
Energy, Entropy and Quantum Tunneling of Protons and Electrons in Brain Mitochondria: Relation to Mitochondrial Impairment in Aging-Related Human Brain Diseases and Therapeutic Measures


{kind=link}
{kind=link}
Abstract
1. Introduction
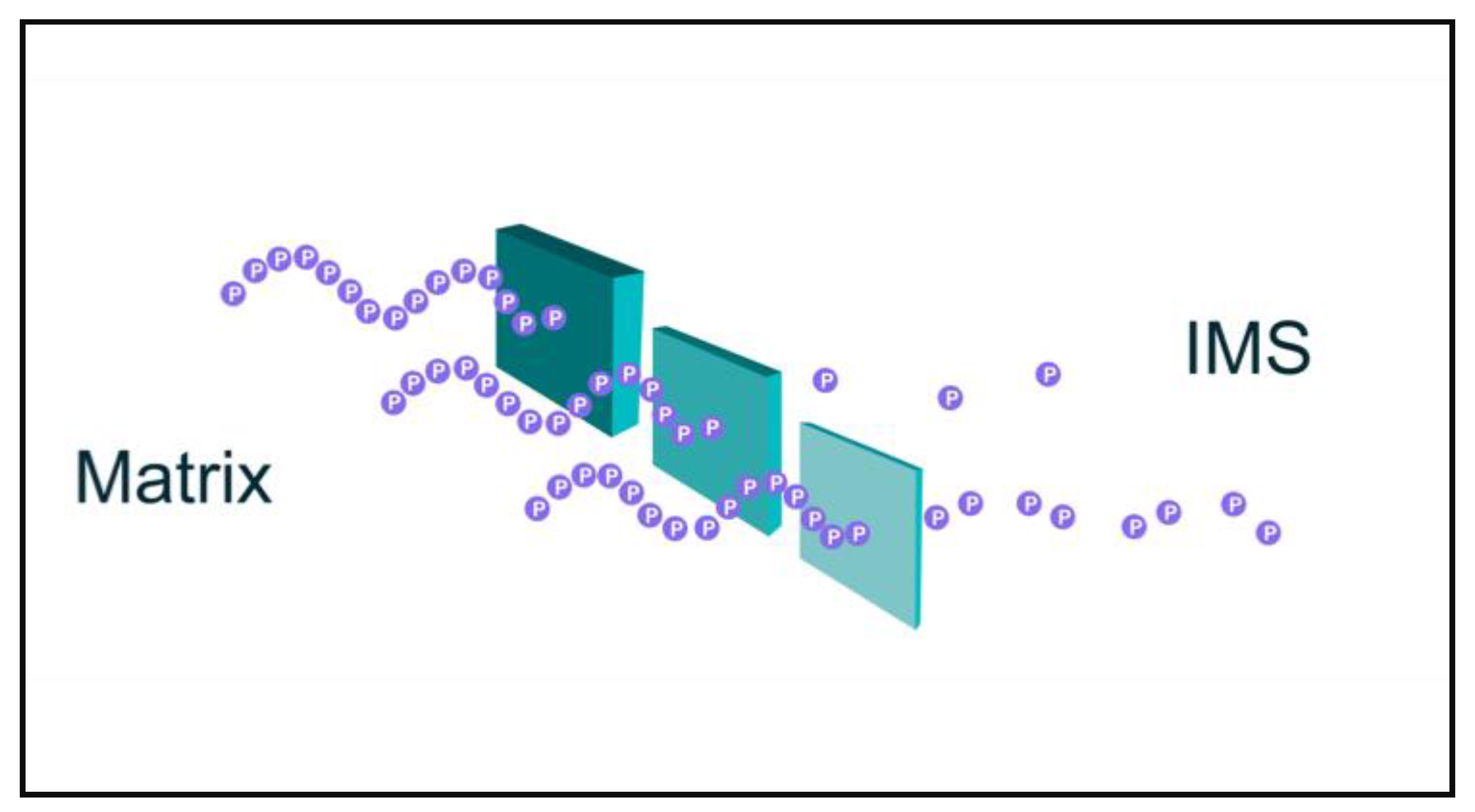
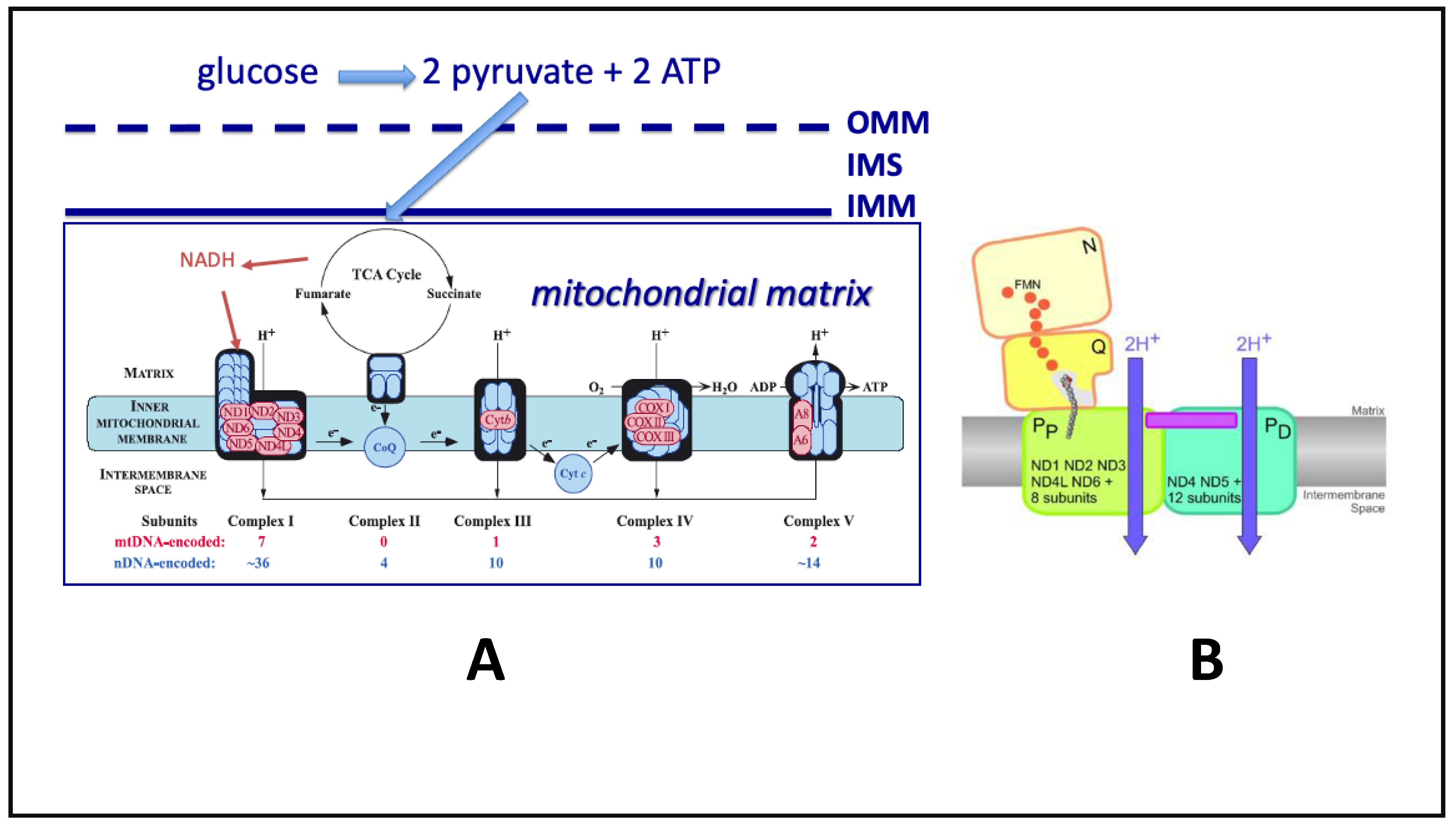
2. Quantum Tunneling of Protons and Electrons in Mitochondria
3. Decoherence
4. Entropy and Neurodegeneration
5. Oxidative Phosphorylation (OXPHOS) Alterations in NDDs
6. Summary of ETC-OXPHOS
7. Brain Mitochondrial Therapeutics
- Correction of mtDNA mutations
- Correction of mtRNA and/or mitochondrial mRNA errors
- Increase in mitochondrial mass leading to increased ETC/OXPHOS capacity to make ATP
- Prevention of epigenetic, nitrative stress (NS) and oxidative stress (OS) damage to ETC/OXPHOS genes or proteins
7.1. Correction of mtDNA Mutations
7.2. Correction of mtRNA and/or Mitochondrial mRNA Errors
7.3. Increase in Mitochondrial Mass Leading to Increased ETC/OXPHOS Capacity to Make ATP
7.4. Prevention of Epigenetic, Nitrative Stress (NS) and Oxidative Stress (OS) Damage to ETC/OXPHOS Genes (Epigenetics) or Proteins (NS and OS)
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Blazey, T.; Snyder, A.Z.; Goyal, M.S.; Vlassenko, A.G.; Raichle, M.E. A systematic meta-analysis of oxygen-to-glucose and oxygen-to-carbohydrate ratios in the resting human brain. PLoS ONE 2018, 13, e0204242. [Google Scholar] [CrossRef] [PubMed]
- Von Bartheld, C.S.; Bahney, J.; Herculano-Houzel, S. The search for true numbers of neurons and glial cells in the human brain: A review of 150 years of cell counting. J. Comp. Neurol. 2016, 524, 3865–3895. [Google Scholar] [CrossRef]
- Löwdin, P.-O. Isotope effect in tunneling and its influence on mutation rates. Mutat. Res. Mol. Mech. Mutagen. 1965, 2, 218–221. [Google Scholar] [CrossRef]
- Srivastava, R. The Role of Proton Transfer on Mutations. Front. Chem. 2019, 7, 536. [Google Scholar] [CrossRef]
- Maudlin, T. Philosophy of Physics: Quantum Theory; Princeton Foundations of Contemporary Philosophy Book, 33; Princeton University Press: Princeton, NJ, USA, 2019. [Google Scholar]
- Dröse, S.; Krack, S.; Sokolova, L.; Zwicker, K.; Barth, H.-D.; Morgner, N.; Heide, H.; Steger, M.; Nübel, E.; Zickermann, V.; et al. Functional Dissection of the Proton Pumping Modules of Mitochondrial Complex I. PLoS Biol. 2011, 9, e1001128. [Google Scholar] [CrossRef] [PubMed]
- Leone, V.; Krah, A.; Faraldo-Gómez, J.D. On the Question of Hydronium Binding to ATP-Synthase Membrane Rotors. Biophys. J. 2010, 99, L53–L55. [Google Scholar] [CrossRef][Green Version]
- Schlosshauer, M. Decoherence and the Quantum-to-Classical Transition; Springer: Berlin/Heidelberg, Germany, 2007. [Google Scholar]
- Bennett, J.P. Medical hypothesis: Neurodegenerative diseases arise from oxidative damage to electron tunneling proteins in mitochondria. Med. Hypotheses 2019, 127, 1–4. [Google Scholar] [CrossRef]
- Trixler, F. Quantum Tunnelling to the Origin and Evolution of Life. Curr. Org. Chem. 2013, 17, 1758–1770. [Google Scholar] [CrossRef]
- Yen, F.; Gao, T. Dielectric Anomaly in Ice near 20 K: Evidence of Macroscopic Quantum Phenomena. J. Phys. Chem. Lett. 2015, 6, 2822–2825. [Google Scholar] [CrossRef] [PubMed]
- Cukierman, S. Et tu, Grotthuss! and other unfinished stories. Biochim. Biophys. Acta (BBA) Bioenerg. 2006, 1757, 876–885. [Google Scholar] [CrossRef] [PubMed]
- Gerwert, K.; Freier, E.; Wolf, S. The role of protein-bound water molecules in microbial rhodopsins. Biochim. Biophys. Acta (BBA) Bioenerg. 2014, 1837, 606–613. [Google Scholar] [CrossRef] [PubMed]
- Hammes-Schiffer, S. Proton-Coupled Electron Transfer: Moving Together and Charging Forward. J. Am. Chem. Soc. 2015, 137, 8860–8871. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Belevich, G.; Gamiz-Hernandez, A.P.; Róg, T.; Vattulainen, I.; Verkhovskaya, M.L.; Wikström, M.; Hummer, G.; Kaila, V.R.I. Redox-induced activation of the proton pump in the respiratory complex I. Proc. Natl. Acad. Sci. USA 2015, 112, 11571–11576. [Google Scholar] [CrossRef] [PubMed]
- Wikstrom, M.; Sharma, V.; Kaila, V.R.; Hosler, J.P.; Hummer, G. New Perspectives on Proton Pumping in Cellular Respiration. Chem. Rev. 2015, 115, 2196–2221. [Google Scholar] [CrossRef] [PubMed]
- Ripple, M.O.; Kim, N.; Springett, R. Mammalian Complex I Pumps 4 Protons per 2 Electrons at High and Physiological Proton Motive Force in Living Cells. J. Biol. Chem. 2013, 288, 5374–5380. [Google Scholar] [CrossRef]
- Signes, A.; Fernandez-Vizarra, E. Assembly of Mammalian Oxidative Phosphorylation Complexes I-V and Supercomplexes. Essays Biochem. 2018, 62, 255–270. [Google Scholar] [PubMed]
- Bennett, S.A.; Tanaz, R.; Cobos, S.N.; Torrente, M.P. Epigenetics in amyotrophic lateral sclerosis: A role for histone post-translational modifications in neurodegenerative disease. Transl. Res. 2019, 204, 19–30. [Google Scholar] [CrossRef]
- Consales, C.; Merla, C.; Marino, C.; Benassi, B. The Epigenetic Component of the Brain Response to Electromagnetic Stimulation in Parkinson’s Disease Patients: A Literature Overview. Bioelectromagnetics 2018, 39, 3–14. [Google Scholar] [CrossRef]
- Phillipson, O.T. Alpha-Synuclein, Epigenetics, Mitochondria, Metabolism, Calcium Traffic, & Circadian Dysfunction in Parkinson’s Disease. An Integrated Strategy for Management. Ageing Res. Rev. 2017, 40, 149–167. [Google Scholar]
- Irwin, M.H.; Moos, W.H.; Faller, D.V.; Steliou, K.; Pinkert, C.A. Epigenetic Treatment of Neurodegenerative Disorders: Alzheimer and Parkinson Diseases. Drug Dev. Res. 2016, 77, 109–123. [Google Scholar] [CrossRef]
- Phang, J.M.; Liu, W.; Hancock, C.N.; Fischer, J.W. Proline Metabolism and Cancer: Emerging Links to Glutamine and Collagen. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Parlato, R.; Liss, B. How Parkinson’s Disease Meets Nucleolar Stress. Biochim. Biophys. Acta 2014, 1842, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Valente, A.X.; Das Neves, R.P.; Oliveira, P.J. Epigenetic engineering to reverse the Parkinson’s expression state. Parkinsonism Relat. Disord. 2012, 18, 717–721. [Google Scholar] [CrossRef]
