktar, E.; Amero, P.; Ma, S.; Ivan, C.; Hu, W.; Wang, Y.; Mangala, L.S.; Dutta, P.; Bhattacharya, P.; et al. Therapeutic efficacy of liposomal Grb2 antisense oligodeoxynucleotide (L-Grb2) in preclinical models of ovarian and uterine cancer. Oncotarget 2020, 11, 2819–2833. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Douillard, J.-Y.; Koralewski, P.; Manegold, C.; Smit, E.F.; Reyes, J.M.; Chang, G.-C.; John, W.J.; Peterson, P.M.; Obasaju, C.K.; et al. Phase III Study of Gemcitabine and Cisplatin With or Without Aprinocarsen, a Protein Kinase C-Alpha Antisense Oligonucleotide, in Patients With Advanced-Stage Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2006, 24, 1428–1434. [Google Scholar] [CrossRef]
- Verheul, H.M.; Pinedo, H.M. The Role of Vascular Endothelial Growth Factor (VEGF) in Tumor Angiogenesis and Early Clinical Development of VEGFReceptor Kinase Inhibitors. Clin. Breast Cancer 2000, 1, S80–S84. [Google Scholar] [CrossRef]
- Ohanian, M.; Ashizawa, A.T.; Garcia-Manero, G.; Pemmaraju, N.; Kadia, T.; Jabbour, E.; Ravandi, F.; Borthakur, G.; Andreeff, M.; Konopleva, M.; et al. Liposomal Grb2 antisense oligodeoxynucleotide (BP1001) in patients with refractory or relapsed haematological malignancies: A single-centre, open-label, dose-escalation, phase 1/1b trial. Lancet Haematol. 2018, 5, e136–e146. [Google Scholar] [CrossRef]
- Iversen, P.L. Structure activity study of clinically observed adverse events and oligomer chemistry. J. Drug Discov. Dev. Deliv. 2016, 3, 1022. [Google Scholar]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gagliardi, M.; Ashizawa, A.T. The Challenges and Strategies of Antisense Oligonucleotide Drug Delivery. Biomedicines 2021, 9, 433. https://doi.org/10.3390/biomedicines9040433
Gagliardi M, Ashizawa AT. The Challenges and Strategies of Antisense Oligonucleotide Drug Delivery. Biomedicines. 2021; 9(4):433. https://doi.org/10.3390/biomedicines9040433
Chicago/Turabian StyleGagliardi, Maria, and Ana Tari Ashizawa. 2021. "The Challenges and Strategies of Antisense Oligonucleotide Drug Delivery" Biomedicines 9, no. 4: 433. https://doi.org/10.3390/biomedicines9040433
APA StyleGagliardi, M., & Ashizawa, A. T. (2021). The Challenges and Strategies of Antisense Oligonucleotide Drug Delivery. Biomedicines, 9(4), 433. https://doi.org/10.3390/biomedicines9040433