
Sarcopenia in Chronic Kidney Disease: Focus on Advanced Glycation End Products as Mediators and Markers of Oxidative Stress

 , , ,
, , , 

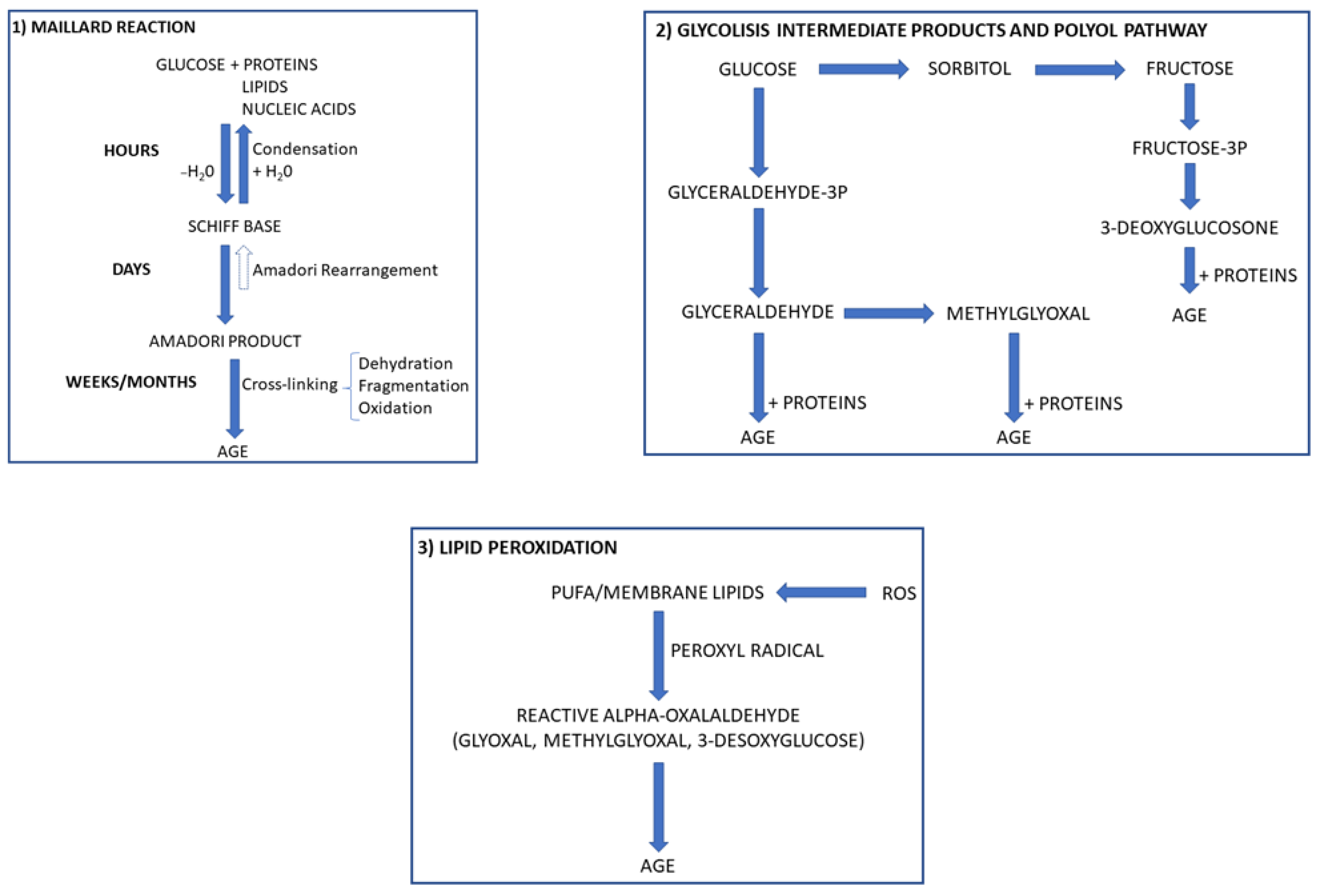{kind=link}
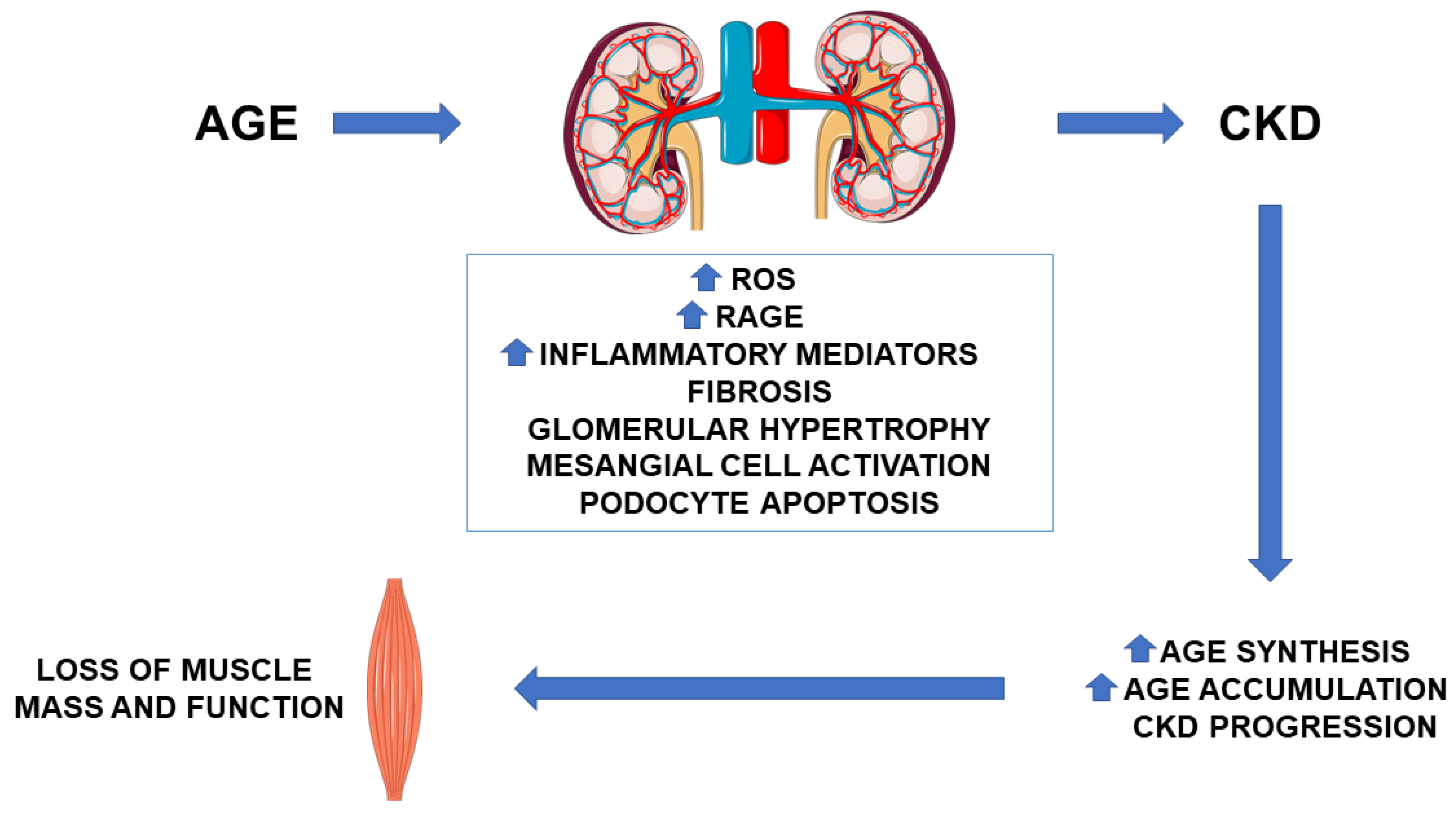{kind=link}
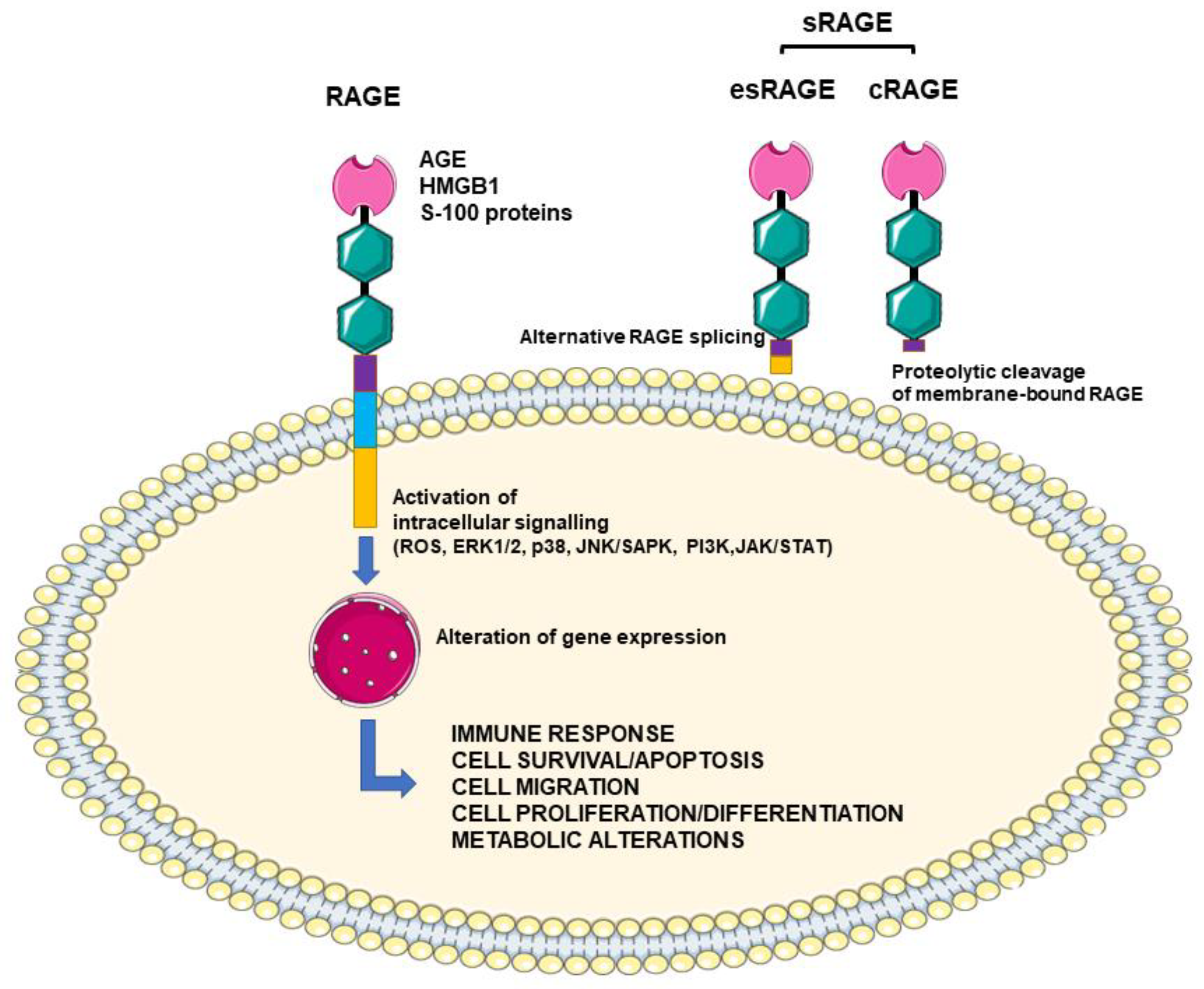{kind=link}
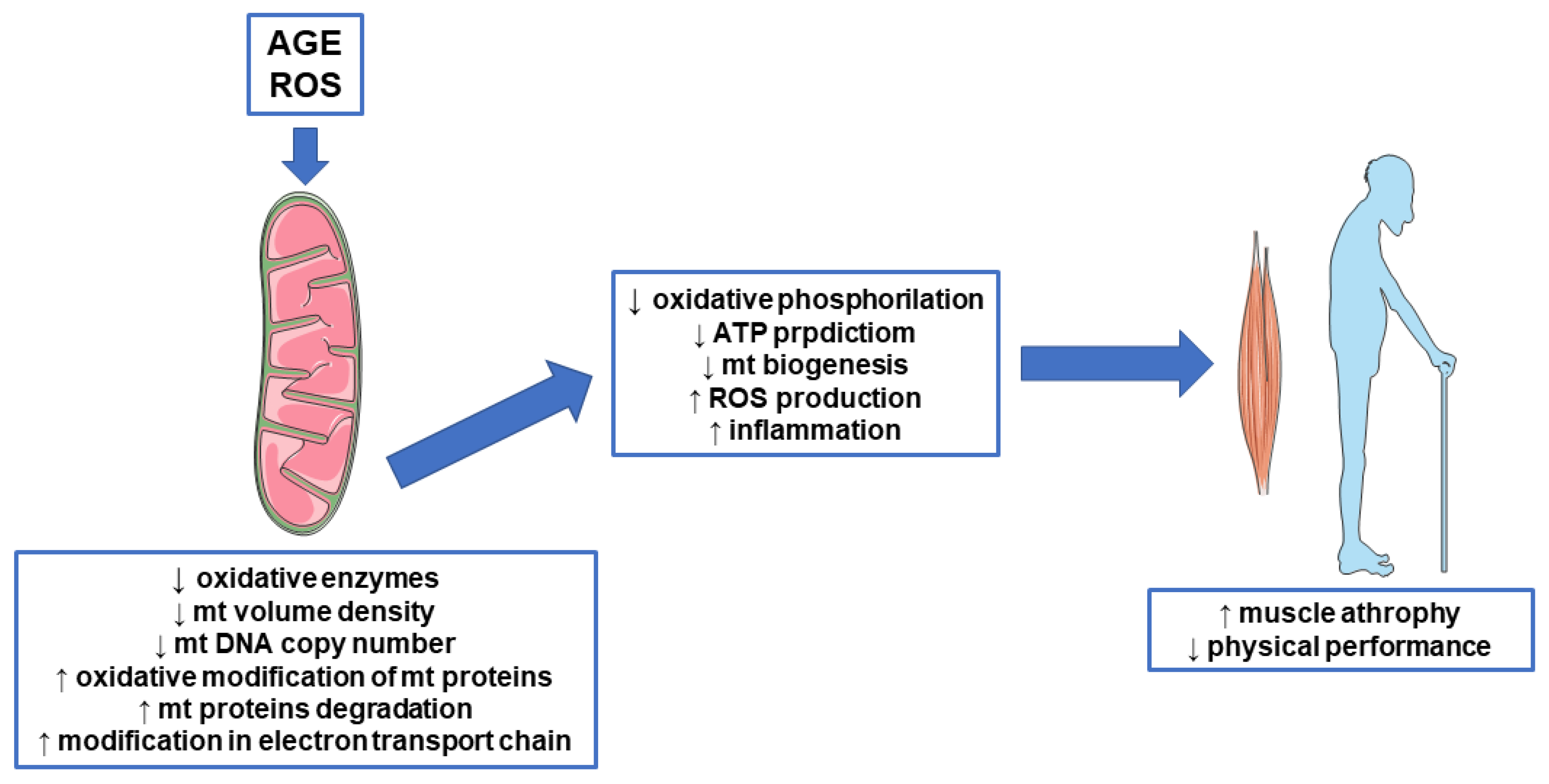{kind=link}
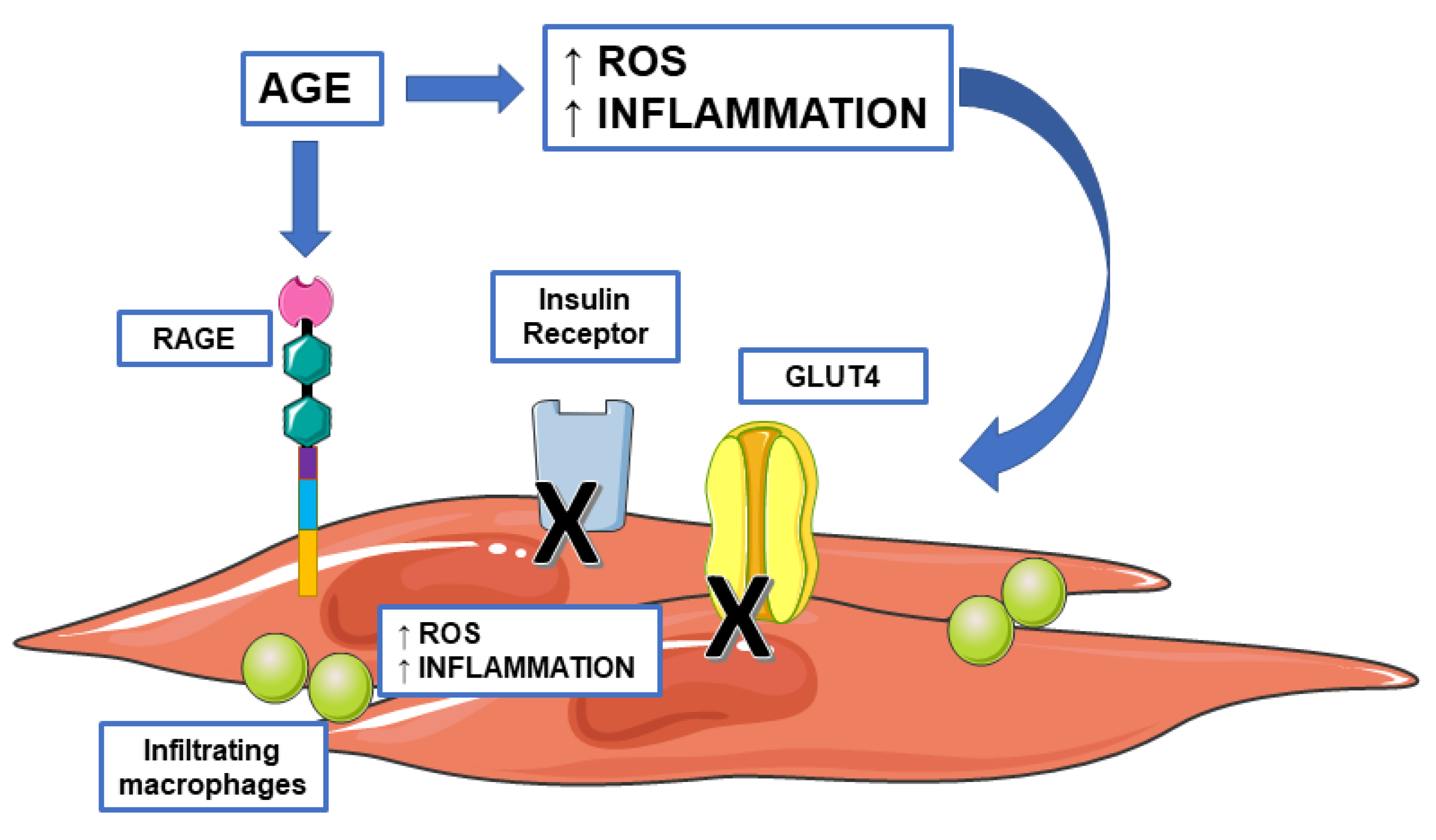{kind=link}
Abstract
1. Sarcopenia in Chronic Kidney Disease: Potential Mechanisms
2. AGE in CKD: Pathogenetic Role
2.1. AGE Synthesis and Accumulation
2.2. Defensive Strategies against AGE
2.3. Effect of AGE on Muscle Function
3. AGE, Mitochondrial Disfunction, and Sarcopenia
3.1. Oxidative Stress and Sarcopenia
3.2. AGE and Mitochondrial Proteins
3.3. AGE and Mitochondrial Biogenesis
3.4. AGE and Mitochondrial Function
4. AGE, Insulin Resistance, and Sarcopenia
4.1. AGE and Inflammation
4.2. AGE and Genes Involved in Insulin Response
5. Therapeutic Intervention: Srage, Anti-Advanced Glycation End Products Agents, and Therapies Targeting Mitochondria
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fahal, I.H. Uraemic sarcopenia: Aetiology and implications. Nephrol. Dial. Transplant. 2014, 29, 1655–1665. [Google Scholar] [CrossRef]
- Esposito, K.; Chiodini, P.; Ceriello, A.; Giugliano, D. A nomogram to estimate the proportion of patients at hemoglobin A1c target <7% with noninsulin antidiabetic drugs in type 2 diabetes: A systematic review of 137 randomized controlled trials with 39,845 patients. Acta Diabetol. 2014, 51, 305–311. [Google Scholar] [CrossRef]
- Pereira, R.A.; Cordeiro, A.C.; Avesani, C.M.; Carrero, J.J.; Lindholm, B.; Amparo, F.C.; Amodeo, C.; Cuppari, L.; Kamimura, M.A. Sarcopenia in chronic kidney disease on conservative therapy: Prevalence and association with mortality. Nephrol. Dial. Transplant. 2015, 30, 1718–1725. [Google Scholar] [CrossRef]
- Hanatani, S.; Izumiya, Y.; Onoue, Y.; Tanaka, T.; Yamamoto, M.; Ishida, T.; Yamamura, S.; Kimura, Y.; Araki, S.; Arima, Y.; et al. Non-invasive testing for sarcopenia predicts future cardiovascular events in patients with chronic kidney disease. Int. J. Cardiol. 2018, 268, 216–221. [Google Scholar] [CrossRef]
- Lai, S.; Muscaritoli, M.; Andreozzi, P.; Sgreccia, A.; De Leo, S.; Mazzaferro, S.; Mitterhofer, A.P.; Pasquali, M.; Protopapa, P.; Spagnoli, A.; et al. Sarcopenia and cardiovascular risk indices in patients with chronic kidney disease on conservative and replacement therapy. Nutrition 2019, 62, 108–114. [Google Scholar] [CrossRef]
- Wang, X.H.; Mitch, W.E. Mechanisms of muscle wasting in chronic kidney disease. Nat. Rev. Nephrol. 2014, 10, 504–516. [Google Scholar] [CrossRef] [PubMed]
- De Souza, V.A.; De Oliveira, D.; Mansur, H.N.; Fernandes, N.M.D.S.; Bastos, M.G. Sarcopenia in Chronic Kidney Disease. Braz. J. Nephrol. 2015, 37, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Enoki, Y.; Maruyama, T. Sarcopenia in Chronic Kidney Disease: Factors, Mechanisms, and Therapeutic Interventions. Biol. Pharm. Bull. 2019, 42, 1437–1445. [Google Scholar] [CrossRef] [PubMed]