- Hurley, M.J.; Dexter, D.T. Voltage-Gated Calcium Channels and Parkinson’s Disease. Pharmacol. Ther. 2012, 133, 324–333. [Google Scholar] [CrossRef]
- Diederich, N.J.; Parent, A. Parkinson’s Disease: Acquired Frailty of Archaic Neural Networks? J. Neurol. Sci. 2012, 314, 143–151. [Google Scholar] [CrossRef]
- Mendelsohn, A.R.; Larrick, J.W. Rapamycin As an Antiaging Therapeutic?: Targeting Mammalian Target of Rapamycin to Treat Hutchinson–Gilford Progeria and Neurodegenerative Diseases. Rejuvenation Res. 2011, 14, 437–441. [Google Scholar] [CrossRef]
- Dos Santos, S.M.; Romeiro, C.F.R.; Rodrigues, C.A.; Cerqueira, A.R.L.; Monteiro, M.C. Mitochondrial Dysfunction and Alpha-Lipoic Acid: Beneficial or Harmful in Alzheimer’s Disease? Oxidative Med. Cell. Longev. 2019, 2019, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ellison, E.M.; Bradley-Whitman, M.A.; Lovell, M.A. Single-Base Resolution Mapping of 5-Hydroxymethylcytosine Modifications in Hippocampus of Alzheimer’s Disease Subjects. J. Mol. Neurosci. 2017, 63, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Vachharajani, V.T.; Liu, T.; Wang, X.; Hoth, J.J.; Yoza, B.K.; McCall, C.E. Sirtuins Link Inflammation and Metabolism. J. Immunol. Res. 2016, 2016, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kamps, J.J.A.G.; Huang, J.; Poater, J.; Xu, C.; Pieters, B.J.G.E.; Dong, A.; Jasmin, M.; Sherman, W.; Beuming, T.; Bickelhaupt, F.M.; et al. Chemical basis for the recognition of trimethyllysine by epigenetic reader proteins. Nat. Commun. 2015, 6, 8911. [Google Scholar] [CrossRef]
- Salminen, A.; Haapasalo, A.; Kauppinen, A.; Kaarniranta, K.; Soininen, H.; Hiltunen, M. Impaired Mitochondrial Energy Metabolism in Alzheimer’s Disease: Impact on Pathogenesis via Disturbed Epigenetic Regulation of Chromatin Landscape. Prog. Neurobiol. 2015, 131, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Daulatzai, M.A. “Boomerang Neuropathology“ of Late-Onset Alzheimer’s Disease Is Shrouded in Harmful “BDDS“: Breathing, Diet, Drinking, and Sleep during Aging. Neurotox. Res. 2015, 28, 55–93. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.J. Epigenetic oxidative redox shift (EORS) theory of aging unifies the free radical and insulin signaling theories. Exp. Gerontol. 2010, 45, 173–179. [Google Scholar] [CrossRef]
- Hata, Y.; Ma, N.; Yoneda, M.; Morimoto, S.; Okano, H.; Murayama, S.; Kawanishi, S.; Kuzuhara, S.; Kokubo, Y. Nitrative Stress and Tau Accumulation in Amyotrophic Lateral Sclerosis/Parkinsonism-Dementia Complex (Als/Pdc) in the Kii Penin-sula, Japan. Front. Neurosci. 2017, 11, 751. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Mackie, K. Interplay of cannabinoid 2 (CB2) receptors with nitric oxide synthases, oxidative and nitrative stress, and cell death during remote neurodegeneration. J. Mol. Med. 2012, 90, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Nardo, G.; Pozzi, S.; Pignataro, M.; Lauranzano, E.; Spano, G.; Garbelli, S.; Mantovani, S.; Marinou, K.; Papetti, L.; Monteforte, M.; et al. Amyotrophic Lateral Sclerosis Multiprotein Biomarkers in Peripheral Blood Mononuclear Cells. PLoS ONE 2011, 6, e25545. [Google Scholar] [CrossRef]
- Chen, K.; Northington, F.J.; Martin, L.J. Inducible nitric oxide synthase is present in motor neuron mitochondria and Schwann cells and contributes to disease mechanisms in ALS mice. Anat. Embryol. 2010, 214, 219–234. [Google Scholar] [CrossRef]
- Basso, M.; Samengo, G.; Nardo, G.; Massignan, T.; D’Alessandro, G.; Tartari, S.; Cantoni, L.; Marino, M.; Cheroni, C.; De Biasi, S.; et al. Characterization of Detergent-Insoluble Proteins in ALS Indicates a Causal Link between Nitrative Stress and Aggregation in Pathogenesis. PLoS ONE 2009, 4, e8130. [Google Scholar] [CrossRef]
- Martin, L.J.; Gertz, B.; Pan, Y.; Price, A.C.; Molkentin, J.D.; Chang, Q. The mitochondrial permeability transition pore in motor neurons: Involvement in the pathobiology of ALS mice. Exp. Neurol. 2009, 218, 333–346. [Google Scholar] [CrossRef]
- Martin, L.J. Transgenic Mice with Human Mutant Genes Causing Parkinson’s Disease and Amyotrophic Lateral Sclerosis Provide Common Insight into Mechanisms of Motor Neuron Selective Vulnerability to Degeneration. Rev. Neurosci. 2007, 18, 115–136. [Google Scholar] [CrossRef]
- Kochman, A.; Kośka, C.; Metodiewa, D. Submolecular adventures of brain tyrosine: What are we searching for now? Amino Acids 2002, 23, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Li, H.; Gao, Z. Copper Binding Induces Nitration of NPY under Nitrative Stress: Complicating the Role of NPY in Alzheimer’s Disease. Chem. Res. Toxicol. 2018, 31, 904–913. [Google Scholar] [CrossRef] [PubMed]
- Majkutewicz, I.; Kurowska, E.; Podlacha, M.; Myślińska, D.; Grembecka, B.; Ruciński, J.; Pierzynowska, K.; Wrona, D. Age-dependent effects of dimethyl fumarate on cognitive and neuropathological features in the streptozotocin-induced rat model of Alzheimer’s disease. Brain Res. 2018, 1686, 19–33. [Google Scholar] [CrossRef]
- Guivernau, B.; Bonet, J.; Valls-Comamala, V.; Bosch-Morato, M.; Godoy, J.A.; Inestrosa, N.C.; Peralvarez-Marin, A.; Fernan-dez-Busquets, X.; Andreu, D.; Oliva, B.; et al. Amyloid-Beta Peptide Nitrotyrosination Stabilizes Oligomers and En-hances Nmdar-Mediated Toxicity. J. Neurosci. 2016, 36, 11693–11703. [Google Scholar] [CrossRef]
- Lu, N.; Li, J.; Gao, Z. Key Roles of Tyr 10 in Cu Bound Abeta Complexes and Its Relevance to Alzheimer’s Disease. Arch. Biochem. Biophys. 2015, 584, 1–9. [Google Scholar] [CrossRef]
- Lu, N.; Li, J.; Tian, R.; Peng, Y.Y. Key Roles of Arg(5), Tyr(10) and His Residues in Abeta-Heme Peroxidase: Relevance to Alzheimer’s Disease. Biochem. Biophys. Res. Commun. 2014, 452, 676–681. [Google Scholar] [CrossRef]
