
Hypoxia/HIF Modulates Immune Responses



Abstract
1. Introduction
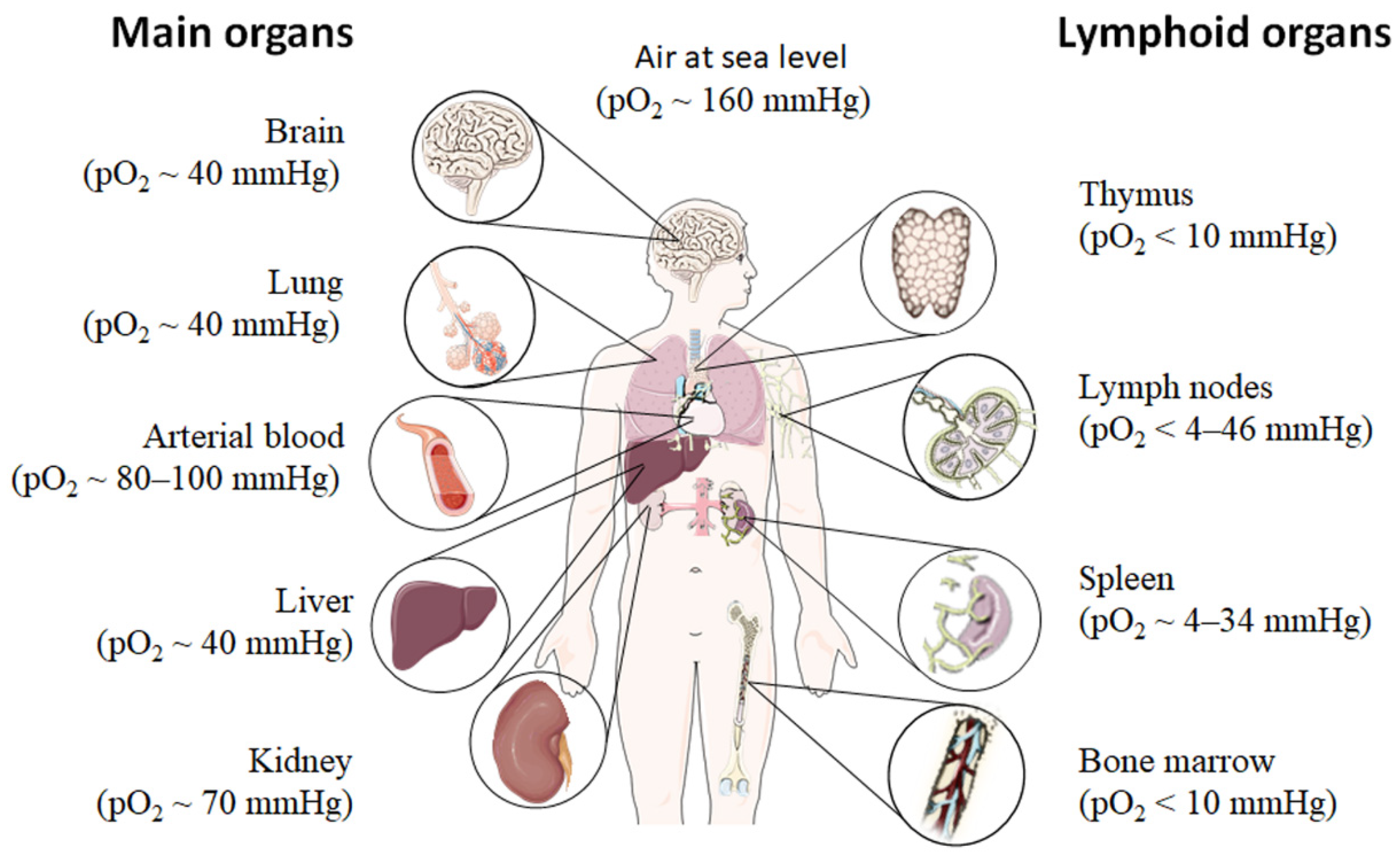
2. Physiological Hypoxia Influences Immunity
3. Pathophysiological Hypoxia Shapes Immune Response
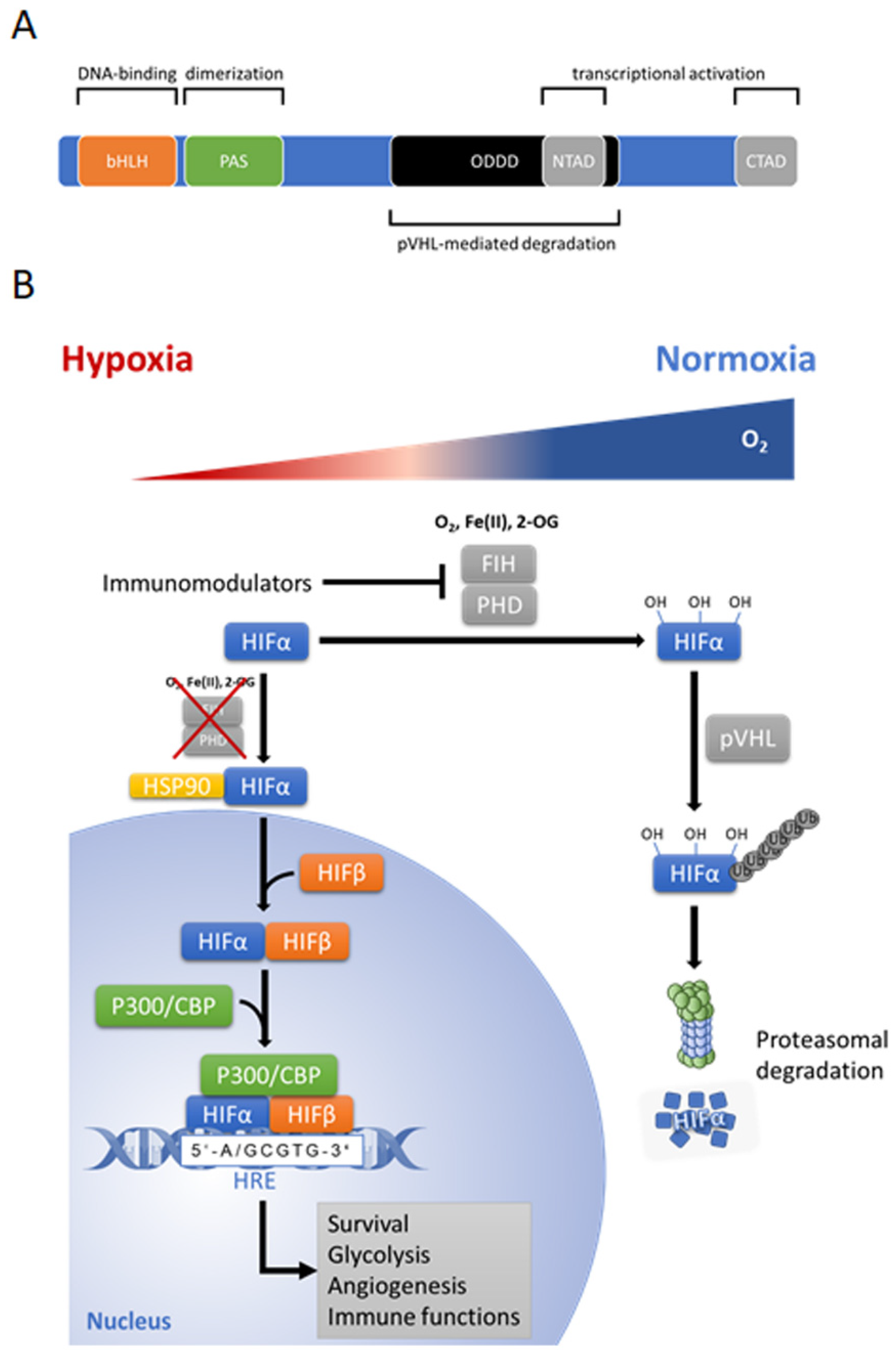
4. Hypoxia-Inducible Factors (HIF’s): Structure, Function, and Regulation
4.1. HIF’s—“Master Regulators” of Cellular Response to Hypoxia
4.2. Cellular Response to Hypoxia Beyond HIF’s
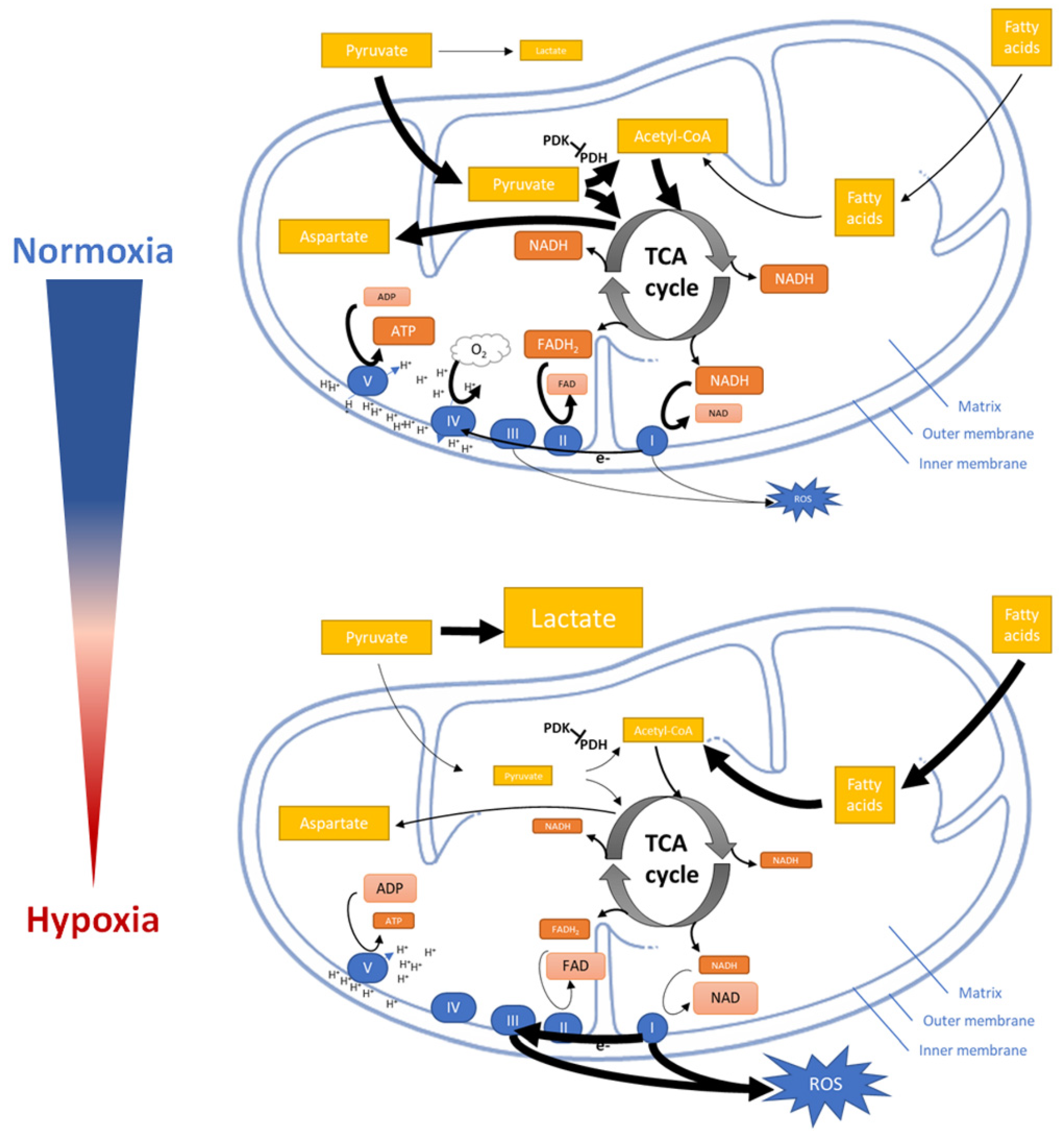
4.3. Mitochondrial Response to Hypoxia
5. Hypoxia-Inducible Factors in the Regulation of Immune Response
5.1. HIFs in Myeloid Cell Function
5.2. HIFs in Adaptive Immunity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | HIF Mediated Effects | Ref. |
|---|---|---|
| CD4+ T cells | HIF-1: survival ↑, glycolysis ↑, Th17 differentiation and effector function↑, Th1 differentiation ↑, effector function ↓ (IFN-γ↓), Treg differentiation↓, Tr1 differentiation ↓ | [192,195,196,197,198,199,200,201,202] |
| HIF-2: Treg differentiation ↓ | [203] | |
| CD8+ T cells | HIF-1: survival ↑ glycolysis ↑ effector function ↑ (anti-viral infection ↑, antitumor capacity ↑; IL-13 ↑) CD8+ TC2 cell differentiation ↑ effector molecule↑ | [27,204,205,206,207] |
| HIF-2: survival ↑ effector function ↑ (IFN-γ and TNF-α ↑) | [208] | |
| B cells | HIF-1: survival ↑, glycolysis ↑, cell cycle ↑, effector function (IgG and IgM antibodies↓, IL-10 ↑), chemotherapeutic antitumor effect ↓ | [209,210,211] |
| HIF-2: unknown |
5.3. HIFs in Innate Lymphoid Immune Response
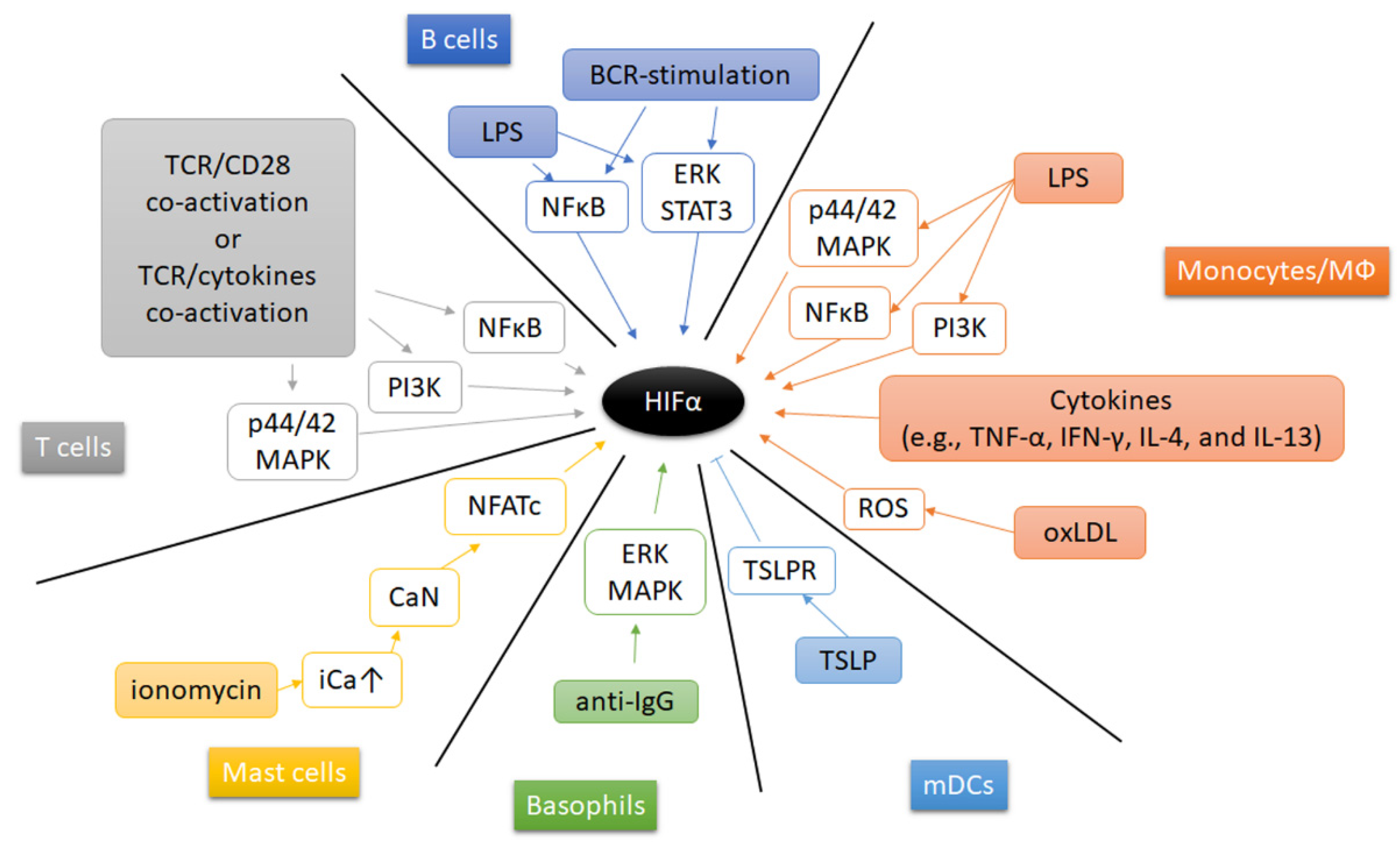
6. Extracellular Signals Regulating HIF’s in Immune Cells Apart from Hypoxia
7. Summary and Outlook: How to Treat by Targeting HIF to Modulate Immunity
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Taylor, C.T.; Colgan, S.P. Regulation of immunity and inflammation by hypoxia in immunological niches. Nat. Rev. Immunol. 2017, 17, 774. [Google Scholar] [CrossRef]
- Beerman, I.; Luis, T.C.; Singbrant, S.; Lo Celso, C.; Mendez-Ferrer, S. The evolving view of the hematopoietic stem cell niche. Exp. Hematol. 2017, 50, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.K.; Zuniga-Pflucker, J.C. An overview of the intrathymic intricacies of T cell development. J. Immunol. 2014, 192, 4017–4023. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.W.; Karakasheva, T.A.; Hicks, P.D.; Bass, A.J.; Rustgi, A.K. The tumor microenvironment in esophageal cancer. Oncogene 2016, 35, 5337–5349. [Google Scholar] [CrossRef]
- Maru, Y. The lung metastatic niche. J. Mol. Med. (Berl) 2015, 93, 1185–1192. [Google Scholar] [CrossRef]
- Biswas, S.; Davis, H.; Irshad, S.; Sandberg, T.; Worthley, D.; Leedham, S. Microenvironmental control of stem cell fate in intestinal homeostasis and disease. J. Pathol. 2015, 237, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Hallenbeck, J.M.; Hansson, G.K.; Becker, K.J. Immunology of ischemic vascular disease: Plaque to attack. Trends Immunol. 2005, 26, 550–556. [Google Scholar] [CrossRef]
- Multhoff, G.; Molls, M.; Radons, J. Chronic inflammation in cancer development. Front. Immunol. 2011, 2, 98. [Google Scholar] [CrossRef]
- Lin, N.; Simon, M.C. Hypoxia-inducible factors: Key regulators of myeloid cells during inflammation. J. Clin. Investig. 2016, 126, 3661–3671. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.T.; Doherty, G.; Fallon, P.G.; Cummins, E.P. Hypoxia-dependent regulation of inflammatory pathways in immune cells. J. Clin. Investig. 2016, 126, 3716–3724. [Google Scholar] [CrossRef]
- Scholz, C.C.; Taylor, C.T. Targeting the HIF pathway in inflammation and immunity. Curr. Opin. Pharmacol. 2013, 13, 646–653. [Google Scholar] [CrossRef] [PubMed]
- McNamee, E.N.; Korns Johnson, D.; Homann, D.; Clambey, E.T. Hypoxia and hypoxia-inducible factors as regulators of T cell development, differentiation, and function. Immunol. Res. 2013, 55, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Carreau, A.; El Hafny-Rahbi, B.; Matejuk, A.; Grillon, C.; Kieda, C. Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J. Cell Mol. Med. 2011, 15, 1239–1253. [Google Scholar] [CrossRef]
- Boveris, D.L.; Boveris, A. Oxygen delivery to the tissues and mitochondrial respiration. Front. Biosci. 2007, 12, 1014–1023. [Google Scholar] [CrossRef]
- Leach, R.M.; Treacher, D.F. Oxygen transport-2. Tissue hypoxia. BMJ 1998, 317, 1370–1373. [Google Scholar] [CrossRef]
- Available online: http://smart.servier.com/ (accessed on 1 March 2021).
- Jiang, B.H.; Semenza, G.L.; Bauer, C.; Marti, H.H. Hypoxia-inducible factor 1 levels vary exponentially over a physiologically relevant range of O2 tension. Am. J. Physiol. 1996, 271, C1172–C1180. [Google Scholar] [CrossRef] [PubMed]
- Glover, L.E.; Lee, J.S.; Colgan, S.P. Oxygen metabolism and barrier regulation in the intestinal mucosa. J. Clin. Investig. 2016, 126, 3680–3688. [Google Scholar] [CrossRef]
- Maxwell, P.H.; Ferguson, D.J.; Nicholls, L.G.; Iredale, J.P.; Pugh, C.W.; Johnson, M.H.; Ratcliffe, P.J. Sites of erythropoietin production. Kidney Int. 1997, 51, 393–401. [Google Scholar] [CrossRef]
- Haase, V.H. Regulation of erythropoiesis by hypoxia-inducible factors. Blood Rev. 2013, 27, 41–53. [Google Scholar] [CrossRef]
- Macklin, P.S.; McAuliffe, J.; Pugh, C.W.; Yamamoto, A. Hypoxia and HIF pathway in cancer and the placenta. Placenta 2017, 56, 8–13. [Google Scholar] [CrossRef]
- Wang, W.; Winlove, C.P.; Michel, C.C. Oxygen partial pressure in outer layers of skin of human finger nail folds. J. Physiol. 2003, 549, 855–863. [Google Scholar] [CrossRef]
- Takubo, K.; Goda, N.; Yamada, W.; Iriuchishima, H.; Ikeda, E.; Kubota, Y.; Shima, H.; Johnson, R.S.; Hirao, A.; Suematsu, M.; et al. Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell 2010, 7, 391–402. [Google Scholar] [CrossRef]
- Parmar, K.; Mauch, P.; Vergilio, J.A.; Sackstein, R.; Down, J.D. Distribution of hematopoietic stem cells in the bone marrow according to regional hypoxia. Proc. Natl. Acad. Sci. USA 2007, 104, 5431–5436. [Google Scholar] [CrossRef]
- Hale, L.P.; Braun, R.D.; Gwinn, W.M.; Greer, P.K.; Dewhirst, M.W. Hypoxia in the thymus: Role of oxygen tension in thymocyte survival. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H1467–H1477. [Google Scholar] [CrossRef]
- Braun, R.D.; Lanzen, J.L.; Snyder, S.A.; Dewhirst, M.W. Comparison of tumor and normal tissue oxygen tension measurements using OxyLite or microelectrodes in rodents. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H2533–H2544. [Google Scholar] [CrossRef]
- Caldwell, C.C.; Kojima, H.; Lukashev, D.; Armstrong, J.; Farber, M.; Apasov, S.G.; Sitkovsky, M.V. Differential effects of physiologically relevant hypoxic conditions on T lymphocyte development and effector functions. J. Immunol. 2001, 167, 6140–6149. [Google Scholar] [CrossRef]
- Huang, J.H.; Cardenas-Navia, L.I.; Caldwell, C.C.; Plumb, T.J.; Radu, C.G.; Rocha, P.N.; Wilder, T.; Bromberg, J.S.; Cronstein, B.N.; Sitkovsky, M.; et al. Requirements for T lymphocyte migration in explanted lymph nodes. J. Immunol. 2007, 178, 7747–7755. [Google Scholar] [CrossRef] [PubMed]
- Eliasson, P.; Rehn, M.; Hammar, P.; Larsson, P.; Sirenko, O.; Flippin, L.A.; Cammenga, J.; Jonsson, J.I. Hypoxia mediates low cell-cycle activity and increases the proportion of long-term-reconstituting hematopoietic stem cells during in vitro culture. Exp. Hematol. 2010, 38, 301.e302–310.e302. [Google Scholar] [CrossRef] [PubMed]
- Hermitte, F.; Brunet de la Grange, P.; Belloc, F.; Praloran, V.; Ivanovic, Z. Very low O2 concentration (0.1%) favors G0 return of dividing CD34+ cells. Stem Cells 2006, 24, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Ivanovic, Z.; Hermitte, F.; Brunet de la Grange, P.; Dazey, B.; Belloc, F.; Lacombe, F.; Vezon, G.; Praloran, V. Simultaneous maintenance of human cord blood SCID-repopulating cells and expansion of committed progenitors at low O2 concentration (3%). Stem Cells 2004, 22, 716–724. [Google Scholar] [CrossRef]
- Cipolleschi, M.G.; Dello Sbarba, P.; Olivotto, M. The role of hypoxia in the maintenance of hematopoietic stem cells. Blood 1993, 82, 2031–2037. [Google Scholar] [CrossRef]
- Cho, S.H.; Raybuck, A.L.; Stengel, K.; Wei, M.; Beck, T.C.; Volanakis, E.; Thomas, J.W.; Hiebert, S.; Haase, V.H.; Boothby, M.R. Germinal centre hypoxia and regulation of antibody qualities by a hypoxia response system. Nature 2016, 537, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Abbott, R.K.; Thayer, M.; Labuda, J.; Silva, M.; Philbrook, P.; Cain, D.W.; Kojima, H.; Hatfield, S.; Sethumadhavan, S.; Ohta, A.; et al. Germinal Center Hypoxia Potentiates Immunoglobulin Class Switch Recombination. J. Immunol. 2016, 197, 4014–4020. [Google Scholar] [CrossRef] [PubMed]