- Sifuentes-Franco, S.; Pacheco-Moisés, F.P.; Rodríguez-Carrizalez, A.D.; Miranda-Díaz, A.G. The Role of Oxidative Stress, Mitochondrial Function, and Autophagy in Diabetic Polyneuropathy. J. Diabetes Res. 2017, 2017, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, Y.; Toyoguchi, T.; Inage, K.; Fujimoto, K.; Orita, S.; Suzuki, M.; Kanamoto, H.; Abe, K.; Norimoto, M.; Umimura, T.; et al. Advanced glycation end products are associated with sarcopenia in older women: Aging marker dynamics. J. Women Aging 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Mori, H.; Kuroda, A.; Ishizu, M.; Ohishi, M.; Takashi, Y.; Otsuka, Y.; Taniguchi, S.; Tamaki, M.; Kurahashi, K.; Yoshida, S.; et al. Association of accumulated advanced glycation end-products with a high prevalence of sarcopenia and dynapenia in patients with type 2 diabetes. J. Diabetes Investig. 2019, 10, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Chiappalupi, S.; Sorci, G.; Vukasinovic, A.; Salvadori, L.; Sagheddu, R.; Coletti, D.; Renga, G.; Romani, L.; Donato, R.; Riuzzi, F. Targeting RAGE prevents muscle wasting and prolongs survival in cancer cachexia. J. Cachexia Sarcopenia Muscle 2020, 11, 929–946. [Google Scholar] [CrossRef] [PubMed]
- Takata, T.; Sakasai-Sakai, A.; Takeuchi, M. Impact of intracellular toxic advanced glycation end-products (TAGE) on murine myoblast cell death. Diabetol. Metab. Syndr. 2020, 12, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Thornalley, P.J. Advanced glycation end products in the pathogenesis of chronic kidney disease. Kidney Int. 2018, 93, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J. Dietary AGEs and ALEs and risk to human health by their interaction with the receptor for advanced glycation endproducts (RAGE)–an introduction. Mol. Nutr. Food Res. 2007, 51, 1107–1110. [Google Scholar] [CrossRef]
- Nowotny, K.; Jung, T.; Höhn, A.; Weber, D.; Grune, T. Advanced Glycation End Products and Oxidative Stress in Type 2 Diabetes Mellitus. Biomolecules 2015, 5, 194–222. [Google Scholar] [CrossRef]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Mel’Nikova, T.I.; Porozov, Y.B.; Terentiev, A.A. Oxidative Stress and Advanced Lipoxidation and Glycation End Products (ALEs and AGEs) in Aging and Age-Related Diseases. Oxidative Med. Cell. Longev. 2019, 2019, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Bucala, R.; Vlassara, H. Advanced glycosylation end products in diabetic renal and vascular disease. Am. J. Kidney Dis. 1995, 26, 875–888. [Google Scholar] [CrossRef]
- Shimoike, T.; Inoguchi, T.; Umeda, F.; Nawata, H.; Kawano, K.; Ochi, H. The meaning of serum levels of advanced glycosylation end products in diabetic nephropathy. Metabolism 2000, 49, 1030–1035. [Google Scholar] [CrossRef]
- Tezuka, Y.; Nakaya, I.; Nakayama, K.; Nakayama, M.; Yahata, M.; Soma, J. Methylglyoxal as a prognostic factor in patients with chronic kidney disease. Nephrology 2019, 24, 943–950. [Google Scholar] [CrossRef]
- Calviño, J.; Cigarran, S.; Gonzalez-Tabares, L.; Menendez, N.; Latorre, J.; Cillero, S.; Millan, B.; Cobelo, C.; Sanjurjo-Amado, A.; Quispe, J.; et al. Advanced glycation end products (AGEs) estimated by skin autofluorescence are related with cardiovascular risk in renal transplant. PLOS ONE 2018, 13, e0201118. [Google Scholar] [CrossRef]