- Weinreb, O.; Amit, T.; Bar-Am, O.; Youdim, M.B. Ladostigil: A Novel Multimodal Neuroprotective Drug with Cholines-terase and Brain-Selective Monoamine Oxidase Inhibitory Activities for Alzheimer’s Disease Treatment. Curr. Drug. Targets 2012, 13, 483–494. [Google Scholar] [CrossRef]
- Giridharan, V.V.; Thandavarayan, R.A.; Konishi, T. Effect of Shengmai-san on Cognitive Performance and Cerebral Oxidative Damage in BALB/c Mice. J. Med. Food 2011, 14, 601–609. [Google Scholar] [CrossRef]
- Quinn, J.F.; Bussiere, J.R.; Hammond, R.S.; Montine, T.J.; Henson, E.; Jones, R.E.; Stackman, R.W., Jr. Chronic Dietary Alpha-Lipoic Acid Reduces Deficits in Hippocampal Memory of Aged Tg2576 Mice. Neurobiol. Aging 2007, 28, 213–225. [Google Scholar] [CrossRef]
- Calabrese, V.; Sultana, R.; Scapagnini, G.; Guagliano, E.; Sapienza, M.; Bella, R.; Kanski, J.; Pennisi, G.; Mancuso, C.; Stella, A.M.; et al. Nitrosative Stress, Cellular Stress Response, and Thiol Homeostasis in Patients with Alzheimer’s Disease. Antioxid. Redox Signal. 2006, 8, 1975–1986. [Google Scholar] [CrossRef] [PubMed]
- Giasson, B.I.; Ischiropoulos, H.; Lee, V.M.; Trojanowski, J.Q. The Relationship between Oxidative/Nitrative Stress and Pathological Inclusions in Alzheimer’s and Parkinson’s Diseases. Free Radic. Biol. Med. 2002, 32, 1264–1275. [Google Scholar] [CrossRef]
- Williamson, K.S.; Gabbita, S.P.; Mou, S.; West, M.; Pye, Q.N.; Markesbery, W.R.; Cooney, R.V.; Grammas, P.; Reimann-Philipp, U.; Floyd, R.A.; et al. The Nitration Product 5-Nitro-Gamma-Tocopherol Is Increased in the Alzheimer Brain. Nitric Oxide 2002, 6, 221–227. [Google Scholar] [CrossRef]
- Giasson, B.I.; Duda, J.E.; Murray, I.V.; Chen, Q.; Souza, J.M.; Hurtig, H.I.; Ischiropoulos, H.; Trojanowski, J.Q.; Lee, V.M. Oxidative Damage Linked to Neurodegeneration by Selective Alpha-Synuclein Nitration in Synucleinopathy Lesions. Science 2000, 290, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Wen, X.; Jiang, H.; Wang, J.; Song, N.; Xie, J. Interactions between Iron and Alpha-Synuclein Pathology in Parkinson’s Disease. Free Radic. Biol. Med. 2019, 141, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Maki, R.A.; Holzer, M.; Motamedchaboki, K.; Malle, E.; Masliah, E.; Marsche, G.; Reynolds, G.F. Human Myeloperoxidase (Hmpo) Is Expressed in Neurons in the Substantia Nigra in Parkinson’s Disease and in the Hmpo-Alpha-Synuclein-A53t Mouse Model, Correlating with Increased Nitration and Aggregation of Alpha-Synuclein and Exacerbation of Motor Impairment. Free Radic. Biol. Med. 2019, 141, 115–140. [Google Scholar]
- He, Y.; Yu, Z.; Chen, S. Alpha-Synuclein Nitration and Its Implications in Parkinson’s Disease. ACS Chem. Neurosci. 2019, 10, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.Q.; Wang, Y.L.; Yuan, B.S.; Yuan, X.; Hou, X.O.; Bian, Y.S.; Liu, C.F.; Hu, L.F. Impaired Cbs-H2s Signaling Axis Contributes to Mptp-Induced Neurodegeneration in a Mouse Model of Parkinson’s Disease. Brain Behav. Immun. 2018, 67, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Collier, T.J.; Kanaan, N.M.; Kordower, J.H. Aging and Parkinson’s Disease: Different Sides of the Same Coin? Mov. Disord. 2017, 32, 983–990. [Google Scholar] [CrossRef]
- Kleinknecht, A.; Popova, B.; Lazaro, D.F.; Pinho, R.; Valerius, O.; Outeiro, T.F.; Braus, G.H. C-Terminal Tyrosine Residue Modifications Modulate the Protective Phosphorylation of Serine 129 of Alpha-Synuclein in a Yeast Model of Parkinson’s Disease. PLoS Genet. 2016, 12, e1006098. [Google Scholar] [CrossRef]
- Dias, V.; Junn, E.; Mouradian, M.M. The Role of Oxidative Stress in Parkinson’s Disease. J. Parkinson’s Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef]
- Schildknecht, S.; Gerding, H.R.; Karreman, C.; Drescher, M.; Lashuel, H.A.; Outeiro, T.F.; Di Monte, D.A.; Leist, M. Oxidative and Nitrative Alpha-Synuclein Modifications and Proteostatic Stress: Implications for Disease Mechanisms and Interventions in Synucleinopathies. J. Neurochem. 2013, 125, 491–511. [Google Scholar] [CrossRef]
- Lee, J.; Giordano, S.; Zhang, J. Autophagy, mitochondria and oxidative stress: Cross-talk and redox signalling. Biochem. J. 2011, 441, 523–540. [Google Scholar] [CrossRef] [PubMed]
- Sian-Hulsmann, J.; Mandel, S.; Youdim, M.B.; Riederer, P. The Relevance of Iron in the Pathogenesis of Parkinson’s Disease. J. Neurochem. 2011, 118, 939–957. [Google Scholar] [CrossRef]
- Kupershmidt, L.; Okun, Z.; Amit, T.; Mandel, S.; Saltsman, I.; Mahammed, A.; Bar-Am, O.; Gross, Z.; Youdim, M.B. Metal-locorroles as Cytoprotective Agents against Oxidative and Nitrative Stress in Cellular Models of Neurodegeneration. J. Neurochem. 2010, 113, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Danielson, S.R.; Andersen, J.K. Oxidative and Nitrative Protein Modifications in Parkinson’s Disease. Free Radic. Biol. Med. 2008, 44, 1787–1794. [Google Scholar] [CrossRef]
- Trostchansky, A.; Rubbo, H. Lipid nitration and formation of lipid-protein adducts: Biological insights. Amino Acids 2006, 32, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Weinstock, M.; Luques, L.; Bejar, C.; Shoham, S. Ladostigil, a novel multifunctional drug for the treatment of dementia co-morbid with depression. Focus Extrapyramidal Dysfunct. 2006, 70, 443–446. [Google Scholar] [CrossRef]
- Jenner, P.; Olanow, C.W. The Pathogenesis of Cell Death in Parkinson’s Disease. Neurology 2006, 66 (Suppl. 4), S24–S36. [Google Scholar] [CrossRef]
- Norris, E.H.; Giasson, B.I. Role of Oxidative Damage in Protein Aggregation Associated with Parkinson’s Disease and Related Disorders. Antioxid. Redox Signal. 2005, 7, 672–684. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, A. Postmortem Studies in Parkinson’s Disease. Dialogues Clin. Neurosci. 2004, 6, 281–293. [Google Scholar] [PubMed]
- Yamin, G.; Uversky, V.N.; Fink, A.L. Nitration Inhibits Fibrillation of Human Alpha-Synuclein in Vitro by Formation of Soluble Oligomers. FEBS Lett. 2003, 542, 147–152. [Google Scholar] [CrossRef]