- Colgan, S.P.; Campbell, E.L.; Kominsky, D.J. Hypoxia and Mucosal Inflammation. Annu. Rev. Pathol. 2016, 11, 77–100. [Google Scholar] [CrossRef] [PubMed]
- Labiano, S.; Palazon, A.; Melero, I. Immune response regulation in the tumor microenvironment by hypoxia. Semin. Oncol. 2015, 42, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Unwith, S.; Zhao, H.; Hennah, L.; Ma, D. The potential role of HIF on tumour progression and dissemination. Int. J. Cancer 2015, 136, 2491–2503. [Google Scholar] [CrossRef]
- Lee, P.; Chandel, N.S.; Simon, M.C. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat. Rev. Mol. Cell Biol. 2020, 21, 268–283. [Google Scholar] [CrossRef]
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-induced angiogenesis: Good and evil. Genes Cancer 2011, 2, 1117–1133. [Google Scholar] [CrossRef]
- Chiu, D.K.; Xu, I.M.; Lai, R.K.; Tse, A.P.; Wei, L.L.; Koh, H.Y.; Li, L.L.; Lee, D.; Lo, R.C.; Wong, C.M.; et al. Hypoxia induces myeloid-derived suppressor cell recruitment to hepatocellular carcinoma through chemokine (C-C motif) ligand 26. Hepatology 2016, 64, 797–813. [Google Scholar] [CrossRef]
- Chiu, D.K.; Tse, A.P.; Xu, I.M.; Di Cui, J.; Lai, R.K.; Li, L.L.; Koh, H.Y.; Tsang, F.H.; Wei, L.L.; Wong, C.M.; et al. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat. Commun 2017, 8, 517. [Google Scholar] [CrossRef]
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.I.; Cheng, P.; Cho, H.I.; Celis, E.; Quiceno, D.G.; Padhya, T.; et al. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010, 207, 2439–2453. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Doedens, A.L.; Stockmann, C.; Rubinstein, M.P.; Liao, D.; Zhang, N.; DeNardo, D.G.; Coussens, L.M.; Karin, M.; Goldrath, A.W.; Johnson, R.S. Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer Res. 2010, 70, 7465–7475. [Google Scholar] [CrossRef]
- Loeffler, D.A.; Keng, P.C.; Baggs, R.B.; Lord, E.M. Lymphocytic infiltration and cytotoxicity under hypoxic conditions in the EMT6 mouse mammary tumor. Int J. Cancer 1990, 45, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Mazurek, J. Diagnostic and therapeutic role of a psychiatrist in a diagnostic and therapeutic team in a de-habituation ward. Psychiatr. Pol. 1975, 9, 349–350. [Google Scholar] [PubMed]
- Yousaf, I.; Kaeppler, J.; Frost, S.; Seymour, L.W.; Jacobus, E.J. Attenuation of the Hypoxia Inducible Factor Pathway after Oncolytic Adenovirus Infection Coincides with Decreased Vessel Perfusion. Cancers 2020, 12, 851. [Google Scholar] [CrossRef] [PubMed]
- Devraj, G.; Beerlage, C.; Brune, B.; Kempf, V.A. Hypoxia and HIF-1 activation in bacterial infections. Microbes Infect. 2017, 19, 144–156. [Google Scholar] [CrossRef]
- Friedrich, D.; Fecher, R.A.; Rupp, J.; Deepe, G.S., Jr. Impact of HIF-1alpha and hypoxia on fungal growth characteristics and fungal immunity. Microbes Infect. 2017, 19, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Werth, N.; Beerlage, C.; Rosenberger, C.; Yazdi, A.S.; Edelmann, M.; Amr, A.; Bernhardt, W.; von Eiff, C.; Becker, K.; Schafer, A.; et al. Activation of hypoxia inducible factor 1 is a general phenomenon in infections with human pathogens. PLoS ONE 2010, 5, e11576. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, K.; Taylor, C.T. The impact of hypoxia on bacterial infection. FEBS J. 2015, 282, 2260–2266. [Google Scholar] [CrossRef]
- Schaible, B.; McClean, S.; Selfridge, A.; Broquet, A.; Asehnoune, K.; Taylor, C.T.; Schaffer, K. Hypoxia modulates infection of epithelial cells by Pseudomonas aeruginosa. PLoS ONE 2013, 8, e56491. [Google Scholar] [CrossRef]
- Zeitouni, N.E.; Chotikatum, S.; von Kockritz-Blickwede, M.; Naim, H.Y. The impact of hypoxia on intestinal epithelial cell functions: Consequences for invasion by bacterial pathogens. Mol. Cell. Pediatr. 2016, 3, 14. [Google Scholar] [CrossRef] [PubMed]
- Schaible, B.; Taylor, C.T.; Schaffer, K. Hypoxia increases antibiotic resistance in Pseudomonas aeruginosa through altering the composition of multidrug efflux pumps. Antimicrob. Agents Chemother. 2012, 56, 2114–2118. [Google Scholar] [CrossRef]
- Karhausen, J.; Haase, V.H.; Colgan, S.P. Inflammatory hypoxia: Role of hypoxia-inducible factor. Cell Cycle 2005, 4, 256–258. [Google Scholar] [CrossRef]
- Campbell, E.L.; Bruyninckx, W.J.; Kelly, C.J.; Glover, L.E.; McNamee, E.N.; Bowers, B.E.; Bayless, A.J.; Scully, M.; Saeedi, B.J.; Golden-Mason, L.; et al. Transmigrating neutrophils shape the mucosal microenvironment through localized oxygen depletion to influence resolution of inflammation. Immunity 2014, 40, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Bone, N.; Jiang, S.; Park, D.W.; Tadie, J.M.; Deshane, J.; Rodriguez, C.A.; Pittet, J.F.; Abraham, E.; Zmijewski, J.W. AMP-Activated Protein Kinase and Glycogen Synthase Kinase 3beta Modulate the Severity of Sepsis-Induced Lung Injury. Mol. Med. 2016, 21, 937–950. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion--from mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Harten, S.K.; Ashcroft, M.; Maxwell, P.H. Prolyl hydroxylase domain inhibitors: A route to HIF activation and neuroprotection. Antioxid. Redox Signal. 2010, 12, 459–480. [Google Scholar] [CrossRef]
- Jian, Z.; Liu, R.; Zhu, X.; Smerin, D.; Zhong, Y.; Gu, L.; Fang, W.; Xiong, X. The Involvement and Therapy Target of Immune Cells After Ischemic Stroke. Front. Immunol. 2019, 10, 2167. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Carmeliet, P. Hypoxia and inflammation. N. Engl. J. Med. 2011, 364, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factor 1: Master regulator of O2 homeostasis. Curr. Opin. Genet. Dev. 1998, 8, 588–594. [Google Scholar] [CrossRef]
- Schodel, J.; Oikonomopoulos, S.; Ragoussis, J.; Pugh, C.W.; Ratcliffe, P.J.; Mole, D.R. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood 2011, 117, e207–e217. [Google Scholar] [CrossRef] [PubMed]
- Wenger, R.H.; Stiehl, D.P.; Camenisch, G. Integration of oxygen signaling at the consensus HRE. Sci. STKE 2005, 2005, re12. [Google Scholar] [CrossRef]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed]
- Hogenesch, J.B.; Chan, W.K.; Jackiw, V.H.; Brown, R.C.; Gu, Y.Z.; Pray-Grant, M.; Perdew, G.H.; Bradfield, C.A. Characterization of a subset of the basic-helix-loop-helix-PAS superfamily that interacts with components of the dioxin signaling pathway. J. Biol. Chem. 1997, 272, 8581–8593. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr.; Ratcliffe, P.J. Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef]
- Gu, Y.Z.; Moran, S.M.; Hogenesch, J.B.; Wartman, L.; Bradfield, C.A. Molecular characterization and chromosomal localization of a third alpha-class hypoxia inducible factor subunit, HIF3alpha. Gene Expr. 1998, 7, 205–213. [Google Scholar] [PubMed]
- Makino, Y.; Uenishi, R.; Okamoto, K.; Isoe, T.; Hosono, O.; Tanaka, H.; Kanopka, A.; Poellinger, L.; Haneda, M.; Morimoto, C. Transcriptional up-regulation of inhibitory PAS domain protein gene expression by hypoxia-inducible factor 1 (HIF-1): A negative feedback regulatory circuit in HIF-1-mediated signaling in hypoxic cells. J. Biol. Chem. 2007, 282, 14073–14082. [Google Scholar] [CrossRef]
- Makino, Y.; Kanopka, A.; Wilson, W.J.; Tanaka, H.; Poellinger, L. Inhibitory PAS domain protein (IPAS) is a hypoxia-inducible splicing variant of the hypoxia-inducible factor-3alpha locus. J. Biol. Chem. 2002, 277, 32405–32408. [Google Scholar] [CrossRef]
- Makino, Y.; Cao, R.; Svensson, K.; Bertilsson, G.; Asman, M.; Tanaka, H.; Cao, Y.; Berkenstam, A.; Poellinger, L. Inhibitory PAS domain protein is a negative regulator of hypoxia-inducible gene expression. Nature 2001, 414, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology (Bethesda) 2009, 24, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G. Proline hydroxylation and gene expression. Annu. Rev. Biochem. 2005, 74, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; White, S.B.; Zhao, Q.; Lee, F.S. HIF-1alpha binding to VHL is regulated by stimulus-sensitive proline hydroxylation. Proc. Natl. Acad. Sci. USA 2001, 98, 9630–9635. [Google Scholar] [CrossRef]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef]
- Lando, D.; Peet, D.J.; Whelan, D.A.; Gorman, J.J.; Whitelaw, M.L. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science 2002, 295, 858–861. [Google Scholar] [CrossRef]
- Mahon, P.C.; Hirota, K.; Semenza, G.L. FIH-1: A novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001, 15, 2675–2686. [Google Scholar] [CrossRef]
- Walmsley, S.R.; Print, C.; Farahi, N.; Peyssonnaux, C.; Johnson, R.S.; Cramer, T.; Sobolewski, A.; Condliffe, A.M.; Cowburn, A.S.; Johnson, N.; et al. Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J. Exp. Med. 2005, 201, 105–115. [Google Scholar] [CrossRef]