- Vlassara, H.; Striker, L.J.; Teichberg, S.; Fuh, H.; Li, Y.M.; Steffes, M. Advanced glycation end products induce glomerular sclerosis and albuminuria in normal rats. Proc. Natl. Acad. Sci. USA 1994, 91, 11704–11708. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, A.; Pires, M.J.; Oliveira, P.A. Pathophysiological Mechanisms of Renal Fibrosis: A Review of Animal Models and Therapeutic Strategies. In Vivo 2017, 31, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced Glycation End Products. Circulation 2006, 114, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Liebisch, M.; Bondeva, T.; Franke, S.; Daniel, C.; Amann, K.; Wolf, G. Activation of the receptor for advanced glycation end products induces nuclear inhibitor of protein phosphatase-1 suppression. Kidney Int. 2014, 86, 103–117. [Google Scholar] [CrossRef]
- Selvin, E.; Halushka, M.K.; Rawlings, A.M.; Hoogeveen, R.C.; Ballantyne, C.M.; Coresh, J.; Astor, B.C. sRAGE and Risk of Diabetes, Cardiovascular Disease, and Death. Diabetes 2013, 62, 2116–2121. [Google Scholar] [CrossRef] [PubMed]
- Dozio, E.; Briganti, S.; Delnevo, A.; Vianello, E.; Ermetici, F.; Secchi, F.; Sardanelli, F.; Morricone, L.; Malavazos, A.E.; Romanelli, M.M.C. Relationship between soluble receptor for advanced glycation end products (sRAGE), body composition and fat distribution in healthy women. Eur. J. Nutr. 2017, 56, 2557–2564. [Google Scholar] [CrossRef] [PubMed]
- Dozio, E.; Corradi, V.; Vianello, E.; Scalzotto, E.; De Cal, M.; Romanelli, M.M.C.; Ronco, C. Increased Levels of sRAGE in Diabetic CKD-G5D Patients: A Potential Protective Mechanism against AGE-Related Upregulation of Fibroblast Growth Factor 23 and Inflammation. Mediat. Inflamm. 2017, 2017, 9845175. [Google Scholar] [CrossRef]
- Dozio, E.; Vianello, E.; Bandera, F.; Longhi, E.; Brizzola, S.; Nebuloni, M.; Romanelli, M.M.C. Soluble Receptor for Advanced Glycation End Products: A Protective Molecule against Intramyocardial Lipid Accumulation in Obese Zucker Rats? Mediat. Inflamm. 2019, 2019, 2712376. [Google Scholar] [CrossRef]
- Dozio, E.; Vianello, E.; Sitzia, C.; Ambrogi, F.; Benedini, S.; Gorini, S.; Rampoldi, B.; Rigolini, R.; Tacchini, L.; Romanelli, M.M.C. Circulating Irisin and esRAGE as Early Biomarkers of Decline of Metabolic Health. J. Clin. Med. 2020, 9, 454. [Google Scholar] [CrossRef]
- Hudson, B.I.; Kalea, A.Z.; Arriero, M.D.M.; Harja, E.; Boulanger, E.; D’Agati, V.; Schmidt, A.M. Interaction of the RAGE Cytoplasmic Domain with Diaphanous-1 Is Required for Ligand-stimulated Cellular Migration through Activation of Rac1 and Cdc42. J. Biol. Chem. 2008, 283, 34457–34468. [Google Scholar] [CrossRef]
- Raucci, A.; Cugusi, S.; Antonelli, A.; Barabino, S.M.; Monti, L.; Bierhaus, A.; Reiss, K.; Saftig, P.; Bianchi, M.E. A soluble form of the receptor for advanced glycation endproducts (RAGE) is produced by proteolytic cleavage of the membrane-bound form by the sheddase a disintegrin and metalloprotease 10 (ADAM10). FASEB J. 2008, 22, 3716–3727. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Thornalley, P.J. Methylglyoxal, glyoxalase 1 and the dicarbonyl proteome. Amino Acids 2010, 42, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P. Glyoxalase I–structure, function and a critical role in the enzymatic defence against glycation. Biochem. Soc. Trans. 2003, 31, 1343–1348. [Google Scholar] [CrossRef]
- Mishra, M.; Prasad, K. AGE–RAGE Stress, Stressors, and Antistressors in Health and Disease. Int. J. Angiol. 2017, 27, 001–012. [Google Scholar] [CrossRef] [PubMed]
- Diesel, W.; Knight, B.K.; Noakes, T.D.; Swanepoel, C.R.; Smit, R.V.Z.; Kaschula, R.O.; Sinclair-Smith, C.C. Morphologic Features of the Myopathy Associated with Chronic Renal Failure. Am. J. Kidney Dis. 1993, 22, 677–684. [Google Scholar] [CrossRef]