- Bozzo, F.; Mirra, A.; Carri, M.T. Oxidative Stress and Mitochondrial Damage in the Pathogenesis of Als: New Perspectives. Neurosci. Lett. 2017, 636, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Bai, Z.; Qin, X.; Cheng, Y. Aberrations in Oxidative Stress Markers in Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. Oxid. Med. Cell. Longev. 2019, 2019, 1712323. [Google Scholar] [CrossRef]
- Butterfield, D.A. Amyloid Beta-Peptide (1-42)-Induced Oxidative Stress and Neurotoxicity: Implications for Neurodegener-ation in Alzheimer’s Disease Brain. A Review. Free Radic. Res. 2002, 36, 1307–1313. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Castegna, A.; Drake, J.; Scapagnini, G.; Calabrese, V. Vitamin E and Neurodegenerative Disorders Associated with Oxidative Stress. Nutr. Neurosci. 2002, 5, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Griffin, S.; Munch, G.; Pasinetti, G.M. Amyloid Beta-Peptide and Amyloid Pathology Are Central to the Oxidative Stress and Inflammatory Cascades under Which Alzheimer’s Disease Brain Exists. J. Alzheimer’s Dis. 2002, 4, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Kanski, J. Methionine Residue 35 Is Critical for the Oxidative Stress and Neurotoxic Properties of Alzheimer’s Amyloid Beta-Peptide 1-42. Peptides 2002, 23, 1299–1309. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Lauderback, C.M. Lipid Peroxidation and Protein Oxidation in Alzheimer’s Disease Brain: Potential Causes and Consequences Involving Amyloid Beta-Peptide-Associated Free Radical Oxidative Stress. Free Radic. Biol. Med. 2002, 32, 1050–1060. [Google Scholar] [CrossRef]
- Chang, Y.-T.; Chang, W.-N.; Tsai, N.-W.; Huang, C.-C.; Kung, C.-T.; Su, Y.-J.; Lin, W.-C.; Cheng, B.-C.; Su, C.-M.; Chiang, Y.-F.; et al. The Roles of Biomarkers of Oxidative Stress and Antioxidant in Alzheimer’s Disease: A Systematic Review. BioMed Res. Int. 2014, 2014, 1–14. [Google Scholar] [CrossRef]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Boil. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Chen, Z.; Zhong, C. Oxidative Stress in Alzheimer’s Disease. Neurosci. Bull. 2014, 30, 271–281. [Google Scholar] [CrossRef]
- Drake, J.; Kanski, J.; Varadarajan, S.; Tsoras, M.; Butterfield, D.A. Elevation of Brain Glutathione by Gamma-Glutamylcysteine Ethyl Ester Protects against Peroxynitrite-Induced Oxidative Stress. J. Neurosci. Res. 2002, 68, 776–784. [Google Scholar] [CrossRef]
- Kanski, J.; Aksenova, M.; Schoneich, C.; Butterfield, D.A. Substitution of Isoleucine-31 by Helical-Breaking Proline Abol-ishes Oxidative Stress and Neurotoxic Properties of Alzheimer’s Amyloid Beta-Peptide. Free Radic. Biol. Med. 2002, 32, 1205–1211. [Google Scholar] [CrossRef]
- Kanski, J.; Varadarajan, S.; Aksenova, M.; Butterfield, D.A. Role of Glycine-33 and Methionine-35 in Alzheimer’s Amyloid Beta-Peptide 1-42-Associated Oxidative Stress and Neurotoxicity. Biochim. Biophys. Acta 2002, 1586, 190–198. [Google Scholar] [CrossRef]
- LaFontaine, M.A.; Mattson, M.P.; Butterfield, D.A. Oxidative Stress in Synaptosomal Proteins from Mutant Presenilin-1 Knock-in Mice: Implications for Familial Alzheimer’s Disease. Neurochem. Res. 2002, 27, 417–421. [Google Scholar] [CrossRef]
- Tönnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.G.; Zhu, X. Oxidative Stress and Mitochondrial Dysfunction in Alzheimer’s Disease. Biochim. Biophys. Acta 2014, 1842, 1240–1247. [Google Scholar] [CrossRef]
- Guo, J.D.; Zhao, X.; Li, Y.; Li, G.R.; Liu, X.L. Damage to Dopaminergic Neurons by Oxidative Stress in Parkinson’s Disease (Review). Int. J. Mol. Med. 2018, 41, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.T. Oxidative Stress and Mitochondrial Dysfunction-Linked Neurodegenerative Disorders. Neurol. Res. 2017, 39, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Khan, Z.; Ali, S.A. Oxidative Stress-Related Biomarkers in Parkinson’s Disease: A Systematic Review and Meta-Analysis. Iran. J. Neurol. 2018, 17, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.; Wong, Y.C.; Ysselstein, D.; Severino, A.; Krainc, D. Synaptic, Mitochondrial, and Lysosomal Dysfunction in Parkinson’s Disease. Trends Neurosci. 2019, 42, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson Disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Raza, C.; Anjum, R.; Shakeel, N.U.A. Parkinson’s Disease: Mechanisms, Translational Models and Management Strate-gies. Life Sci. 2019, 226, 77–90. [Google Scholar] [CrossRef]
- Trist, B.G.; Hare, D.J.; Double, K.L. Oxidative Stress in the Aging Substantia Nigra and the Etiology of Parkinson’s Disease. Aging Cell 2019, 18, e13031. [Google Scholar] [CrossRef]
- Wei, Z.; Li, X.; Li, X.; Liu, Q.; Cheng, Y. Oxidative Stress in Parkinson’s Disease: A Systematic Review and Meta-Analysis. Front. Mol. Neurosci. 2018, 11, 236. [Google Scholar] [CrossRef]
- Santibanez-Koref, M.; Griffin, H.; Turnbull, D.M.; Chinnery, P.F.; Herbert, M.; Hudson, F. Assessing Mitochondrial Heteroplasmy Using Next Generation Sequencing: A Note of Caution. Mitochondrion 2019, 46, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Craven, L.; Murphy, J.; Turnbull, D.M.; Taylor, R.W.; Gorman, G.S.; McFarland, R. Scientific and Ethical Issues in Mitochondrial Donation. New Bioeth. 2018, 24, 57–73. [Google Scholar] [CrossRef]
- Chinnery, P.F.; Craven, L.; Mitalipov, S.; Stewart, J.B.; Herbert, M.; Turnbull, D.M. The challenges of mitochondrial replacement. PLoS Genet. 2014, 10, e1004315. [Google Scholar] [CrossRef]
- Craven, L.; Murphy, J.L.; Turnbull, D.M. Mitochondrial donation—Hope for families with mitochondrial DNA disease. Emerg. Top. Life Sci. 2020, 4, 151–154. [Google Scholar] [CrossRef]
- Hyslop, L.A.; Blakeley, P.; Craven, L.; Richardson, J.; Fogarty, N.M.E.; Fragouli, E.; Lamb, M.; Wamaitha, S.E.; Prathalingam, N.; Zhang, Q.; et al. Towards clinical application of pronuclear transfer to prevent mitochondrial DNA disease. Nature 2016, 534, 383–386. [Google Scholar] [CrossRef]
- Pickett, S.J.; Blain, A.; Ng, Y.S.; Wilson, I.J.; Taylor, R.W.; McFarland, R.; Turnbull, D.M.; Gorman, G.S. Mitochondrial Donation—Which Women Could Benefit? New Engl. J. Med. 2019, 380, 1971–1972. [Google Scholar] [CrossRef]