- Cramer, T.; Yamanishi, Y.; Clausen, B.E.; Forster, I.; Pawlinski, R.; Mackman, N.; Haase, V.H.; Jaenisch, R.; Corr, M.; Nizet, V.; et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell 2003, 112, 645–657. [Google Scholar] [CrossRef]
- Jantsch, J.; Chakravortty, D.; Turza, N.; Prechtel, A.T.; Buchholz, B.; Gerlach, R.G.; Volke, M.; Glasner, J.; Warnecke, C.; Wiesener, M.S.; et al. Hypoxia and hypoxia-inducible factor-1 alpha modulate lipopolysaccharide-induced dendritic cell activation and function. J. Immunol. 2008, 180, 4697–4705. [Google Scholar] [CrossRef]
- Burrows, N.; Maxwell, P.H. Hypoxia and B cells. Exp. Cell Res. 2017, 356, 197–203. [Google Scholar] [CrossRef]
- Tao, J.H.; Barbi, J.; Pan, F. Hypoxia-inducible factors in T lymphocyte differentiation and function. A Review in the Theme: Cellular Responses to Hypoxia. Am. J. Physiol. Cell Physiol. 2015, 309, C580–C589. [Google Scholar] [CrossRef] [PubMed]
- Palazon, A.; Goldrath, A.W.; Nizet, V.; Johnson, R.S. HIF transcription factors, inflammation, and immunity. Immunity 2014, 41, 518–528. [Google Scholar] [CrossRef]
- Zinkernagel, A.S.; Johnson, R.S.; Nizet, V. Hypoxia inducible factor (HIF) function in innate immunity and infection. J. Mol. Med. (Berl.) 2007, 85, 1339–1346. [Google Scholar] [CrossRef]
- Saxena, K.; Jolly, M.K. Acute vs. Chronic vs. Cyclic Hypoxia: Their Differential Dynamics, Molecular Mechanisms, and Effects on Tumor Progression. Biomolecules 2019, 9, 339. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.H.; Lee, C.H.; Liang, J.A.; Yu, C.Y.; Shyu, W.C. Cycling hypoxia increases U87 glioma cell radioresistance via ROS induced higher and long-term HIF-1 signal transduction activity. Oncol. Rep. 2010, 24, 1629–1636. [Google Scholar] [CrossRef] [PubMed]
- Galanis, A.; Pappa, A.; Giannakakis, A.; Lanitis, E.; Dangaj, D.; Sandaltzopoulos, R. Reactive oxygen species and HIF-1 signalling in cancer. Cancer Lett. 2008, 266, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Nanduri, J.; Vaddi, D.R.; Khan, S.A.; Wang, N.; Makerenko, V.; Prabhakar, N.R. Xanthine oxidase mediates hypoxia-inducible factor-2alpha degradation by intermittent hypoxia. PLoS ONE 2013, 8, e75838. [Google Scholar] [CrossRef]
- Nanduri, J.; Wang, N.; Yuan, G.; Khan, S.A.; Souvannakitti, D.; Peng, Y.J.; Kumar, G.K.; Garcia, J.A.; Prabhakar, N.R. Intermittent hypoxia degrades HIF-2alpha via calpains resulting in oxidative stress: Implications for recurrent apnea-induced morbidities. Proc. Natl. Acad. Sci. USA 2009, 106, 1199–1204. [Google Scholar] [CrossRef]
- Ravi, R.; Mookerjee, B.; Bhujwalla, Z.M.; Sutter, C.H.; Artemov, D.; Zeng, Q.; Dillehay, L.E.; Madan, A.; Semenza, G.L.; Bedi, A. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha. Genes Dev. 2000, 14, 34–44. [Google Scholar]
- Isaacs, J.S.; Jung, Y.J.; Mimnaugh, E.G.; Martinez, A.; Cuttitta, F.; Neckers, L.M. Hsp90 regulates a von Hippel Lindau-independent hypoxia-inducible factor-1 alpha-degradative pathway. J. Biol. Chem. 2002, 277, 29936–29944. [Google Scholar] [CrossRef]
- Selfridge, A.C.; Cavadas, M.A.; Scholz, C.C.; Campbell, E.L.; Welch, L.C.; Lecuona, E.; Colgan, S.P.; Barrett, K.E.; Sporn, P.H.; Sznajder, J.I.; et al. Hypercapnia Suppresses the HIF-dependent Adaptive Response to Hypoxia. J. Biol. Chem. 2016, 291, 11800–11808. [Google Scholar] [CrossRef]
- Hubbi, M.E.; Gilkes, D.M.; Hu, H.; Kshitiz; Ahmed, I.; Semenza, G.L. Cyclin-dependent kinases regulate lysosomal degradation of hypoxia-inducible factor 1alpha to promote cell-cycle progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3325–E3334. [Google Scholar] [CrossRef]
- Hubbi, M.E.; Semenza, G.L. An essential role for chaperone-mediated autophagy in cell cycle progression. Autophagy 2015, 11, 850–851. [Google Scholar] [CrossRef]
- Bernardi, R.; Guernah, I.; Jin, D.; Grisendi, S.; Alimonti, A.; Teruya-Feldstein, J.; Cordon-Cardo, C.; Simon, M.C.; Rafii, S.; Pandolfi, P.P. PML inhibits HIF-1alpha translation and neoangiogenesis through repression of mTOR. Nature 2006, 442, 779–785. [Google Scholar] [CrossRef]
- Brugarolas, J.B.; Vazquez, F.; Reddy, A.; Sellers, W.R.; Kaelin, W.G., Jr. TSC2 regulates VEGF through mTOR-dependent and-independent pathways. Cancer Cell 2003, 4, 147–158. [Google Scholar] [CrossRef]
- Hudson, C.C.; Liu, M.; Chiang, G.G.; Otterness, D.M.; Loomis, D.C.; Kaper, F.; Giaccia, A.J.; Abraham, R.T. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol. Cell Biol. 2002, 22, 7004–7014. [Google Scholar] [CrossRef] [PubMed]
- Sacks, D.; Baxter, B.; Campbell, B.C.; Carpenter, J.S.; Cognard, C.; Dippel, D.; Eesa, M.; Fischer, U.; Hausegger, K.; Hirsch, J.A.; et al. Multisociety Consensus Quality Improvement Revised Consensus Statement for Endovascular Therapy of Acute Ischemic Stroke. Int. J. Stroke 2018, 13, 612–632. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.Y.; Hughes, R.; Murdoch, C.; Coffelt, S.B.; Biswas, S.K.; Harris, A.L.; Johnson, R.S.; Imityaz, H.Z.; Simon, M.C.; Fredlund, E.; et al. Hypoxia-inducible factors 1 and 2 are important transcriptional effectors in primary macrophages experiencing hypoxia. Blood 2009, 114, 844–859. [Google Scholar] [CrossRef]
- Rius, J.; Guma, M.; Schachtrup, C.; Akassoglou, K.; Zinkernagel, A.S.; Nizet, V.; Johnson, R.S.; Haddad, G.G.; Karin, M. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature 2008, 453, 807–811. [Google Scholar] [CrossRef]
- Jung, Y.J.; Isaacs, J.S.; Lee, S.; Trepel, J.; Neckers, L. IL-1beta-mediated up-regulation of HIF-1alpha via an NFkappaB/COX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesis. FASEB J. 2003, 17, 2115–2117. [Google Scholar] [CrossRef]
- Zhou, J.; Schmid, T.; Brune, B. Tumor necrosis factor-alpha causes accumulation of a ubiquitinated form of hypoxia inducible factor-1alpha through a nuclear factor-kappaB-dependent pathway. Mol. Biol. Cell 2003, 14, 2216–2225. [Google Scholar] [CrossRef]
- Jung, Y.; Isaacs, J.S.; Lee, S.; Trepel, J.; Liu, Z.G.; Neckers, L. Hypoxia-inducible factor induction by tumour necrosis factor in normoxic cells requires receptor-interacting protein-dependent nuclear factor kappa B activation. Biochem. J. 2003, 370, 1011–1017. [Google Scholar] [CrossRef]
- Nishi, K.; Oda, T.; Takabuchi, S.; Oda, S.; Fukuda, K.; Adachi, T.; Semenza, G.L.; Shingu, K.; Hirota, K. LPS induces hypoxia-inducible factor 1 activation in macrophage-differentiated cells in a reactive oxygen species-dependent manner. Antioxid. Redox Signal. 2008, 10, 983–995. [Google Scholar] [CrossRef]
- Brunelle, J.K.; Bell, E.L.; Quesada, N.M.; Vercauteren, K.; Tiranti, V.; Zeviani, M.; Scarpulla, R.C.; Chandel, N.S. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005, 1, 409–414. [Google Scholar] [CrossRef]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef]
- Lee, G.; Won, H.S.; Lee, Y.M.; Choi, J.W.; Oh, T.I.; Jang, J.H.; Choi, D.K.; Lim, B.O.; Kim, Y.J.; Park, J.W.; et al. Oxidative Dimerization of PHD2 is Responsible for its Inactivation and Contributes to Metabolic Reprogramming via HIF-1alpha Activation. Sci. Rep. 2016, 6, 18928. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.N.; Xi, M.M.; Guo, Y.; Hai, C.X.; Yang, W.L.; Qin, X.J. NADPH oxidase-mitochondria axis-derived ROS mediate arsenite-induced HIF-1alpha stabilization by inhibiting prolyl hydroxylases activity. Toxicol. Lett. 2014, 224, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Bonello, S.; Zahringer, C.; BelAiba, R.S.; Djordjevic, T.; Hess, J.; Michiels, C.; Kietzmann, T.; Gorlach, A. Reactive oxygen species activate the HIF-1alpha promoter via a functional NFkappaB site. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Masson, N.; Singleton, R.S.; Sekirnik, R.; Trudgian, D.C.; Ambrose, L.J.; Miranda, M.X.; Tian, Y.M.; Kessler, B.M.; Schofield, C.J.; Ratcliffe, P.J. The FIH hydroxylase is a cellular peroxide sensor that modulates HIF transcriptional activity. EMBO Rep. 2012, 13, 251–257. [Google Scholar] [CrossRef]
- Mateo, J.; Garcia-Lecea, M.; Cadenas, S.; Hernandez, C.; Moncada, S. Regulation of hypoxia-inducible factor-1alpha by nitric oxide through mitochondria-dependent and -independent pathways. Biochem. J. 2003, 376, 537–544. [Google Scholar] [CrossRef]
- Sebastiani, G.D.; Passiu, G. The current outlook in the therapy of autoimmune diseases. Ann. Ital. Med. Int. 1992, 7, 95–101. [Google Scholar]
- Berchner-Pfannschmidt, U.; Tug, S.; Trinidad, B.; Oehme, F.; Yamac, H.; Wotzlaw, C.; Flamme, I.; Fandrey, J. Nuclear oxygen sensing: Induction of endogenous prolyl-hydroxylase 2 activity by hypoxia and nitric oxide. J. Biol. Chem. 2008, 283, 31745–31753. [Google Scholar] [CrossRef] [PubMed]
- Berchner-Pfannschmidt, U.; Yamac, H.; Trinidad, B.; Fandrey, J. Nitric oxide modulates oxygen sensing by hypoxia-inducible factor 1-dependent induction of prolyl hydroxylase 2. J. Biol. Chem. 2007, 282, 1788–1796. [Google Scholar] [CrossRef] [PubMed]