- Yabuuchi, J.; Ueda, S.; Yamagishi, S.-I.; Nohara, N.; Nagasawa, H.; Wakabayashi, K.; Matsui, T.; Yuichiro, H.; Kadoguchi, T.; Otsuka, T.; et al. Association of advanced glycation end products with sarcopenia and frailty in chronic kidney disease. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Mastrocola, R.; Nigro, D.; Chiazza, F.; Medana, C.; Bello, F.D.; Boccuzzi, G.; Collino, M.; Aragno, M. Fructose-derived advanced glycation end-products drive lipogenesis and skeletal muscle reprogramming via SREBP-1c dysregulation in mice. Free. Radic. Biol. Med. 2016, 91, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.-Y.; Yang, R.-S.; Sheu, M.-L.; Chan, D.-C.; Yang, T.-H.; Tsai, K.-S.; Chiang, C.-K.; Liu, S.-H. Advanced glycation end-products induce skeletal muscle atrophy and dysfunction in diabetic mice via a RAGE-mediated, AMPK-down-regulated, Akt pathway. J. Pathol. 2016, 238, 470–482. [Google Scholar] [CrossRef]
- Mastrocola, R.; Collino, M.; Nigro, D.; Chiazza, F.; D’Antona, G.; Aragno, M.; Minetto, M.A. Accumulation of Advanced Glycation End-Products and Activation of the SCAP/SREBP Lipogenetic Pathway Occur in Diet-Induced Obese Mouse Skeletal Muscle. PLOS ONE 2015, 10, e0119587. [Google Scholar] [CrossRef]
- Davis, H.M.; Essex, A.L.; Valdez, S.; Deosthale, P.J.; Aref, M.W.; Allen, M.R.; Bonetto, A.; Plotkin, L.I. Short-term pharmacologic RAGE inhibition differentially affects bone and skeletal muscle in middle-aged mice. Bone 2019, 124, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Kubo, A.; Sugioka, Y.; Mitsui, R.; Fukuhara, N.; Nihei, F.; Takeda, Y. Relationship between advanced glycation end-product accumulation and low skeletal muscle mass in Japanese men and women. Geriatr. Gerontol. Int. 2017, 17, 785–790. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Li, C.; Liu, C.; Lin, C.; Lin, W.; Li, T.; Lin, C. Relationship among urinary advanced glycation-end products, skeletal muscle mass and physical performance in community-dwelling older adults. Geriatr. Gerontol. Int. 2019, 19, 1017–1022. [Google Scholar] [CrossRef] [PubMed]
- Semba, R.D.; Bandinelli, S.; Sun, K.; Guralnik, J.M.; Ferrucci, L. Relationship of an advanced glycation end product, plasma carboxymethyl-lysine, with slow walking speed in older adults: The InCHIANTI study. Graefe’s Arch. Clin. Exp. Ophthalmol. 2010, 108, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, L.D.F.; Araújo, A.B.; Quadros, K.R.D.S.; Carbonara, C.E.M.; Dertkigil, S.S.J.; Sposito, A.C.; de Oliveira, R.B. AGEs accumulation is related to muscle degeneration and vascular calcification in peritoneal dialysis patients. Braz. J. Nephrol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Dalal, M.; Ferrucci, L.; Sun, K.; Beck, J.; Fried, L.P.; Semba, R.D. Elevated Serum Advanced Glycation End Products and Poor Grip Strength in Older Community-Dwelling Women. J. Gerontol. Ser. A Boil. Sci. Med. Sci. 2009, 64, 132–137. [Google Scholar] [CrossRef]
- Whitson, H.E.; Arnold, A.M.; Yee, L.M.; Mukamal, K.J.; Kizer, J.R.; Djousse, L.; Ix, J.H.; Siscovick, D.; Tracy, R.P.; Thielke, S.M.; et al. Serum Carboxymethyl-Lysine, Disability, and Frailty in Older Persons: The Cardiovascular Health Study. J. Gerontol. Ser. A Boil. Sci. Med. Sci. 2014, 69, 710–716. [Google Scholar] [CrossRef]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.-S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef]