- Richardson, J.; Irving, L.; Hyslop, L.A.; Choudhary, M.; Murdoch, A.; Turnbull, D.M.; Herbert, M. Concise Reviews: Assisted Reproductive Technologies to Prevent Transmission of Mitochondrial DNA Disease. Stem Cells 2015, 33, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Amato, P.; Tachibana, M.; Sparman, M.; Mitalipov, S. Three-Parent in Vitro Fertilization: Gene Replacement for the Prevention of Inherited Mitochondrial Diseases. Fertil. Steril. 2014, 101, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Byrne, J.A.; Pedersen, D.A.; Clepper, L.L.; Nelson, M.; Sanger, W.G.; Gokhale, S.; Wolf, D.P.; Mitalipov, S.M. Producing primate embryonic stem cells by somatic cell nuclear transfer. Nature 2007, 450, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Folmes, C.D.L.; Ma, H.; Mitalipov, S.; Terzic, A. Mitochondria in pluripotent stem cells: Stemness regulators and disease targets. Curr. Opin. Genet. Dev. 2016, 38, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kang, E.; Wu, J.; Gutierrez, N.M.; Koski, A.; Tippner-Hedges, R.; Agaronyan, K.; Platero-Luengo, A.; Martinez-Redondo, P.; Ma, H.; Lee, Y.; et al. Mitochondrial Re-placement in Human Oocytes Carrying Pathogenic Mitochondrial DNA Mutations. Nature 2016, 540, 270–275. [Google Scholar] [CrossRef]
- Ma, H.; Folmes, C.D.L.; Wu, J.; Morey, R.; Mora-Castilla, S.; Ocampo, A.; Ma, L.; Poulton, J.; Wang, X.; Ahmed, R.; et al. Metabolic rescue in pluripotent cells from patients with mtDNA disease. Nat. Cell Biol. 2015, 524, 234–238. [Google Scholar] [CrossRef]
- Ma, H.; Marti Gutierrez, N.; Morey, R.; Van Dyken, C.; Kang, E.; Hayama, T.; Lee, Y.; Li, Y.; Tippner-Hedges, R.; Wolf, D.P.; et al. Incompatibility between Nuclear and Mitochondrial Genomes Contributes to an Interspecies Re-productive Barrier. Cell Metab. 2016, 24, 283–294. [Google Scholar] [CrossRef]
- Ma, H.; O’Neil, R.C.; Gutierrez, N.M.; Hariharan, M.; Zhang, Z.Z.; He, Y.; Cinnioglu, C.; Kayali, R.; Kang, E.; Lee, Y.; et al. Functional Human Oocytes Generated by Transfer of Polar Body Genomes. Cell Stem Cell 2017, 20, 112–119. [Google Scholar] [CrossRef]
- Mitalipov, S.; Wolf, D.P. Clinical and Ethical Implications of Mitochondrial Gene Transfer. Trends Endocrinol. Metab. 2014, 25, 5–7. [Google Scholar] [CrossRef]
- Tachibana, M.; Amato, P.; Sparman, M.; Gutierrez, N.M.; Tippner-Hedges, R.; Ma, H.; Kang, E.; Fulati, A.; Lee, H.-S.; Sritanaudomchai, H.; et al. Human Embryonic Stem Cells Derived by Somatic Cell Nuclear Transfer. Cell 2013, 153, 1228–1238. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Amato, P.; Sparman, M.; Woodward, J.; Sanchis, D.M.; Ma, H.; Gutierrez, N.M.; Tippner-Hedges, R.; Kang, E.; Lee, H.-S.; et al. Towards germline gene therapy of inherited mitochondrial diseases. Nat. Cell Biol. 2013, 493, 627–631. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Sparman, M.; Mitalipov, S. Chromosome transfer in mature oocytes. Fertil. Steril. 2012, 97, e16. [Google Scholar] [CrossRef]
- Tachibana, M.; Sparman, M.; Sritanaudomchai, H.; Ma, H.; Clepper, L.; Woodward, J.; Li, Y.; Ramsey, C.; Kolotushkina, O.; Mitalipov, S. Mitochondrial gene replacement in primate offspring and embryonic stem cells. Nature 2009, 461, 367–372. [Google Scholar] [CrossRef]
- Wolf, D.P.; Mitalipov, N.; Mitalipov, S. Mitochondrial replacement therapy in reproductive medicine. Trends Mol. Med. 2015, 21, 68–76. [Google Scholar] [CrossRef]
- Wolf, D.P.; Mitalipov, S. Mitochondrial Replacement Therapies Can Circumvent Mtdna-Based Disease Transmission. Cell Metab. 2014, 20, 6–8. [Google Scholar] [CrossRef][Green Version]
- Zhao, M.T.; Chen, H.; Liu, Q.; Shao, N.Y.; Sayed, N.; Wo, H.T.; Zhang, J.Z.; Ong, S.G.; Liu, C.; Kim, Y.; et al. Molecular and Functional Resemblance of Differentiated Cells Derived from Isogenic Human Ipscs and Scnt-Derived Escs. Proc. Natl. Acad. Sci. USA 2017, 114, E11111–E11120. [Google Scholar] [CrossRef] [PubMed]
- Elson, J.L.; Swalwell, H.; Blakely, E.L.; McFarland, R.; Taylor, R.W.; Turnbull, D.M. Pathogenic mitochondrial tRNA mutations—Which mutations are inherited and why? Hum. Mutat. 2009, 30, E984–E992. [Google Scholar] [CrossRef] [PubMed]
- Yarham, J.W.; Elson, J.L.; Blakely, E.L.; McFarland, R.; Taylor, R.W. Mitochondrial tRNA mutations and disease. Wiley Interdiscip. Rev. RNA 2010, 1, 304–324. [Google Scholar] [CrossRef]
- Ribas de Pouplana, L. The Mitochondrial Trna Conundrum. Nat. Rev. Mol. Cell Biol. 2020, 21, 361. [Google Scholar] [CrossRef]
- Richter, U.; Evans, M.E.; Clark, W.C.; Marttinen, P.; Shoubridge, E.A.; Suomalainen, A.; Wredenberg, A.; Wedell, A.; Pan, T.; Battersby, B.J. Rna Modification Landscape of the Human Mitochondrial Trna(Lys) Regulates Protein Synthesis. Nat. Commun. 2018, 9, 3966. [Google Scholar] [CrossRef]
- Karicheva, O.Z.; Kolesnikova, O.A.; Schirtz, T.; Vysokikh, M.Y.; Mager-Heckel, A.-M.; Lombès, A.; Boucheham, A.; Krasheninnikov, I.A.; Martin, R.P.; Entelis, N.; et al. Correction of the consequences of mitochondrial 3243A>G mutation in the MT-TL1 gene causing the MELAS syndrome by tRNA import into mitochondria. Nucleic Acids Res. 2011, 39, 8173–8186. [Google Scholar] [CrossRef]
- Wang, G.; Shimada, E.; Zhang, J.; Hong, J.S.; Smith, G.M.; Teitell, M.A.; Koehler, C.M. Correcting human mitochondrial mutations with targeted RNA import. Proc. Natl. Acad. Sci. USA 2012, 109, 4840–4845. [Google Scholar] [CrossRef]
- Alfadhel, M.; Nashabat, M.; Abu Ali, Q.; Hundallah, K. Mitochondrial iron-sulfur cluster biogenesis from molecular understanding to clinical disease. Neuroscience 2017, 22, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Artika, I.M. Current understanding of structure, function and biogenesis of yeast mitochondrial ATP synthase. J. Bioenerg. Biomembr. 2019, 51, 315–328. [Google Scholar] [CrossRef]