- Cummins, E.P.; Taylor, C.T. Hypoxia-responsive transcription factors. Pflugers Arch. 2005, 450, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.T.; Cummins, E.P. The role of NF-kappaB in hypoxia-induced gene expression. Ann. N. Y. Acad. Sci. 2009, 1177, 178–184. [Google Scholar] [CrossRef]
- Cummins, E.P.; Berra, E.; Comerford, K.M.; Ginouves, A.; Fitzgerald, K.T.; Seeballuck, F.; Godson, C.; Nielsen, J.E.; Moynagh, P.; Pouyssegur, J.; et al. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc. Natl. Acad. Sci. USA 2006, 103, 18154–18159. [Google Scholar] [CrossRef]
- Bertero, T.; Rezzonico, R.; Pottier, N.; Mari, B. Impact of MicroRNAs in the Cellular Response to Hypoxia. Int. Rev. Cell Mol. Biol. 2017, 333, 91–158. [Google Scholar] [CrossRef]
- Nallamshetty, S.; Chan, S.Y.; Loscalzo, J. Hypoxia: A master regulator of microRNA biogenesis and activity. Free Radic. Biol. Med. 2013, 64, 20–30. [Google Scholar] [CrossRef]
- Shehade, H.; Acolty, V.; Moser, M.; Oldenhove, G. Cutting Edge: Hypoxia-Inducible Factor 1 Negatively Regulates Th1 Function. J. Immunol. 2015, 195, 1372–1376. [Google Scholar] [CrossRef]
- Wang, H.; Flach, H.; Onizawa, M.; Wei, L.; McManus, M.T.; Weiss, A. Negative regulation of Hif1a expression and TH17 differentiation by the hypoxia-regulated microRNA miR-210. Nat. Immunol. 2014, 15, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Zeng, J.; Yuan, J.; Deng, X.; Huang, Y.; Chen, L.; Zhang, P.; Feng, H.; Liu, Z.; Wang, Z.; et al. MicroRNA-210 overexpression promotes psoriasis-like inflammation by inducing Th1 and Th17 cell differentiation. J. Clin. Investig. 2018, 128, 2551–2568. [Google Scholar] [CrossRef] [PubMed]
- Feldman, D.E.; Chauhan, V.; Koong, A.C. The unfolded protein response: A novel component of the hypoxic stress response in tumors. Mol. Cancer Res. 2005, 3, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Jackson, R.J.; Hellen, C.U.; Pestova, T.V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010, 11, 113–127. [Google Scholar] [CrossRef]
- Brewer, J.W.; Diehl, J.A. PERK mediates cell-cycle exit during the mammalian unfolded protein response. Proc. Natl. Acad. Sci. USA 2000, 97, 12625–12630. [Google Scholar] [CrossRef] [PubMed]
- Moore, C.E.; Mikolajek, H.; Regufe da Mota, S.; Wang, X.; Kenney, J.W.; Werner, J.M.; Proud, C.G. Elongation Factor 2 Kinase Is Regulated by Proline Hydroxylation and Protects Cells during Hypoxia. Mol. Cell Biol. 2015, 35, 1788–1804. [Google Scholar] [CrossRef]
- Feng, T.; Yamamoto, A.; Wilkins, S.E.; Sokolova, E.; Yates, L.A.; Munzel, M.; Singh, P.; Hopkinson, R.J.; Fischer, R.; Cockman, M.E.; et al. Optimal translational termination requires C4 lysyl hydroxylation of eRF1. Mol. Cell 2014, 53, 645–654. [Google Scholar] [CrossRef]
- Romero-Ruiz, A.; Bautista, L.; Navarro, V.; Heras-Garvin, A.; March-Diaz, R.; Castellano, A.; Gomez-Diaz, R.; Castro, M.J.; Berra, E.; Lopez-Barneo, J.; et al. Prolyl hydroxylase-dependent modulation of eukaryotic elongation factor 2 activity and protein translation under acute hypoxia. J. Biol. Chem. 2012, 287, 9651–9658. [Google Scholar] [CrossRef]
- Connolly, E.; Braunstein, S.; Formenti, S.; Schneider, R.J. Hypoxia inhibits protein synthesis through a 4E-BP1 and elongation factor 2 kinase pathway controlled by mTOR and uncoupled in breast cancer cells. Mol. Cell Biol. 2006, 26, 3955–3965. [Google Scholar] [CrossRef]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Arsham, A.M.; Howell, J.J.; Simon, M.C. A novel hypoxia-inducible factor-independent hypoxic response regulating mammalian target of rapamycin and its targets. J. Biol. Chem. 2003, 278, 29655–29660. [Google Scholar] [CrossRef] [PubMed]
- Sofer, A.; Lei, K.; Johannessen, C.M.; Ellisen, L.W. Regulation of mTOR and cell growth in response to energy stress by REDD1. Mol. Cell Biol. 2005, 25, 5834–5845. [Google Scholar] [CrossRef] [PubMed]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G., Jr. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004, 18, 2893–2904. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Cash, T.P.; Jones, R.G.; Keith, B.; Thompson, C.B.; Simon, M.C. Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol. Cell 2006, 21, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Van der Kwast, T.H.; Vellanki, R.N.; Foltz, W.D.; McKee, T.D.; Sonenberg, N.; Pandolfi, P.P.; Koritzinsky, M.; Wouters, B.G. The mTOR Targets 4E-BP1/2 Restrain Tumor Growth and Promote Hypoxia Tolerance in PTEN-driven Prostate Cancer. Mol. Cancer Res. 2018, 16, 682–695. [Google Scholar] [CrossRef] [PubMed]
- King, H.A.; Cobbold, L.C.; Willis, A.E. The role of IRES trans-acting factors in regulating translation initiation. Biochem. Soc. Trans. 2010, 38, 1581–1586. [Google Scholar] [CrossRef]
- Kozak, M. A second look at cellular mRNA sequences said to function as internal ribosome entry sites. Nucleic Acids Res. 2005, 33, 6593–6602. [Google Scholar] [CrossRef]
- Ho, J.J.D.; Wang, M.; Audas, T.E.; Kwon, D.; Carlsson, S.K.; Timpano, S.; Evagelou, S.L.; Brothers, S.; Gonzalgo, M.L.; Krieger, J.R.; et al. Systemic Reprogramming of Translation Efficiencies on Oxygen Stimulus. Cell Rep. 2016, 14, 1293–1300. [Google Scholar] [CrossRef]
- Uniacke, J.; Perera, J.K.; Lachance, G.; Francisco, C.B.; Lee, S. Cancer cells exploit eIF4E2-directed synthesis of hypoxia response proteins to drive tumor progression. Cancer Res. 2014, 74, 1379–1389. [Google Scholar] [CrossRef]
- Uniacke, J.; Holterman, C.E.; Lachance, G.; Franovic, A.; Jacob, M.D.; Fabian, M.R.; Payette, J.; Holcik, M.; Pause, A.; Lee, S. An oxygen-regulated switch in the protein synthesis machinery. Nature 2012, 486, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.A.; Palazzo, A.F. Localization of mRNAs to the endoplasmic reticulum. Wiley Interdiscip. Rev. RNA 2014, 5, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef]
- Iyer, N.V.; Kotch, L.E.; Agani, F.; Leung, S.W.; Laughner, E.; Wenger, R.H.; Gassmann, M.; Gearhart, J.D.; Lawler, A.M.; Yu, A.Y.; et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998, 12, 149–162. [Google Scholar] [CrossRef]
- Garcia-Bermudez, J.; Baudrier, L.; La, K.; Zhu, X.G.; Fidelin, J.; Sviderskiy, V.O.; Papagiannakopoulos, T.; Molina, H.; Snuderl, M.; Lewis, C.A.; et al. Aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumours. Nat. Cell Biol. 2018, 20, 775–781. [Google Scholar] [CrossRef]
- Schulz, T.J.; Zarse, K.; Voigt, A.; Urban, N.; Birringer, M.; Ristow, M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007, 6, 280–293. [Google Scholar] [CrossRef]
- Bell, E.L.; Klimova, T.A.; Eisenbart, J.; Schumacker, P.T.; Chandel, N.S. Mitochondrial reactive oxygen species trigger hypoxia-inducible factor-dependent extension of the replicative life span during hypoxia. Mol. Cell Biol. 2007, 27, 5737–5745. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, N.; Hughes, B.; Bigras, E.; Shoubridge, E.; Hekimi, S. Evolutionary conservation of the clk-1-dependent mechanism of longevity: Loss of mclk1 increases cellular fitness and lifespan in mice. Genes Dev. 2005, 19, 2424–2434. [Google Scholar] [CrossRef]
- Solaini, G.; Baracca, A.; Lenaz, G.; Sgarbi, G. Hypoxia and mitochondrial oxidative metabolism. Biochim. Biophys. Acta 2010, 1797, 1171–1177. [Google Scholar] [CrossRef]
- Hernansanz-Agustin, P.; Choya-Foces, C.; Carregal-Romero, S.; Ramos, E.; Oliva, T.; Villa-Pina, T.; Moreno, L.; Izquierdo-Alvarez, A.; Cabrera-Garcia, J.D.; Cortes, A.; et al. Na(+) controls hypoxic signalling by the mitochondrial respiratory chain. Nature 2020, 586, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhang, D.; Hu, D.; Zhou, X.; Zhou, Y. The role of mitochondria in NLRP3 inflammasome activation. Mol. Immunol. 2018, 103, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Scimia, M.C.; Wilkinson, D.; Trelles, R.D.; Wood, M.R.; Bowtell, D.; Dillin, A.; Mercola, M.; Ronai, Z.A. Fine-tuning of Drp1/Fis1 availability by AKAP121/Siah2 regulates mitochondrial adaptation to hypoxia. Mol. Cell 2011, 44, 532–544. [Google Scholar] [CrossRef] [PubMed]
- Safronova, O.; Pluemsampant, S.; Nakahama, K.; Morita, I. Regulation of chemokine gene expression by hypoxia via cooperative activation of NF-kappaB and histone deacetylase. Int. J. Biochem. Cell Biol. 2009, 41, 2270–2280. [Google Scholar] [CrossRef] [PubMed]
- Koumenis, C.; Naczki, C.; Koritzinsky, M.; Rastani, S.; Diehl, A.; Sonenberg, N.; Koromilas, A.; Wouters, B.G. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2alpha. Mol. Cell Biol. 2002, 22, 7405–7416. [Google Scholar] [CrossRef] [PubMed]
- Krzywinska, E.; Stockmann, C. Hypoxia, Metabolism and Immune Cell Function. Biomedicines 2018, 6, 56. [Google Scholar] [CrossRef]