- Adey, D.; Kumar, R.; McCarthy, J.T.; Nair, K.S. Reduced synthesis of muscle proteins in chronic renal failure. Am. J. Physiol. Metab. 2000, 278, E219–E225. [Google Scholar] [CrossRef]
- Gamboa, J.L.; Billings, F.T.; Bojanowski, M.T.; Gilliam, L.A.; Yu, C.; Roshanravan, B.; Roberts, L.J.; Himmelfarb, J.; Ikizler, T.A.; Brown, N.J. Mitochondrial dysfunction and oxidative stress in patients with chronic kidney disease. Physiol. Rep. 2016, 4, e12780. [Google Scholar] [CrossRef]
- Roshanravan, B.; Kestenbaum, B.; Gamboa, J.; Jubrias, S.A.; Ayers, E.; Curtin, L.; Himmelfarb, J.; de Boer, I.H.; Conley, K.E. CKD and Muscle Mitochondrial Energetics. Am. J. Kidney Dis. 2016, 68, 658–659. [Google Scholar] [CrossRef]
- Gouspillou, G.; Hepple, R.T. Editorial: Mitochondria in Skeletal Muscle Health, Aging and Diseases. Front. Physiol. 2016, 7, 446. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Freire, M.; Scalzo, P.; D’Agostino, J.; Moore, Z.A.; Diaz-Ruiz, A.; Fabbri, E.; Zane, A.; Chen, B.; Becker, K.G.; Lehrmann, E.; et al. Skeletal muscle ex vivo mitochondrial respiration parallels decline in vivo oxidative capacity, cardiorespiratory fitness, and muscle strength: The Baltimore Longitudinal Study of Aging. Aging Cell 2018, 17, e12725. [Google Scholar] [CrossRef]
- Migliavacca, E.; Tay, S.K.H.; Patel, H.P.; Sonntag, T.; Civiletto, G.; McFarlane, C.; Forrester, T.; Barton, S.J.; Leow, M.K.; Antoun, E.; et al. Mitochondrial oxidative capacity and NAD+ biosynthesis are reduced in human sarcopenia across ethnicities. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Talbert, E.E.; Adhihetty, P.J. Reactive oxygen and nitrogen species as intracellular signals in skeletal muscle. J. Physiol. 2011, 589, 2129–2138. [Google Scholar] [CrossRef]
- Dai, D.-F.; Chiao, Y.A.; Marcinek, D.J.; Szeto, H.H.; Rabinovitch, P.S. Mitochondrial oxidative stress in aging and healthspan. Longev. Healthspan 2014, 3, 6. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Wang, L.; Delguste, F.; Durand, A.; Guilbaud, A.; Rousselin, C.; Schmidt, A.M.; Tessier, F.; Boulanger, E.; Neviere, R. Advanced glycation end products receptor RAGE controls myocardial dysfunction and oxidative stress in high-fat fed mice by sustaining mitochondrial dynamics and autophagy-lysosome pathway. Free. Radic. Biol. Med. 2017, 112, 397–410. [Google Scholar] [CrossRef]
- Quirós, P.M.; Langer, T.; López-Otín, C. New roles for mitochondrial proteases in health, ageing and disease. Nat. Rev. Mol. Cell Biol. 2015, 16, 345–359. [Google Scholar] [CrossRef]
- Koltai, E.; Hart, N.; Taylor, A.W.; Goto, S.; Ngo, J.K.; Davies, K.J.A.; Radak, Z. Age-associated declines in mitochondrial biogenesis and protein quality control factors are minimized by exercise training. Am. J. Physiol. Integr. Comp. Physiol. 2012, 303, R127–R134. [Google Scholar] [CrossRef]
- Zhou, H.; Yuan, D.; Gao, W.; Tian, J.; Sun, H.; Yu, S.; Wang, J.; Sun, L. Loss of high-temperature requirement protein A2 protease activity induces mitonuclear imbalance via differential regulation of mitochondrial biogenesis in sarcopenia. IUBMB Life 2020, 72, 1659–1679. [Google Scholar] [CrossRef]
- Popov, L. Mitochondrial biogenesis: An update. J. Cell. Mol. Med. 2020, 24, 4892–4899. [Google Scholar] [CrossRef]
- Safdar, A.; Hamadeh, M.J.; Kaczor, J.J.; Raha, S.; Debeer, J.; Tarnopolsky, M.A. Aberrant Mitochondrial Homeostasis in the Skeletal Muscle of Sedentary Older Adults. PLOS ONE 2010, 5, e10778. [Google Scholar] [CrossRef]