- Babbitt, S.E.; Sutherland, M.C.; San Francisco, B.; Mendez, D.L.; Kranz, R.G. Mitochondrial cytochrome c biogenesis: No longer an enigma. Trends Biochem. Sci. 2015, 40, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Barros, M.H.; McStay, G.P. Modular biogenesis of mitochondrial respiratory complexes. Mitochondrion 2020, 50, 94–114. [Google Scholar] [CrossRef] [PubMed]
- Bishop, D.J.; Botella, J.; Genders, A.J.; Lee, M.J.-C.; Saner, N.J.; Kuang, J.; Yan, X.; Granata, C. High-Intensity Exercise and Mitochondrial Biogenesis: Current Controversies and Future Research Directions. Physiology 2019, 34, 56–70. [Google Scholar] [CrossRef] [PubMed]
- Bouchez, C.; Devin, A. Mitochondrial Biogenesis and Mitochondrial Reactive Oxygen Species (Ros): A Complex Rela-tionship Regulated by the Camp/Pka Signaling Pathway. Cells 2019, 8, 287. [Google Scholar] [CrossRef] [PubMed]
- Bragoszewski, P.; Turek, M.; Chacinska, A. Control of mitochondrial biogenesis and function by the ubiquitin–proteasome system. Open Biol. 2017, 7. [Google Scholar] [CrossRef]
- Bykov, Y.S.; Rapaport, D.; Herrmann, J.M.; Schuldiner, M. Cytosolic Events in the Biogenesis of Mitochondrial Proteins. Trends Biochem. Sci. 2020, 45, 650–667. [Google Scholar] [CrossRef]
- Cameron, R.B.; Beeson, C.C.; Schnellmann, R.G. Development of Therapeutics that Induce Mitochondrial Biogenesis for the Treatment of Acute and Chronic Degenerative Diseases. J. Med. Chem. 2016, 59, 10411–10434. [Google Scholar] [CrossRef]
- Carelli, V.; Maresca, A.; Caporali, L.; Trifunov, S.; Zanna, C.; Rugolo, M. Mitochondria: Biogenesis and mitophagy balance in segregation and clonal expansion of mitochondrial DNA mutations. Int. J. Biochem. Cell Biol. 2015, 63, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Cherry, A.D.; Piantadosi, C.A. Regulation of Mitochondrial Biogenesis and Its Intersection with Inflammatory Responses. Antioxid. Redox Signal. 2015, 22, 965–976. [Google Scholar] [CrossRef]
- Craig, D.M.; Ashcroft, S.P.; Belew, M.Y.; Stocks, B.; Currell, K.; Baar, K.; Philp, A. Utilizing small nutrient compounds as enhancers of exercise-induced mitochondrial biogenesis. Front. Physiol. 2015, 6, 296. [Google Scholar] [CrossRef] [PubMed]
- Davinelli, S.; De Stefani, D.; De Vivo, I.; Scapagnini, G. Polyphenols as Caloric Restriction Mimetics Regulating Mitochondrial Biogenesis and Mitophagy. Trends Endocrinol. Metab. 2020, 31, 536–550. [Google Scholar] [CrossRef]
- De Oliveira, M.R.; Jardim, F.R.; Setzer, W.N.; Nabavi, S.M. Curcumin, mitochondrial biogenesis, and mitophagy: Exploring recent data and indicating future needs. Biotechnol. Adv. 2016, 34, 813–826. [Google Scholar] [CrossRef]
- De Oliveira, M.R.; Nabavi, S.F.; Manayi, A.; Daglia, M.; Hajheydari, Z. Resveratrol and the mitochondria: From triggering the intrinsic apoptotic pathway to inducing mitochondrial biogenesis, a mechanistic view. Biochim. Biophys. Acta (BBA) Gen. Subj. 2016, 1860, 727–745. [Google Scholar] [CrossRef]
- Doan, K.N.; Ellenrieder, L.; Becker, T. Mitochondrial porin links protein biogenesis to metabolism. Curr. Genet. 2019, 65, 899–903. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W., 2nd; Vega, R.B.; Kelly, D.P. Mitochondrial Biogenesis and Dynamics in the Developing and Diseased Heart. Genes Dev. 2015, 29, 1981–1991. [Google Scholar] [CrossRef] [PubMed]
- Drwesh, L.; Rapaport, D. Biogenesis Pathways of Alpha-Helical Mitochondrial Outer Membrane Proteins. Biol. Chem. 2020, 401, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Dutkiewicz, R.; Nowak, M. Molecular chaperones involved in mitochondrial iron–sulfur protein biogenesis. JBIC J. Biol. Inorg. Chem. 2018, 23, 569–579. [Google Scholar] [CrossRef]
- Edens, B.M.; Miller, N.; Ma, Y.-C. Impaired Autophagy and Defective Mitochondrial Function: Converging Paths on the Road to Motor Neuron Degeneration. Front. Cell. Neurosci. 2016, 10, 44. [Google Scholar] [CrossRef]
- Edwards, R.; Gerlich, S.; Tokatlidis, K. The biogenesis of mitochondrial intermembrane space proteins. Biol. Chem. 2020, 401, 737–747. [Google Scholar] [CrossRef]
- Ellenrieder, L.; Martensson, C.U.; Becker, T. Biogenesis of Mitochondrial Outer Membrane Proteins, Problems and Diseases. Biol. Chem. 2015, 396, 1199–1213. [Google Scholar] [CrossRef]
- Erlich, A.T.; Tryon, L.D.; Crilly, M.J.; Memme, J.M.; Moosavi, Z.S.M.; Oliveira, A.N.; Beyfuss, K.; Hood, D.A. Function of specialized regulatory proteins and signaling pathways in exercise-induced muscle mitochondrial biogenesis. Integr. Med. Res. 2016, 5, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J.; Scorza, F.A. Effects of antiepileptic drugs on mitochondrial functions, morphology, kinetics, biogenesis, and survival. Epilepsy Res. 2017, 136, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Fontecha-Barriuso, M.; Martin-Sanchez, D.; Martinez-Moreno, J.M.; Monsalve, M.; Ramos, A.M.; Sanchez-Niño, M.D.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A.B. The Role of PGC-1α and Mitochondrial Biogenesis in Kidney Diseases. Biomolecules 2020, 10, 347. [Google Scholar] [CrossRef] [PubMed]
- Franz, A.; Kevei, É.; Hoppe, T. Double-edged alliance: Mitochondrial surveillance by the UPS and autophagy. Curr. Opin. Cell Biol. 2015, 37, 18–27. [Google Scholar] [CrossRef]
- Fu, W.; Liu, Y.; Yin, H. Mitochondrial Dynamics: Biogenesis, Fission, Fusion, and Mitophagy in the Regulation of Stem Cell Behaviors. Stem Cells Int. 2019, 2019, 1–15. [Google Scholar] [CrossRef]
- Golpich, M.; Amini, E.; Mohamed, Z.; Ali, R.A.; Ibrahim, N.M.; Ahmadiani, A. Mitochondrial Dysfunction and Biogenesis in Neurodegenerative diseases: Pathogenesis and Treatment. CNS Neurosci. Ther. 2017, 23, 5–22. [Google Scholar] [CrossRef]
- Gomes, J.V.P.; Rigolon, T.C.B.; da Silveira Souza, M.S.; Alvarez-Leite, J.I.; Lucia, C.M.D.; Martino, H.S.D.; Rosa, C.D.O.B. Anti-obesity Effects of Anthocyanins on Mitochondrial Biogenesis, Inflammation, and Oxidative Stress: A Systematic Review. Nutrition 2019, 66, 192–202. [Google Scholar] [CrossRef]
- Granata, C.; Jamnick, N.A.; Bishop, D.J. Principles of Exercise Prescription, and How They Influence Exercise-Induced Changes of Transcription Factors and Other Regulators of Mitochondrial Biogenesis. Sports Med. 2018, 48, 1541–1559. [Google Scholar] [CrossRef]