- Amulic, B.; Cazalet, C.; Hayes, G.L.; Metzler, K.D.; Zychlinsky, A. Neutrophil function: From mechanisms to disease. Annu. Rev. Immunol. 2012, 30, 459–489. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Wera, O.; Lancellotti, P.; Oury, C. The Dual Role of Neutrophils in Inflammatory Bowel Diseases. J. Clin. Med. 2016, 5, 118. [Google Scholar] [CrossRef]
- Harris, A.J.; Thompson, A.R.; Whyte, M.K.; Walmsley, S.R. HIF-mediated innate immune responses: Cell signaling and therapeutic implications. Hypoxia (Auckl) 2014, 2, 47–58. [Google Scholar] [CrossRef]
- Peyssonnaux, C.; Datta, V.; Cramer, T.; Doedens, A.; Theodorakis, E.A.; Gallo, R.L.; Hurtado-Ziola, N.; Nizet, V.; Johnson, R.S. HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J. Clin. Investig. 2005, 115, 1806–1815. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.A.; Elks, P.M.; Marriott, H.M.; Eamsamarng, S.; Higgins, K.R.; Lewis, A.; Williams, L.; Parmar, S.; Shaw, G.; McGrath, E.E.; et al. Hypoxia-inducible factor 2alpha regulates key neutrophil functions in humans, mice, and zebrafish. Blood 2014, 123, 366–376. [Google Scholar] [CrossRef]
- Mollerherm, H.; von Kockritz-Blickwede, M.; Branitzki-Heinemann, K. Antimicrobial Activity of Mast Cells: Role and Relevance of Extracellular DNA Traps. Front. Immunol. 2016, 7, 265. [Google Scholar] [CrossRef]
- Crotty Alexander, L.E.; Akong-Moore, K.; Feldstein, S.; Johansson, P.; Nguyen, A.; McEachern, E.K.; Nicatia, S.; Cowburn, A.S.; Olson, J.; Cho, J.Y.; et al. Myeloid cell HIF-1alpha regulates asthma airway resistance and eosinophil function. J. Mol. Med. (Berl.) 2013, 91, 637–644. [Google Scholar] [CrossRef]
- Sumbayev, V.V.; Yasinska, I.; Oniku, A.E.; Streatfield, C.L.; Gibbs, B.F. Involvement of hypoxia-inducible factor-1 in the inflammatory responses of human LAD2 mast cells and basophils. PLoS ONE 2012, 7, e34259. [Google Scholar] [CrossRef]
- Nissim Ben Efraim, A.H.; Eliashar, R.; Levi-Schaffer, F. Hypoxia modulates human eosinophil function. Clin. Mol. Allergy 2010, 8, 10. [Google Scholar] [CrossRef]
- Sumbayev, V.V.; Nicholas, S.A.; Streatfield, C.L.; Gibbs, B.F. Involvement of hypoxia-inducible factor-1 HiF(1alpha) in IgE-mediated primary human basophil responses. Eur. J. Immunol. 2009, 39, 3511–3519. [Google Scholar] [CrossRef] [PubMed]
- Gondin, J.; Theret, M.; Duhamel, G.; Pegan, K.; Mathieu, J.R.; Peyssonnaux, C.; Cuvellier, S.; Latroche, C.; Chazaud, B.; Bendahan, D.; et al. Myeloid HIFs are dispensable for resolution of inflammation during skeletal muscle regeneration. J. Immunol. 2015, 194, 3389–3399. [Google Scholar] [CrossRef] [PubMed]
- Takeda, N.; O’Dea, E.L.; Doedens, A.; Kim, J.W.; Weidemann, A.; Stockmann, C.; Asagiri, M.; Simon, M.C.; Hoffmann, A.; Johnson, R.S. Differential activation and antagonistic function of HIF-{alpha} isoforms in macrophages are essential for NO homeostasis. Genes Dev. 2010, 24, 491–501. [Google Scholar] [CrossRef]
- Peyssonnaux, C.; Cejudo-Martin, P.; Doedens, A.; Zinkernagel, A.S.; Johnson, R.S.; Nizet, V. Cutting edge: Essential role of hypoxia inducible factor-1alpha in development of lipopolysaccharide-induced sepsis. J. Immunol. 2007, 178, 7516–7519. [Google Scholar] [CrossRef]
- Dehn, S.; DeBerge, M.; Yeap, X.Y.; Yvan-Charvet, L.; Fang, D.; Eltzschig, H.K.; Miller, S.D.; Thorp, E.B. HIF-2alpha in Resting Macrophages Tempers Mitochondrial Reactive Oxygen Species To Selectively Repress MARCO-Dependent Phagocytosis. J. Immunol. 2016, 197, 3639–3649. [Google Scholar] [CrossRef]
- Fluck, K.; Breves, G.; Fandrey, J.; Winning, S. Hypoxia-inducible factor 1 in dendritic cells is crucial for the activation of protective regulatory T cells in murine colitis. Mucosal Immunol. 2016, 9, 379–390. [Google Scholar] [CrossRef]
- Wobben, R.; Husecken, Y.; Lodewick, C.; Gibbert, K.; Fandrey, J.; Winning, S. Role of hypoxia inducible factor-1alpha for interferon synthesis in mouse dendritic cells. Biol. Chem. 2013, 394, 495–505. [Google Scholar] [CrossRef]
- Kohler, T.; Reizis, B.; Johnson, R.S.; Weighardt, H.; Forster, I. Influence of hypoxia-inducible factor 1alpha on dendritic cell differentiation and migration. Eur. J. Immunol. 2012, 42, 1226–1236. [Google Scholar] [CrossRef]
- Naldini, A.; Morena, E.; Pucci, A.; Miglietta, D.; Riboldi, E.; Sozzani, S.; Carraro, F. Hypoxia affects dendritic cell survival: Role of the hypoxia-inducible factor-1alpha and lipopolysaccharide. J. Cell Physiol. 2012, 227, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Kong, T.; Eltzschig, H.K.; Karhausen, J.; Colgan, S.P.; Shelley, C.S. Leukocyte adhesion during hypoxia is mediated by HIF-1-dependent induction of beta2 integrin gene expression. Proc. Natl. Acad. Sci. USA 2004, 101, 10440–10445. [Google Scholar] [CrossRef]
- Mantovani, A.; Biswas, S.K.; Galdiero, M.R.; Sica, A.; Locati, M. Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 2013, 229, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Galli, S.J.; Borregaard, N.; Wynn, T.A. Phenotypic and functional plasticity of cells of innate immunity: Macrophages, mast cells and neutrophils. Nat. Immunol. 2011, 12, 1035–1044. [Google Scholar] [CrossRef]
- Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; O’Neill, L.A. Reprogramming mitochondrial metabolism in macrophages as an anti-inflammatory signal. Eur. J. Immunol. 2016, 46, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Jha, A.K.; Huang, S.C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Prados, J.C.; Traves, P.G.; Cuenca, J.; Rico, D.; Aragones, J.; Martin-Sanz, P.; Cascante, M.; Bosca, L. Substrate fate in activated macrophages: A comparison between innate, classic, and alternative activation. J. Immunol. 2010, 185, 605–614. [Google Scholar] [CrossRef]
- O’Neill, L.A.; Hardie, D.G. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature 2013, 493, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Imtiyaz, H.Z.; Williams, E.P.; Hickey, M.M.; Patel, S.A.; Durham, A.C.; Yuan, L.J.; Hammond, R.; Gimotty, P.A.; Keith, B.; Simon, M.C. Hypoxia-inducible factor 2alpha regulates macrophage function in mouse models of acute and tumor inflammation. J. Clin. Investig. 2010, 120, 2699–2714. [Google Scholar] [CrossRef]
- Kelly, B.; O’Neill, L.A. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef]
- Everts, B.; Amiel, E.; Huang, S.C.; Smith, A.M.; Chang, C.H.; Lam, W.Y.; Redmann, V.; Freitas, T.C.; Blagih, J.; van der Windt, G.J.; et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKvarepsilon supports the anabolic demands of dendritic cell activation. Nat. Immunol. 2014, 15, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Masopust, D.; Schenkel, J.M. The integration of T cell migration, differentiation and function. Nat. Rev. Immunol. 2013, 13, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Biju, M.P.; Neumann, A.K.; Bensinger, S.J.; Johnson, R.S.; Turka, L.A.; Haase, V.H. Vhlh gene deletion induces Hif-1-mediated cell death in thymocytes. Mol. Cell Biol. 2004, 24, 9038–9047. [Google Scholar] [CrossRef]
- Gaber, T.; Chen, Y.; Krauss, P.L.; Buttgereit, F. Metabolism of T Lymphocytes in Health and Disease. Int. Rev. Cell Mol. Biol. 2019, 342, 95–148. [Google Scholar] [CrossRef]
- Gaber, T.; Strehl, C.; Buttgereit, F. Metabolic regulation of inflammation. Nat. Rev. Rheumatol. 2017, 13, 267–279. [Google Scholar] [CrossRef]
- Makino, Y.; Nakamura, H.; Ikeda, E.; Ohnuma, K.; Yamauchi, K.; Yabe, Y.; Poellinger, L.; Okada, Y.; Morimoto, C.; Tanaka, H. Hypoxia-inducible factor regulates survival of antigen receptor-driven T cells. J. Immunol. 2003, 171, 6534–6540. [Google Scholar] [CrossRef]
- Brockmann, L.; Soukou, S.; Steglich, B.; Czarnewski, P.; Zhao, L.; Wende, S.; Bedke, T.; Ergen, C.; Manthey, C.; Agalioti, T.; et al. Molecular and functional heterogeneity of IL-10-producing CD4(+) T cells. Nat. Commun. 2018, 9, 5457. [Google Scholar] [CrossRef] [PubMed]
- Mascanfroni, I.D.; Takenaka, M.C.; Yeste, A.; Patel, B.; Wu, Y.; Kenison, J.E.; Siddiqui, S.; Basso, A.S.; Otterbein, L.E.; Pardoll, D.M.; et al. Metabolic control of type 1 regulatory T cell differentiation by AHR and HIF1-alpha. Nat. Med. 2015, 21, 638–646. [Google Scholar] [CrossRef]
- Pot, C.; Apetoh, L.; Awasthi, A.; Kuchroo, V.K. Induction of regulatory Tr1 cells and inhibition of T(H)17 cells by IL-27. Semin. Immunol. 2011, 23, 438–445. [Google Scholar] [CrossRef]
- Dang, E.V.; Barbi, J.; Yang, H.Y.; Jinasena, D.; Yu, H.; Zheng, Y.; Bordman, Z.; Fu, J.; Kim, Y.; Yen, H.R.; et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell 2011, 146, 772–784. [Google Scholar] [CrossRef]
- Shi, L.Z.; Wang, R.; Huang, G.; Vogel, P.; Neale, G.; Green, D.R.; Chi, H. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med. 2011, 208, 1367–1376. [Google Scholar] [CrossRef] [PubMed]