- Garcia, S.; Nissanka, N.; Mareco, E.A.; Rossi, S.; Peralta, S.; Díaz, F.; Rotundo, R.L.; Carvalho, R.F.; Moraes, C.T. Overexpression of PGC-1α in aging muscle enhances a subset of young-like molecular patterns. Aging Cell 2018, 17, e12707. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, M.; Miyashita, K.; Wakino, S.; Mitsuishi, M.; Hayashi, K.; Itoh, H. Chronic kidney disease reduces muscle mitochondria and exercise endurance and its exacerbation by dietary protein through inactivation of pyruvate dehydrogenase. Kidney Int. 2014, 85, 1330–1339. [Google Scholar] [CrossRef] [PubMed]
- Enoki, Y.; Watanabe, H.; Arake, R.; Fujimura, R.; Ishiodori, K.; Imafuku, T.; Nishida, K.; Sugimoto, R.; Nagao, S.; Miyamura, S.; et al. Potential therapeutic interventions for chronic kidney disease-associated sarcopenia via indoxyl sulfate-induced mitochondrial dysfunction. J. Cachexia Sarcopenia Muscle 2017, 8, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.H.; Yue, F.; Saw, S.K.; Foguth, R.; Cannon, J.R.; Shannahan, J.H.; Kuang, S.; Sabbaghi, A.; Carroll, C.C. Advanced Glycation End-Products Suppress Mitochondrial Function and Proliferative Capacity of Achilles Tendon-Derived Fibroblasts. Sci. Rep. 2019, 9, 1–17. [Google Scholar] [CrossRef]
- Lo, M.-C.; Chen, M.-H.; Lee, W.-S.; Lu, C.-I.; Chang, C.-R.; Kao, S.-H.; Lee, H.-M. Nε-(carboxymethyl) lysine-induced mitochondrial fission and mitophagy cause decreased insulin secretion from β-cells. Am. J. Physiol. Metab. 2015, 309, E829–E839. [Google Scholar] [CrossRef]
- Mao, Y.X.; Cai, W.J.; Sun, X.Y.; Dai, P.P.; Li, X.M.; Wang, Q.; Huang, X.L.; He, B.; Wang, P.P.; Wu, G.; et al. RAGE-dependent mitochondria pathway: A novel target of silibinin against apoptosis of osteoblastic cells induced by advanced glycation end products. Cell Death Dis. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Proud, C. Regulation of protein synthesis by insulin. Biochem. Soc. Trans. 2006, 34, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Siew, E.; Pupim, L.; Majchrzak, K.; Shintani, A.; Flakoll, P.; Ikizler, T. Insulin resistance is associated with skeletal muscle protein breakdown in non-diabetic chronic hemodialysis patients. Kidney Int. 2007, 71, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Pupim, L.B.; Flakoll, P.J.; Majchrzak, K.M.; Guy, D.L.A.; Stenvinkel, P.; Ikizler, T.A. Increased muscle protein breakdown in chronic hemodialysis patients with type 2 diabetes mellitus. Kidney Int. 2005, 68, 1857–1865. [Google Scholar] [CrossRef]
- Sandu, O.; Song, K.; Cai, W.; Zheng, F.; Uribarri, J.; Vlassara, H. Insulin Resistance and Type 2 Diabetes in High-Fat-Fed Mice Are Linked to High Glycotoxin Intake. Diabetes 2005, 54, 2314–2319. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.E.; Dordevic, A.L.; Tan, S.M.; Ryan, L.; Coughlan, M.T. Dietary Advanced Glycation End Products and Risk Factors for Chronic Disease: A Systematic Review of Randomised Controlled Trials. Nutrients 2016, 8, 125. [Google Scholar] [CrossRef] [PubMed]
- Peppa, M.; Uribarri, J.; Cai, W.; Lu, M.; Vlassara, H. Glycoxidation and inflammation in renal failure patients. Am. J. Kidney Dis. 2004, 43, 690–695. [Google Scholar] [CrossRef] [PubMed]