- Grevel, A.; Pfanner, N.; Becker, T. Coupling of import and assembly pathways in mitochondrial protein biogenesis. Biol. Chem. 2019, 401, 117–129. [Google Scholar] [CrossRef]
- Groennebaek, T.; Vissing, K. Impact of Resistance Training on Skeletal Muscle Mitochondrial Biogenesis, Content, and Function. Front. Physiol. 2017, 8, 713. [Google Scholar] [CrossRef]
- Gupta, A.; Becker, T. Mechanisms and pathways of mitochondrial outer membrane protein biogenesis. Biochim. Biophys. Acta (BBA) Bioenerg. 2021, 1862, 148323. [Google Scholar] [CrossRef] [PubMed]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction between the Nrf2 and PGC-1α Signaling Pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Duan, Y.; Yao, K.; Li, F.; Hou, Y.; Wu, G.; Yin, Y. Beta-Hydroxy-Beta-Methylbutyrate, Mitochondrial Biogenesis, and Skeletal Muscle Health. Amino Acids 2016, 48, 653–664. [Google Scholar] [CrossRef]
- Herrmann, J.M.; Riemer, J. Apoptosis inducing factor and mitochondrial NADH dehydrogenases: Redox-controlled gear boxes to switch between mitochondrial biogenesis and cell death. Biol. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Hey-Mogensen, M.; Clausen, T.R. Targeting Mitochondrial Biogenesis and Mitochondrial Substrate Utilization to Treat Obesity and Insulin Resistance, Respectively—Two Data-Driven Hypotheses. Curr. Diabetes Rev. 2017, 13, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Hiraumi, Y.; Huang, C.; Andres, A.M.; Xiong, Y.; Ramil, J.; Gottlieb, R.A. Myogenic Progenitor Cell Differentiation Is Dependent on Modulation of Mitochondrial Biogenesis through Autophagy. In Etiology and Morphogenesis of Congenital Heart Disease: From Gene Function and Cellular Interaction to Morphology; Nakanishi, T., Markwald, R.R., Baldwin, H.S., Keller, B.B., Srivastava, D., Yamagishi, H., Eds.; Springer: Tokyo, Japan, 2016. [Google Scholar]
- Hood, D.A.; Tryon, L.D.; Carter, H.N.; Kim, Y.; Chen, C.C. Unravelling the Mechanisms Regulating Muscle Mitochondrial Biogenesis. Biochem. J. 2016, 473, 2295–2314. [Google Scholar] [CrossRef]
- Horten, P.; Colina-Tenorio, L.; Rampelt, H. Biogenesis of Mitochondrial Metabolite Carriers. Biomolecules 2020, 10, 1008. [Google Scholar] [CrossRef]
- Islam, H.; Edgett, B.A.; Gurd, B.J. Coordination of mitochondrial biogenesis by PGC-1α in human skeletal muscle: A reevaluation. Metabolism 2018, 79, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Islam, H.; Hood, D.A.; Gurd, B.J. Looking beyond PGC-1α: Emerging regulators of exercise-induced skeletal muscle mitochondrial biogenesis and their activation by dietary compounds. Appl. Physiol. Nutr. Metab. 2020, 45, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Jamwal, S.; Blackburn, J.K.; Elsworth, J.D. PPARgamma/Pgc1alpha Signaling as a Potential Therapeutic Target for Mitochondrial Biogenesis in Neurodegenerative Disorders. Pharmacol. Ther. 2020, 107705. [Google Scholar] [CrossRef]
- Kandezi, N.; Mohammadi, M.; Ghaffari, M.; Gholami, M.; Motaghinejad, M.; Safari, S. Novel Insight to Neuroprotective Potential of Curcumin: A Mechanistic Review of Possible Involvement of Mitochondrial Biogenesis and PI3/Akt/ GSK3 or PI3/Akt/CREB/BDNF Signaling Pathways. Int. J. Mol. Cell. Med. 2020, 9, 1–32. [Google Scholar]
- Kiyama, T.; Chen, C.K.; Wang, S.W.; Pan, P.; Ju, Z.; Wang, J.; Takada, S.; Klein, W.H.; Mao, C.A. Essential Roles of Mitochondrial Biogenesis Regulator Nrf1 in Retinal Development and Homeostasis. Mol. Neurodegener. 2018, 13, 56. [Google Scholar] [CrossRef] [PubMed]
- Kondadi, A.K.; Anand, R.; Reichert, A.S. Functional Interplay between Cristae Biogenesis, Mitochondrial Dynamics and Mitochondrial DNA Integrity. Int. J. Mol. Sci. 2019, 20, 4311. [Google Scholar] [CrossRef]
- Krämer, L.; Groh, C.; Herrmann, J.M. The proteasome: Friend and foe of mitochondrial biogenesis. FEBS Lett. 2020. [Google Scholar] [CrossRef]
- Li, P.A.; Hou, X.; Hao, S. Mitochondrial biogenesis in neurodegeneration. J. Neurosci. Res. 2017, 95, 2025–2029. [Google Scholar] [CrossRef]
- Lill, R.; Freibert, S.-A. Mechanisms of Mitochondrial Iron-Sulfur Protein Biogenesis. Annu. Rev. Biochem. 2020, 89, 471–499. [Google Scholar] [CrossRef]
- Lima, T.I.; Araujo, H.N.; Menezes, E.S.; Sponton, C.H.; Araújo, M.B.; Bomfim, L.H.; Queiroz, A.L.; Passos, M.A.; DE Sousa, T.A.; Hirabara, S.M.; et al. Role of microRNAs on the Regulation of Mitochondrial Biogenesis and Insulin Signaling in Skeletal Muscle. J. Cell. Physiol. 2016, 232, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Widlund, H.R.; Puigserver, P. PGC-1 Coactivators: Shepherding the Mitochondrial Biogenesis of Tumors. Trends Cancer 2016, 2, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Manganas, P.; MacPherson, L.; Tokatlidis, K. Oxidative protein biogenesis and redox regulation in the mitochondrial intermembrane space. Cell Tissue Res. 2017, 367, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Mansilla, N.; Racca, S.; Gras, D.E.; Gonzalez, D.H.; Welchen, E. The Complexity of Mitochondrial Complex IV: An Update of Cytochrome c Oxidase Biogenesis in Plants. Int. J. Mol. Sci. 2018, 19, 662. [Google Scholar] [CrossRef]
- McKie, G.L.; Wright, D.C. Biochemical Adaptations in White Adipose Tissue Following Aerobic Exercise: From Mitochondrial Biogenesis to Browning. Biochem. J. 2020, 477, 1061–1081. [Google Scholar] [CrossRef] [PubMed]
- Melber, A.; Winge, D.R. Steps toward Understanding Mitochondrial Fe/S Cluster Biogenesis. Methods Enzym. 2018, 599, 265–292. [Google Scholar] [CrossRef]
- Miao, L.; Shen, X.; Whiteman, M.; Xin, H.; Shen, Y.; Xin, X.; Moore, P.K.; Zhu, Y.-Z. Hydrogen Sulfide Mitigates Myocardial Infarction via Promotion of Mitochondrial Biogenesis-Dependent M2 Polarization of Macrophages. Antioxid. Redox Signal. 2016, 25, 268–281. [Google Scholar] [CrossRef]
- Moulin, C.; Caumont-Sarcos, A.; Ieva, R. Mitochondrial Presequence Import: Multiple Regulatory Knobs Fine-Tune Mitochondrial Biogenesis and Homeostasis. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 930–944. [Google Scholar] [CrossRef]