- Roncarolo, M.G.; Gregori, S.; Battaglia, M.; Bacchetta, R.; Fleischhauer, K.; Levings, M.K. Interleukin-10-secreting type 1 regulatory T cells in rodents and humans. Immunol. Rev. 2006, 212, 28–50. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.H.; Raybuck, A.L.; Blagih, J.; Kemboi, E.; Haase, V.H.; Jones, R.G.; Boothby, M.R. Hypoxia-inducible factors in CD4(+) T cells promote metabolism, switch cytokine secretion, and T cell help in humoral immunity. Proc. Natl. Acad. Sci. USA 2019, 116, 8975–8984. [Google Scholar] [CrossRef]
- Singh, Y.; Garden, O.A.; Lang, F.; Cobb, B.S. MicroRNAs regulate T-cell production of interleukin-9 and identify hypoxia-inducible factor-2alpha as an important regulator of T helper 9 and regulatory T-cell differentiation. Immunology 2016, 149, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Palazon, A.; Tyrakis, P.A.; Macias, D.; Velica, P.; Rundqvist, H.; Fitzpatrick, S.; Vojnovic, N.; Phan, A.T.; Loman, N.; Hedenfalk, I.; et al. An HIF-1alpha/VEGF-A Axis in Cytotoxic T Cells Regulates Tumor Progression. Cancer Cell 2017, 32, 669–683. [Google Scholar] [CrossRef] [PubMed]
- Tyrakis, P.A.; Palazon, A.; Macias, D.; Lee, K.L.; Phan, A.T.; Velica, P.; You, J.; Chia, G.S.; Sim, J.; Doedens, A.; et al. S-2-hydroxyglutarate regulates CD8(+) T-lymphocyte fate. Nature 2016, 540, 236–241. [Google Scholar] [CrossRef]
- Gubser, P.M.; Bantug, G.R.; Razik, L.; Fischer, M.; Dimeloe, S.; Hoenger, G.; Durovic, B.; Jauch, A.; Hess, C. Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nat. Immunol. 2013, 14, 1064–1072. [Google Scholar] [CrossRef] [PubMed]
- Finlay, D.K.; Rosenzweig, E.; Sinclair, L.V.; Feijoo-Carnero, C.; Hukelmann, J.L.; Rolf, J.; Panteleyev, A.A.; Okkenhaug, K.; Cantrell, D.A. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J. Exp. Med. 2012, 209, 2441–2453. [Google Scholar] [CrossRef]
- Kim, J.H.; Han, J.W.; Choi, Y.J.; Rha, M.S.; Koh, J.Y.; Kim, K.H.; Kim, C.G.; Lee, Y.J.; Kim, A.R.; Park, J.; et al. Functions of human liver CD69(+)CD103(-)CD8(+) T cells depend on HIF-2alpha activity in healthy and pathologic livers. J. Hepatol. 2020, 72, 1170–1181. [Google Scholar] [CrossRef]
- Meng, X.; Grotsch, B.; Luo, Y.; Knaup, K.X.; Wiesener, M.S.; Chen, X.X.; Jantsch, J.; Fillatreau, S.; Schett, G.; Bozec, A. Hypoxia-inducible factor-1alpha is a critical transcription factor for IL-10-producing B cells in autoimmune disease. Nat. Commun. 2018, 9, 251. [Google Scholar] [CrossRef] [PubMed]
- Kojima, H.; Gu, H.; Nomura, S.; Caldwell, C.C.; Kobata, T.; Carmeliet, P.; Semenza, G.L.; Sitkovsky, M.V. Abnormal B lymphocyte development and autoimmunity in hypoxia-inducible factor 1alpha -deficient chimeric mice. Proc. Natl. Acad. Sci. USA 2002, 99, 2170–2174. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Li, R.; Yang, Y.; Shi, C.; Shen, Y.; Lu, C.; Chen, Y.; Zhou, W.; Lin, A.; Yu, L.; et al. Specific Decrease in B-Cell-Derived Extracellular Vesicles Enhances Post-Chemotherapeutic CD8(+) T Cell Responses. Immunity 2019, 50, 738–750. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; McKenzie, B.S.; Zhou, L.; Tadokoro, C.E.; Lepelley, A.; Lafaille, J.J.; Cua, D.J.; Littman, D.R. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 2006, 126, 1121–1133. [Google Scholar] [CrossRef]
- Kryczek, I.; Zhao, E.; Liu, Y.; Wang, Y.; Vatan, L.; Szeliga, W.; Moyer, J.; Klimczak, A.; Lange, A.; Zou, W. Human TH17 cells are long-lived effector memory cells. Sci. Transl. Med. 2011, 3, 104ra100. [Google Scholar] [CrossRef] [PubMed]
- Clambey, E.T.; McNamee, E.N.; Westrich, J.A.; Glover, L.E.; Campbell, E.L.; Jedlicka, P.; de Zoeten, E.F.; Cambier, J.C.; Stenmark, K.R.; Colgan, S.P.; et al. Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc. Natl. Acad. Sci. USA 2012, 109, E2784–E2793. [Google Scholar] [CrossRef]
- Ben-Shoshan, J.; Maysel-Auslender, S.; Mor, A.; Keren, G.; George, J. Hypoxia controls CD4+CD25+ regulatory T-cell homeostasis via hypoxia-inducible factor-1alpha. Eur. J. Immunol. 2008, 38, 2412–2418. [Google Scholar] [CrossRef] [PubMed]
- Georgiev, P.; Belikoff, B.G.; Hatfield, S.; Ohta, A.; Sitkovsky, M.V.; Lukashev, D. Genetic deletion of the HIF-1alpha isoform I.1 in T cells enhances antibacterial immunity and improves survival in a murine peritonitis model. Eur. J. Immunol. 2013, 43, 655–666. [Google Scholar] [CrossRef]
- Thiel, M.; Caldwell, C.C.; Kreth, S.; Kuboki, S.; Chen, P.; Smith, P.; Ohta, A.; Lentsch, A.B.; Lukashev, D.; Sitkovsky, M.V. Targeted deletion of HIF-1alpha gene in T cells prevents their inhibition in hypoxic inflamed tissues and improves septic mice survival. PLoS ONE 2007, 2, e853. [Google Scholar] [CrossRef]
- Lukashev, D.; Klebanov, B.; Kojima, H.; Grinberg, A.; Ohta, A.; Berenfeld, L.; Wenger, R.H.; Ohta, A.; Sitkovsky, M. Cutting edge: Hypoxia-inducible factor 1alpha and its activation-inducible short isoform I.1 negatively regulate functions of CD4+ and CD8+ T lymphocytes. J. Immunol. 2006, 177, 4962–4965. [Google Scholar] [CrossRef] [PubMed]
- Higashiyama, M.; Hokari, R.; Hozumi, H.; Kurihara, C.; Ueda, T.; Watanabe, C.; Tomita, K.; Nakamura, M.; Komoto, S.; Okada, Y.; et al. HIF-1 in T cells ameliorated dextran sodium sulfate-induced murine colitis. J. Leukoc. Biol. 2012, 91, 901–909. [Google Scholar] [CrossRef]
- Lee, J.H.; Elly, C.; Park, Y.; Liu, Y.C. E3 Ubiquitin Ligase VHL Regulates Hypoxia-Inducible Factor-1alpha to Maintain Regulatory T Cell Stability and Suppressive Capacity. Immunity 2015, 42, 1062–1074. [Google Scholar] [CrossRef]
- Harty, J.T.; Tvinnereim, A.R.; White, D.W. CD8+ T cell effector mechanisms in resistance to infection. Annu. Rev. Immunol. 2000, 18, 275–308. [Google Scholar] [CrossRef]
- Sukumar, M.; Liu, J.; Ji, Y.; Subramanian, M.; Crompton, J.G.; Yu, Z.; Roychoudhuri, R.; Palmer, D.C.; Muranski, P.; Karoly, E.D.; et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J. Clin. Investig. 2013, 123, 4479–4488. [Google Scholar] [CrossRef]
- Pollizzi, K.N.; Patel, C.H.; Sun, I.H.; Oh, M.H.; Waickman, A.T.; Wen, J.; Delgoffe, G.M.; Powell, J.D. mTORC1 and mTORC2 selectively regulate CD8(+) T cell differentiation. J. Clin. Investig. 2015, 125, 2090–2108. [Google Scholar] [CrossRef] [PubMed]
- Frebel, H.; Nindl, V.; Schuepbach, R.A.; Braunschweiler, T.; Richter, K.; Vogel, J.; Wagner, C.A.; Loffing-Cueni, D.; Kurrer, M.; Ludewig, B.; et al. Programmed death 1 protects from fatal circulatory failure during systemic virus infection of mice. J. Exp. Med. 2012, 209, 2485–2499. [Google Scholar] [CrossRef]
- Medzhitov, R.; Schneider, D.S.; Soares, M.P. Disease tolerance as a defense strategy. Science 2012, 335, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Doedens, A.L.; Phan, A.T.; Stradner, M.H.; Fujimoto, J.K.; Nguyen, J.V.; Yang, E.; Johnson, R.S.; Goldrath, A.W. Hypoxia-inducible factors enhance the effector responses of CD8(+) T cells to persistent antigen. Nat. Immunol. 2013, 14, 1173–1182. [Google Scholar] [CrossRef]
- Ning, F.; Takeda, K.; Schedel, M.; Domenico, J.; Joetham, A.; Gelfand, E.W. Hypoxia Enhances CD8+ Tc2 Dependent Airway Hyperresponsiveness and Inflammation Through Hypoxia Inducible Factor 1alpha. J. Allergy Clin. Immunol. 2019. [Google Scholar] [CrossRef]
- Lund, F.E.; Randall, T.D. Effector and regulatory B cells: Modulators of CD4+ T cell immunity. Nat. Rev. Immunol. 2010, 10, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, W.; Lakkis, F.G.; Chalasani, G. B Cells, Antibodies, and More. Clin. J. Am. Soc. Nephrol. 2016, 11, 137–154. [Google Scholar] [CrossRef] [PubMed]
- Rosser, E.C.; Mauri, C. Regulatory B cells: Origin, phenotype, and function. Immunity 2015, 42, 607–612. [Google Scholar] [CrossRef]
- Lee, K.M.; Stott, R.T.; Zhao, G.; SooHoo, J.; Xiong, W.; Lian, M.M.; Fitzgerald, L.; Shi, S.; Akrawi, E.; Lei, J.; et al. TGF-beta-producing regulatory B cells induce regulatory T cells and promote transplantation tolerance. Eur. J. Immunol. 2014, 44, 1728–1736. [Google Scholar] [CrossRef] [PubMed]
- Shen, P.; Roch, T.; Lampropoulou, V.; O’Connor, R.A.; Stervbo, U.; Hilgenberg, E.; Ries, S.; Dang, V.D.; Jaimes, Y.; Daridon, C.; et al. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature 2014, 507, 366–370. [Google Scholar] [CrossRef]
- Kalampokis, I.; Yoshizaki, A.; Tedder, T.F. IL-10-producing regulatory B cells (B10 cells) in autoimmune disease. Arthritis Res. Ther. 2013, 15 (Suppl. 1), S1. [Google Scholar] [CrossRef] [PubMed]
- Mauri, C.; Bosma, A. Immune regulatory function of B cells. Annu. Rev. Immunol. 2012, 30, 221–241. [Google Scholar] [CrossRef]
- Pieper, K.; Grimbacher, B.; Eibel, H. B-cell biology and development. J. Allergy Clin. Immunol. 2013, 131, 959–971. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.H.; Lin, H.; Zheng, H.; Kim, K.S.; Kim, J.Y.; Chun, Y.S.; Park, J.W.; Nam, J.H.; Kim, W.K.; Zhang, Y.H.; et al. HIF-1alpha-mediated upregulation of TASK-2 K(+) channels augments Ca(2)(+) signaling in mouse B cells under hypoxia. J. Immunol. 2014, 193, 4924–4933. [Google Scholar] [CrossRef]