- Vlassara, H.; Cai, W.; Goodman, S.; Pyzik, R.; Yong, A.; Chen, X.; Zhu, L.; Neade, T.; Beeri, M.; Silverman, J.M.; et al. Protection against Loss of Innate Defenses in Adulthood by Low Advanced Glycation End Products (AGE) Intake: Role of the Antiinflammatory AGE Receptor-1. J. Clin. Endocrinol. Metab. 2009, 94, 4483–4491. [Google Scholar] [CrossRef] [PubMed]
- Piroddi, M.; Palazzetti, I.; Quintaliani, G.; Pilolli, F.; Montaldi, M.; Valentina, V.; Libetta, C.; Galli, F. Circulating Levels and Dietary Intake of the Advanced Glycation End-product Marker Carboxymethyl Lysine in Chronic Kidney Disease Patients on Conservative Predialysis Therapy: A Pilot Study. J. Ren. Nutr. 2011, 21, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Rashid, G.; Benchetrit, S.; Fishman, D.; Bernheim, J. Effect of advanced glycation end-products on gene expression and synthesis of TNF-α and endothelial nitric oxide synthase by endothelial cells. Kidney Int. 2004, 66, 1099–1106. [Google Scholar] [CrossRef]
- Ruan, H.; Hacohen, N.; Golub, T.R.; Van Parijs, L.; Lodish, H.F. Tumor Necrosis Factor- Suppresses Adipocyte-Specific Genes and Activates Expression of Preadipocyte Genes in 3T3-L1 Adipocytes: Nuclear Factor- B Activation by TNF- Is Obligatory. Diabetes 2002, 51, 1319–1336. [Google Scholar] [CrossRef]
- Riuzzi, F.; Beccafico, S.; Sagheddu, R.; Chiappalupi, S.; Giambanco, I.; Bereshchenko, O.; Riccardi, C.; Sorci, G.; Donato, R. Levels of S100B protein drive the reparative process in acute muscle injury and muscular dystrophy. Sci. Rep. 2017, 7, 12537. [Google Scholar] [CrossRef]
- Matulewicz, N.; Karczewska-Kupczewska, M. Insulin resistance and chronic inflammation. Postępy Hig. Med. Doświadczalnej 2016, 70, 1245–1258. [Google Scholar]
- Miele, C.; Riboulet, A.; Maitan, M.A.; Oriente, F.; Romano, C.; Formisano, P.; Giudicelli, J.; Beguinot, F.; Van Obberghen, E. Human Glycated Albumin Affects Glucose Metabolism in L6 Skeletal Muscle Cells by Impairing Insulin-induced Insulin Receptor Substrate (IRS) Signaling through a Protein Kinase Cα-mediated Mechanism. J. Biol. Chem. 2003, 278, 47376–47387. [Google Scholar] [CrossRef]
- Unoki, H.; Bujo, H.; Yamagishi, S.-I.; Takeuchi, M.; Imaizumi, T.; Saito, Y.; Unoki-Kubota, H. Advanced glycation end products attenuate cellular insulin sensitivity by increasing the generation of intracellular reactive oxygen species in adipocytes. Diabetes Res. Clin. Pract. 2007, 76, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Pinto-Junior, D.C.; Silva, K.S.; Michalani, M.L.; Yonamine, C.Y.; Esteves, J.V.; Fabre, N.T.; Thieme, K.; Catanozi, S.; Okamoto, M.M.; Seraphim, P.M.; et al. Advanced glycation end products-induced insulin resistance involves repression of skeletal muscle GLUT4 expression. Sci. Rep. 2018, 8, 8109. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, S.M.; Dong, H.-J.; Li, Z.; Cai, W.; Altomonte, J.; Thung, S.N.; Zeng, F.; Fisher, E.A.; Vlassara, H. Improved Insulin Sensitivity Is Associated with Restricted Intake of Dietary Glycoxidation Products in the db/db Mouse. Diabetes 2002, 51, 2082–2089. [Google Scholar] [CrossRef]
- Kim, T.N.; Park, M.S.; Lee, E.J.; Chung, H.S.; Yoo, H.J.; Kang, H.J.; Song, W.; Baik, S.H.; Choi, K.M. The association of low muscle mass with soluble receptor for advanced glycation end products (sRAGE): The Korean Sarcopenic Obesity Study (KSOS). Diabetes/Metab. Res. Rev. 2018, 34, e2974. [Google Scholar] [CrossRef] [PubMed]