- Goda, N.; Ryan, H.E.; Khadivi, B.; McNulty, W.; Rickert, R.C.; Johnson, R.S. Hypoxia-inducible factor 1alpha is essential for cell cycle arrest during hypoxia. Mol. Cell Biol. 2003, 23, 359–369. [Google Scholar] [CrossRef]
- Qian, T.; Hong, J.; Wang, L.; Wang, Z.; Lu, Z.; Li, Y.; Liu, R.; Chu, Y. Regulation of CD11b by HIF-1alpha and the STAT3 signaling pathway contributes to the immunosuppressive function of B cells in inflammatory bowel disease. Mol. Immunol. 2019, 111, 162–171. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, H.; Liu, R.; Qian, T.; Liu, J.; Huang, E.; Lu, Z.; Zhao, C.; Wang, L.; Chu, Y. Peyer’s patches-derived CD11b(+) B cells recruit regulatory T cells through CXCL9 in dextran sulphate sodium-induced colitis. Immunology 2018, 155, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jiang, X.; Liu, R.; Wang, L.; Qian, T.; Zheng, Y.; Deng, Y.; Huang, E.; Xu, F.; Wang, J.Y.; et al. B cells expressing CD11b effectively inhibit CD4+ T-cell responses and ameliorate experimental autoimmune hepatitis in mice. Hepatology 2015, 62, 1563–1575. [Google Scholar] [CrossRef]
- Jellusova, J.; Cato, M.H.; Apgar, J.R.; Ramezani-Rad, P.; Leung, C.R.; Chen, C.; Richardson, A.D.; Conner, E.M.; Benschop, R.J.; Woodgett, J.R.; et al. Gsk3 is a metabolic checkpoint regulator in B cells. Nat. Immunol. 2017, 18, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Caro-Maldonado, A.; Wang, R.; Nichols, A.G.; Kuraoka, M.; Milasta, S.; Sun, L.D.; Gavin, A.L.; Abel, E.D.; Kelsoe, G.; Green, D.R.; et al. Metabolic reprogramming is required for antibody production that is suppressed in anergic but exaggerated in chronically BAFF-exposed B cells. J. Immunol. 2014, 192, 3626–3636. [Google Scholar] [CrossRef] [PubMed]
- Shikhagaie, M.M.; Germar, K.; Bal, S.M.; Ros, X.R.; Spits, H. Innate lymphoid cells in autoimmunity: Emerging regulators in rheumatic diseases. Nat. Rev. Rheumatol. 2017, 13, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Li, D.; Zhang, X.; Wan, Q.; Zhang, W.; Zheng, M.; Zou, L.; Elly, C.; Lee, J.H.; Liu, Y.C. E3 Ligase VHL Promotes Group 2 Innate Lymphoid Cell Maturation and Function via Glycolysis Inhibition and Induction of Interleukin-33 Receptor. Immunity 2018, 48, 258–270. [Google Scholar] [CrossRef]
- Ni, J.; Wang, X.; Stojanovic, A.; Zhang, Q.; Wincher, M.; Buhler, L.; Arnold, A.; Correia, M.P.; Winkler, M.; Koch, P.S.; et al. Single-Cell RNA Sequencing of Tumor-Infiltrating NK Cells Reveals that Inhibition of Transcription Factor HIF-1alpha Unleashes NK Cell Activity. Immunity 2020, 52, 1075.e1078–1087.e1078. [Google Scholar] [CrossRef] [PubMed]
- Krzywinska, E.; Kantari-Mimoun, C.; Kerdiles, Y.; Sobecki, M.; Isagawa, T.; Gotthardt, D.; Castells, M.; Haubold, J.; Millien, C.; Viel, T.; et al. Loss of HIF-1alpha in natural killer cells inhibits tumour growth by stimulating non-productive angiogenesis. Nat. Commun. 2017, 8, 1597. [Google Scholar] [CrossRef] [PubMed]
- Spits, H.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.; Mebius, R.E.; et al. Innate lymphoid cells--a proposal for uniform nomenclature. Nat. Rev. Immunol. 2013, 13, 145–149. [Google Scholar] [CrossRef]
- Spits, H.; Bernink, J.H.; Lanier, L. NK cells and type 1 innate lymphoid cells: Partners in host defense. Nat. Immunol. 2016, 17, 758–764. [Google Scholar] [CrossRef]
- Viel, S.; Marcais, A.; Guimaraes, F.S.; Loftus, R.; Rabilloud, J.; Grau, M.; Degouve, S.; Djebali, S.; Sanlaville, A.; Charrier, E.; et al. TGF-beta inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci. Signal. 2016, 9, ra19. [Google Scholar] [CrossRef] [PubMed]
- Berchem, G.; Noman, M.Z.; Bosseler, M.; Paggetti, J.; Baconnais, S.; Le Cam, E.; Nanbakhsh, A.; Moussay, E.; Mami-Chouaib, F.; Janji, B.; et al. Hypoxic tumor-derived microvesicles negatively regulate NK cell function by a mechanism involving TGF-beta and miR23a transfer. Oncoimmunology 2016, 5, e1062968. [Google Scholar] [CrossRef]
- Sarkar, S.; Germeraad, W.T.; Rouschop, K.M.; Steeghs, E.M.; van Gelder, M.; Bos, G.M.; Wieten, L. Hypoxia induced impairment of NK cell cytotoxicity against multiple myeloma can be overcome by IL-2 activation of the NK cells. PLoS ONE 2013, 8, e64835. [Google Scholar] [CrossRef]
- Marcais, A.; Cherfils-Vicini, J.; Viant, C.; Degouve, S.; Viel, S.; Fenis, A.; Rabilloud, J.; Mayol, K.; Tavares, A.; Bienvenu, J.; et al. The metabolic checkpoint kinase mTOR is essential for IL-15 signaling during the development and activation of NK cells. Nat. Immunol. 2014, 15, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, R.P.; Loftus, R.M.; Keating, S.E.; Liou, K.T.; Biron, C.A.; Gardiner, C.M.; Finlay, D.K. mTORC1-dependent metabolic reprogramming is a prerequisite for NK cell effector function. J. Immunol. 2014, 193, 4477–4484. [Google Scholar] [CrossRef] [PubMed]
- Keating, S.E.; Zaiatz-Bittencourt, V.; Loftus, R.M.; Keane, C.; Brennan, K.; Finlay, D.K.; Gardiner, C.M. Metabolic Reprogramming Supports IFN-gamma Production by CD56bright NK Cells. J. Immunol. 2016, 196, 2552–2560. [Google Scholar] [CrossRef]
- Velasquez, S.Y.; Killian, D.; Schulte, J.; Sticht, C.; Thiel, M.; Lindner, H.A. Short Term Hypoxia Synergizes with Interleukin 15 Priming in Driving Glycolytic Gene Transcription and Supports Human Natural Killer Cell Activities. J. Biol. Chem. 2016, 291, 12960–12977. [Google Scholar] [CrossRef] [PubMed]
- Blouin, C.C.; Page, E.L.; Soucy, G.M.; Richard, D.E. Hypoxic gene activation by lipopolysaccharide in macrophages: Implication of hypoxia-inducible factor 1alpha. Blood 2004, 103, 1124–1130. [Google Scholar] [CrossRef]
- Palsson-McDermott, E.M.; Curtis, A.M.; Goel, G.; Lauterbach, M.A.; Sheedy, F.J.; Gleeson, L.E.; van den Bosch, M.W.; Quinn, S.R.; Domingo-Fernandez, R.; Johnston, D.G.; et al. Pyruvate kinase M2 regulates Hif-1alpha activity and IL-1beta induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. 2015, 21, 65–80. [Google Scholar] [CrossRef]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Dabritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 2016, 167, 457–470. [Google Scholar] [CrossRef]
- Luo, W.; Hu, H.; Chang, R.; Zhong, J.; Knabel, M.; O’Meally, R.; Cole, R.N.; Pandey, A.; Semenza, G.L. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 2011, 145, 732–744. [Google Scholar] [CrossRef]
- Shirai, T.; Nazarewicz, R.R.; Wallis, B.B.; Yanes, R.E.; Watanabe, R.; Hilhorst, M.; Tian, L.; Harrison, D.G.; Giacomini, J.C.; Assimes, T.L.; et al. The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease. J. Exp. Med. 2016, 213, 337–354. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Li, Z.; Li, H.; Lu, Y.; Wu, H.; Li, Z. Pyruvate kinase M2 accelerates pro-inflammatory cytokine secretion and cell proliferation induced by lipopolysaccharide in colorectal cancer. Cell Signal. 2015, 27, 1525–1532. [Google Scholar] [CrossRef]
- Yang, L.; Xie, M.; Yang, M.; Yu, Y.; Zhu, S.; Hou, W.; Kang, R.; Lotze, M.T.; Billiar, T.R.; Wang, H.; et al. PKM2 regulates the Warburg effect and promotes HMGB1 release in sepsis. Nat. Commun. 2014, 5, 4436. [Google Scholar] [CrossRef] [PubMed]
- Andersson, U.; Wang, H.; Palmblad, K.; Aveberger, A.C.; Bloom, O.; Erlandsson-Harris, H.; Janson, A.; Kokkola, R.; Zhang, M.; Yang, H.; et al. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J. Exp. Med. 2000, 192, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Albina, J.E.; Mastrofrancesco, B.; Vessella, J.A.; Louis, C.A.; Henry, W.L., Jr.; Reichner, J.S. HIF-1 expression in healing wounds: HIF-1alpha induction in primary inflammatory cells by TNF-alpha. Am. J. Physiol. Cell Physiol. 2001, 281, C1971–C1977. [Google Scholar] [CrossRef] [PubMed]
- Shatrov, V.A.; Sumbayev, V.V.; Zhou, J.; Brune, B. Oxidized low-density lipoprotein (oxLDL) triggers hypoxia-inducible factor-1alpha (HIF-1alpha) accumulation via redox-dependent mechanisms. Blood 2003, 101, 4847–4849. [Google Scholar] [CrossRef] [PubMed]
- Menegaut, L.; Thomas, C.; Jalil, A.; Julla, J.B.; Magnani, C.; Ceroi, A.; Basmaciyan, L.; Dumont, A.; Le Goff, W.; Mathew, M.J.; et al. Interplay between Liver X Receptor and Hypoxia Inducible Factor 1alpha Potentiates Interleukin-1beta Production in Human Macrophages. Cell Rep. 2020, 31, 107665. [Google Scholar] [CrossRef]
- Walczak-Drzewiecka, A.; Ratajewski, M.; Wagner, W.; Dastych, J. HIF-1alpha is up-regulated in activated mast cells by a process that involves calcineurin and NFAT. J. Immunol. 2008, 181, 1665–1672. [Google Scholar] [CrossRef]
- Elder, M.J.; Webster, S.J.; Fitzmaurice, T.J.; Shaunak, A.S.D.; Steinmetz, M.; Chee, R.; Mallat, Z.; Cohen, E.S.; Williams, D.L.; Gaston, J.S.H.; et al. Dendritic Cell-Derived TSLP Negatively Regulates HIF-1alpha and IL-1beta During Dectin-1 Signaling. Front. Immunol. 2019, 10, 921. [Google Scholar] [CrossRef]