- Jud, P.; Sourij, H. Therapeutic options to reduce advanced glycation end products in patients with diabetes mellitus: A review. Diabetes Res. Clin. Pract. 2019, 148, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Bongarzone, S.; Savickas, V.; Luzi, F.; Gee, A.D. Targeting the Receptor for Advanced Glycation Endproducts (RAGE): A Medicinal Chemistry Perspective. J. Med. Chem. 2017, 60, 7213–7232. [Google Scholar] [CrossRef]
- Melov, S.; Tarnopolsky, M.A.; Beckman, K.; Felkey, K.; Hubbard, A. Resistance Exercise Reverses Aging in Human Skeletal Muscle. PLoS ONE 2007, 2, e465. [Google Scholar] [CrossRef]
- Tarnopolsky, M.A.T.A. Mitochondrial DNA shifting in older adults following resistance exercise trainingThis paper article is one of a selection of papers published in this Special Issue, entitled 14th International Biochemistry of Exercise Conference–Muscles as Molecular and Metabolic Machines, and has undergone the Journal’s usual peer review process. Appl. Physiol. Nutr. Metab. 2009, 34, 348–354. [Google Scholar] [CrossRef]
- Castaneda, C.; Gordon, P.L.; Uhlin, K.L.; Levey, A.S.; Kehayias, J.J.; Dwyer, J.T.; Fielding, R.A.; Roubenoff, R.; Singh, M.F. Resistance Training to Counteract the Catabolism of a Low-Protein Diet in Patients with Chronic Renal Insufficiency. Ann. Intern. Med. 2001, 135, 965–976. [Google Scholar] [CrossRef]
- Castaneda, C.; Gordon, P.L.; Parker, R.C.; Uhlin, K.L.; Roubenoff, R.; Levey, A.S. Resistance training to reduce the malnutrition-inflammation complex syndrome of chronic kidney disease. Am. J. Kidney Dis. 2004, 43, 607–616. [Google Scholar] [CrossRef]
- Balakrishnan, V.S.; Rao, M.; Menon, V.; Gordon, P.L.; Pilichowska, M.; Castaneda, F.; Castaneda-Sceppa, C. Resistance Training Increases Muscle Mitochondrial Biogenesis in Patients with Chronic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2010, 5, 996–1002. [Google Scholar] [CrossRef] [PubMed]
- Granata, S.; Gassa, A.D.; Tomei, P.; Lupo, A.; Zaza, G. Mitochondria: A new therapeutic target in chronic kidney disease. Nutr. Metab. 2015, 12, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Fougère, B.; Van Kan, G.A.; Vellas, B.; Cesari, M. Redox Systems, Antioxidants and Sarcopenia. Curr. Protein Pept. Sci. 2018, 19, 643–648. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dozio, E.; Vettoretti, S.; Lungarella, G.; Messa, P.; Corsi Romanelli, M.M. Sarcopenia in Chronic Kidney Disease: Focus on Advanced Glycation End Products as Mediators and Markers of Oxidative Stress. Biomedicines 2021, 9, 405. https://doi.org/10.3390/biomedicines9040405
Dozio E, Vettoretti S, Lungarella G, Messa P, Corsi Romanelli MM. Sarcopenia in Chronic Kidney Disease: Focus on Advanced Glycation End Products as Mediators and Markers of Oxidative Stress. Biomedicines. 2021; 9(4):405. https://doi.org/10.3390/biomedicines9040405
Chicago/Turabian StyleDozio, Elena, Simone Vettoretti, Giuseppe Lungarella, Piergiorgio Messa, and Massimiliano M. Corsi Romanelli. 2021. "Sarcopenia in Chronic Kidney Disease: Focus on Advanced Glycation End Products as Mediators and Markers of Oxidative Stress" Biomedicines 9, no. 4: 405. https://doi.org/10.3390/biomedicines9040405
APA StyleDozio, E., Vettoretti, S., Lungarella, G., Messa, P., & Corsi Romanelli, M. M. (2021). Sarcopenia in Chronic Kidney Disease: Focus on Advanced Glycation End Products as Mediators and Markers of Oxidative Stress. Biomedicines, 9(4), 405. https://doi.org/10.3390/biomedicines9040405