- Sena, L.A.; Li, S.; Jairaman, A.; Prakriya, M.; Ezponda, T.; Hildeman, D.A.; Wang, C.R.; Schumacker, P.T.; Licht, J.D.; Perlman, H.; et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 2013, 38, 225–236. [Google Scholar] [CrossRef]
- Semenza, G.L. Pharmacologic Targeting of Hypoxia-Inducible Factors. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 379–403. [Google Scholar] [CrossRef]
- Karhausen, J.; Furuta, G.T.; Tomaszewski, J.E.; Johnson, R.S.; Colgan, S.P.; Haase, V.H. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J. Clin. Investig. 2004, 114, 1098–1106. [Google Scholar] [CrossRef]
- Cummins, E.P.; Seeballuck, F.; Keely, S.J.; Mangan, N.E.; Callanan, J.J.; Fallon, P.G.; Taylor, C.T. The hydroxylase inhibitor dimethyloxalylglycine is protective in a murine model of colitis. Gastroenterology 2008, 134, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Tambuwala, M.M.; Manresa, M.C.; Cummins, E.P.; Aversa, V.; Coulter, I.S.; Taylor, C.T. Targeted delivery of the hydroxylase inhibitor DMOG provides enhanced efficacy with reduced systemic exposure in a murine model of colitis. J. Control. Release 2015, 217, 221–227. [Google Scholar] [CrossRef]
- Robinson, A.; Keely, S.; Karhausen, J.; Gerich, M.E.; Furuta, G.T.; Colgan, S.P. Mucosal protection by hypoxia-inducible factor prolyl hydroxylase inhibition. Gastroenterology 2008, 134, 145–155. [Google Scholar] [CrossRef]
- Gupta, R.; Chaudhary, A.R.; Shah, B.N.; Jadhav, A.V.; Zambad, S.P.; Gupta, R.C.; Deshpande, S.; Chauthaiwale, V.; Dutt, C. Therapeutic treatment with a novel hypoxia-inducible factor hydroxylase inhibitor (TRC160334) ameliorates murine colitis. Clin. Exp. Gastroenterol. 2014, 7, 13–23. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Keely, S.; Campbell, E.L.; Baird, A.W.; Hansbro, P.M.; Shalwitz, R.A.; Kotsakis, A.; McNamee, E.N.; Eltzschig, H.K.; Kominsky, D.J.; Colgan, S.P. Contribution of epithelial innate immunity to systemic protection afforded by prolyl hydroxylase inhibition in murine colitis. Mucosal Immunol. 2014, 7, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Marks, E.; Naudin, C.; Nolan, G.; Goggins, B.J.; Burns, G.; Mateer, S.W.; Latimore, J.K.; Minahan, K.; Plank, M.; Foster, P.S.; et al. Regulation of IL-12p40 by HIF controls Th1/Th17 responses to prevent mucosal inflammation. Mucosal Immunol. 2017, 10, 1224–1236. [Google Scholar] [CrossRef]
- Xue, X.; Ramakrishnan, S.; Anderson, E.; Taylor, M.; Zimmermann, E.M.; Spence, J.R.; Huang, S.; Greenson, J.K.; Shah, Y.M. Endothelial PAS domain protein 1 activates the inflammatory response in the intestinal epithelium to promote colitis in mice. Gastroenterology 2013, 145, 831–841. [Google Scholar] [CrossRef]
- Shah, Y.M. The role of hypoxia in intestinal inflammation. Mol. Cell Pediatr. 2016, 3, 1. [Google Scholar] [CrossRef]
- Mimouna, S.; Goncalves, D.; Barnich, N.; Darfeuille-Michaud, A.; Hofman, P.; Vouret-Craviari, V. Crohn disease-associated Escherichia coli promote gastrointestinal inflammatory disorders by activation of HIF-dependent responses. Gut Microbes 2011, 2, 335–346. [Google Scholar] [CrossRef]
- Guan, S.Y.; Leng, R.X.; Tao, J.H.; Li, X.P.; Ye, D.Q.; Olsen, N.; Zheng, S.G.; Pan, H.F. Hypoxia-inducible factor-1alpha: A promising therapeutic target for autoimmune diseases. Expert Opin. Ther. Targets 2017, 21, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Walsh, J.C.; Lebedev, A.; Aten, E.; Madsen, K.; Marciano, L.; Kolb, H.C. The clinical importance of assessing tumor hypoxia: Relationship of tumor hypoxia to prognosis and therapeutic opportunities. Antioxid. Redox Signal. 2014, 21, 1516–1554. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Zhao, G. Small molecules targeting HIF-1alpha pathway for cancer therapy in recent years. Bioorg. Med. Chem. 2020, 28, 115235. [Google Scholar] [CrossRef] [PubMed]
- Albadari, N.; Deng, S.; Li, W. The transcriptional factors HIF-1 and HIF-2 and their novel inhibitors in cancer therapy. Expert Opin. Drug Discov. 2019, 14, 667–682. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Yu, Q.; Zhang, X. Allosteric inhibition of HIF-2alpha as a novel therapy for clear cell renal cell carcinoma. Drug Discov. Today 2019, 24, 2332–2340. [Google Scholar] [CrossRef]
- Fallah, J.; Rini, B.I. HIF Inhibitors: Status of Current Clinical Development. Curr. Oncol. Rep. 2019, 21, 6. [Google Scholar] [CrossRef]
- Wu, D.; Su, X.; Lu, J.; Li, S.; Hood, B.L.; Vasile, S.; Potluri, N.; Diao, X.; Kim, Y.; Khorasanizadeh, S.; et al. Bidirectional modulation of HIF-2 activity through chemical ligands. Nat. Chem. Biol. 2019, 15, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Courtney, K.D.; Infante, J.R.; Lam, E.T.; Figlin, R.A.; Rini, B.I.; Brugarolas, J.; Zojwalla, N.J.; Lowe, A.M.; Wang, K.; Wallace, E.M.; et al. Phase I Dose-Escalation Trial of PT2385, a First-in-Class Hypoxia-Inducible Factor-2alpha Antagonist in Patients With Previously Treated Advanced Clear Cell Renal Cell Carcinoma. J. Clin. Oncol. 2018, 36, 867–874. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Plimack, E.R.; Bauer, T.M.; Merchan, J.R.; Papadopoulos, K.P.; McDermott, D.F.; Michaelson, M.D.; Appleman, L.J.; Thamake, S.; Zojwalla, N.J.; et al. Phase I/II study of the oral HIF-2 α inhibitor MK-6482 in patients with advanced clear cell renal cell carcinoma (RCC). J. Clin. Oncol. 2020, 38, 611. [Google Scholar] [CrossRef]
- Xu, R.; Wang, K.; Rizzi, J.P.; Huang, H.; Grina, J.A.; Schlachter, S.T.; Wang, B.; Wehn, P.M.; Yang, H.; Dixon, D.D.; et al. 3-[(1S,2S,3R)-2,3-Difluoro-1-hydroxy-7-methylsulfonylindan-4-yl]oxy-5-fluorobenzo nitrile (PT2977), a Hypoxia-Inducible Factor 2alpha (HIF-2alpha) Inhibitor for the Treatment of Clear Cell Renal Cell Carcinoma. J. Med. Chem. 2019, 62, 6876–6893. [Google Scholar] [CrossRef] [PubMed]





| Cell type | HIF Mediated Effects | Ref. |
|---|---|---|
| eutrophil granulocytes | HIF-1: survival ↑, glycolysis ↑, effector function ↑, (adhesion ↑, migration ↑, production of antimicrobial peptides ↑, formation of DNA traps ↑, phagocytosis ↑, oxidative burst ↑) | [9,80,81,162,163] |
| HIF-2: survival ↑ | [164] | |
| Mast cells, basophils and eosinophils | HIF-1: survival ↑, effector function ↑ (synthesis of IL-8, TNF-α, VEGF, and IL-4 following TLR and/or IgE activation), eosinophil chemotaxis ↑ | [165,166,167,168,169] |
| HIF-2: eosinophil chemotaxis ↓ | [166] | |
| Monocytes/Macrophages | HIF-1: survival ↑, effector function ↑ (release pro-inflammatory TNF-α, IL-1β, IL-12 and IL-6, iNOS activity ↑ and NO production, phagocytosis)↑, glycolysis ↑, M1 polarization | [81,170,171,172] |
| HIF-2: arginase-1 ↑, M2 polarization, phagocytosis ↓, proinflammatory cytokine/chemokine (IL-1β, IL-12, TNF-α, IL-6, IFN-γ, and CXCL2) expression, migration | [163,170,171,173] | |
| Dendritic cells | HIF-1: survival ↑, glycolysis ↑, induction of regulatory T cells ↑, effector function ↑ (migration ↑, proper DC-mediated cytotoxic T cell activation, differentiation ↑) | [174,175,176,177] |
| HIF-2: unknown |
| Cell Type | HIF Mediated Effects | Ref. |
|---|---|---|
| Innate lymphoid cells (ILC2) | HIF-1: late-stage maturation and function of ILC2s via targeting IL-33-ST2 pathway | [244] |
| HIF-2: unknown | ||
| NK cells | HIF-1: tumor growth ↑ (angiogenesis↑, VEGFR-1↑) tumor cell killing ↑, INF-γ ↓, OCR/ECAR ratio ↓ | [245,246] |
| HIF-2: unknown |
| Compound/Drug Name | Objective | Type of Cancer | Phase | NCT Number |
|---|---|---|---|---|
| PT2977 (MK6482) (Inhibition of HIF-2 heterodimerization) | Tumor response | advanced solid tumors/ccRCC /specified solid tumors/glioblastoma (GBM) | I | NCT02974738 |
| ORR | Advanced or metastatic ccRCC | II | NCT03634540 | |
| ORR | VHL-Associated Renal Cell Carcinoma | II | NCT03401788 | |
| PT2385 (Inhibition of HIF-2 heterodimerization) | MTD (Part 1: PT2385; Part 2: PT2385 + nivolumab; Part 3: PT2385 + cabozantinib) | Advanced ccRCC | I | NCT02293980 |
| Tumor radiographic response | Recurrent GBM | II | NCT03216499 | |
| ORR | VHL disease-associated ccRCC | II | NCT03108066 | |
| ARO-HIF2 (Neutralization of HIF2A mRNA) | AEs; RP2D | ccRCC | I | NCT04169711 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Gaber, T. Hypoxia/HIF Modulates Immune Responses. Biomedicines 2021, 9, 260. https://doi.org/10.3390/biomedicines9030260
Chen Y, Gaber T. Hypoxia/HIF Modulates Immune Responses. Biomedicines. 2021; 9(3):260. https://doi.org/10.3390/biomedicines9030260
Chicago/Turabian StyleChen, Yuling, and Timo Gaber. 2021. "Hypoxia/HIF Modulates Immune Responses" Biomedicines 9, no. 3: 260. https://doi.org/10.3390/biomedicines9030260
APA StyleChen, Y., & Gaber, T. (2021). Hypoxia/HIF Modulates Immune Responses. Biomedicines, 9(3), 260. https://doi.org/10.3390/biomedicines9030260