- Ndi, M.; Marin-Buera, L.; Salvatori, R.; Singh, A.P.; Ott, M. Biogenesis of the bc1 Complex of the Mitochondrial Respiratory Chain. J. Mol. Biol. 2018, 430, 3892–3905. [Google Scholar] [CrossRef]
- Nirwane, A.; Majumdar, A. Understanding Mitochondrial Biogenesis through Energy Sensing Pathways and Its Translation in Cardio-Metabolic Health. Arch. Physiol. Biochem. 2018, 124, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Nowinski, S.M.; Van Vranken, J.G.; Dove, K.K.; Rutter, J. Impact of Mitochondrial Fatty Acid Synthesis on Mitochondrial Biogenesis. Curr. Biol. 2018, 28, R1212–R1219. [Google Scholar] [CrossRef] [PubMed]
- Ogunbona, O.B.; Claypool, S.M. Emerging Roles in the Biogenesis of Cytochrome C Oxidase for Members of the Mitochondrial Carrier Family. Front. Cell. Dev. Biol. 2019, 7, 3. [Google Scholar] [CrossRef]
- Ortega, S.P.; Chouchani, E.T.; Boudina, S. Stress turns on the heat: Regulation of mitochondrial biogenesis and UCP1 by ROS in adipocytes. Adipocyte 2016, 6, 56–61. [Google Scholar] [CrossRef]
- Panajatovic, M.; Singh, F.; Duthaler, U.; Krahenbuhl, S.; Bouitbir, J. Role of Pgc-1-Alpha-Associated Mitochondrial Biogenesis in Statin-Induced Myotoxicity. Eur. Cardiol. 2020, 15, e35. [Google Scholar] [CrossRef] [PubMed]
- Perry, C.G.; Hawley, J.A. Molecular Basis of Exercise-Induced Skeletal Muscle Mitochondrial Biogenesis: Historical Advances, Current Knowledge, and Future Challenges. Cold Spring Harb. Perspect. Med. 2017, 8, a029686. [Google Scholar] [CrossRef]
- Pfanner, N.; Warscheid, B.; Wiedemann, N. Mitochondrial proteins: From biogenesis to functional networks. Nat. Rev. Mol. Cell Biol. 2019, 20, 267–284. [Google Scholar] [CrossRef]
- Picca, A.; Lezza, A.M. Regulation of Mitochondrial Biogenesis through Tfam-Mitochondrial DNA Interactions: Useful Insights from Aging and Calorie Restriction Studies. Mitochondrion 2015, 25, 67–75. [Google Scholar] [CrossRef]
- Ploumi, C.; Daskalaki, I.; Tavernarakis, N. Mitochondrial biogenesis and clearance: A balancing act. FEBS J. 2016, 284, 183–195. [Google Scholar] [CrossRef]
- Popov, L. Mitochondrial biogenesis: An update. J. Cell. Mol. Med. 2020, 24, 4892–4899. [Google Scholar] [CrossRef] [PubMed]
- Praharaj, P.P.; Panigrahi, D.P.; Bhol, C.S.; Patra, S.; Mishra, S.R.; Mahapatra, K.K.; Behera, B.P.; Singh, A.; Patil, S.; Bhutia, S.K. Mitochondrial rewiring through mitophagy and mitochondrial biogenesis in cancer stem cells: A potential target for anti-CSC cancer therapy. Cancer Lett. 2021, 498, 217–228. [Google Scholar] [CrossRef]
- Rainey, N.E.; Moustapha, A.; Petit, P.X. Curcumin, a Multifaceted Hormetic Agent, Mediates an Intricate Crosstalk between Mitochondrial Turnover, Autophagy, and Apoptosis. Oxidative Med. Cell. Longev. 2020, 2020, 1–23. [Google Scholar] [CrossRef]
- Scholpa, N.E.; Schnellmann, R.G. Mitochondrial-Based Therapeutics for the Treatment of Spinal Cord Injury: Mitochondrial Biogenesis as a Potential Pharmacological Target. J. Pharmacol. Exp. Ther. 2017, 363, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Sheremet, N.L.; Andreeva, N.A.; Shmel’kova, M.S.; Tsigankova, P.G. Mitochondrial biogenesis in hereditary optic neuropathies. Вестник Офтальмологии 2019, 135, 85–91. [Google Scholar] [CrossRef]
- Simmons, E.C.; Scholpa, N.E.; Schnellmann, R.G. Mitochondrial biogenesis as a therapeutic target for traumatic and neurodegenerative CNS diseases. Exp. Neurol. 2020, 329, 113309. [Google Scholar] [CrossRef]
- Skuratovskaia, D.; Komar, A.; Vulf, M.; Litvinova, L. Mitochondrial destiny in type 2 diabetes: The effects of oxidative stress on the dynamics and biogenesis of mitochondria. PeerJ 2020, 8, e9741. [Google Scholar] [CrossRef] [PubMed]
- Smiles, W.J.; Camera, D.M. The Guardian of the Genome P53 Regulates Exercise-Induced Mitochondrial Plasticity Beyond Organelle Biogenesis. Acta Physiol. 2018, 222, e13004. [Google Scholar] [CrossRef]
- Tamura, Y.; Endo, T. Role of Intra- and Inter-mitochondrial Membrane Contact Sites in Yeast Phospholipid Biogenesis. Single Mol. Single Cell Seq. 2017, 997, 121–133. [Google Scholar] [CrossRef]
- Tang, J.X.; Thompson, K.; Taylor, R.W.; Olahova, M. Mitochondrial Oxphos Biogenesis: Co-Regulation of Protein Synthesis, Import, and Assembly Pathways. Int. J. Mol. Sci. 2020, 21, 3820. [Google Scholar] [CrossRef] [PubMed]
- Thornton, C.; Jones, A.; Nair, S.; Aabdien, A.; Mallard, C.; Hagberg, H. Mitochondrial dynamics, mitophagy and biogenesis in neonatal hypoxic-ischaemic brain injury. FEBS Lett. 2017, 592, 812–830. [Google Scholar] [CrossRef]
- Timón-Gómez, A.; Nývltová, E.; Abriata, L.A.; Vila, A.J.; Hosler, J.; Barrientos, A. Mitochondrial cytochrome c oxidase biogenesis: Recent developments. Semin. Cell Dev. Biol. 2018, 76, 163–178. [Google Scholar] [CrossRef] [PubMed]
- Tong, M.; Sadoshima, J. Mitochondrial autophagy in cardiomyopathy. Curr. Opin. Genet. Dev. 2016, 38, 8–15. [Google Scholar] [CrossRef]
- Van Haute, L.; Powell, C.A.; Minczuk, M. Dealing with an Unconventional Genetic Code in Mitochondria: The Biogenesis and Pathogenic Defects of the 5-Formylcytosine Modification in Mitochondrial tRNAMet. Biomolecules 2017, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- VanderVeen, B.N.; Fix, D.K.; Carson, J.A. Disrupted Skeletal Muscle Mitochondrial Dynamics, Mitophagy, and Biogenesis during Cancer Cachexia: A Role for Inflammation. Oxid. Med. Cell. Longev. 2017, 2017, 3292087. [Google Scholar] [CrossRef] [PubMed]
- Vega, R.B.; Horton, J.L.; Kelly, D.P. Maintaining Ancient Organelles: Mitochondrial Biogenesis and Maturation. Circ. Res. 2015, 116, 1820–1834. [Google Scholar] [CrossRef]
- Paz, M.V.; Cotán, D.; Garrido-Maraver, J.; Cordero, M.D.; Oropesa-Ávila, M.; De La Mata, M.; Pavón, A.D.; De Lavera, I.; Alcocer-Gómez, E.; Sánchez-Alcázar, J.A. Targeting autophagy and mitophagy for mitochondrial diseases treatment. Expert Opin. Ther. Targets 2015, 20, 487–500. [Google Scholar] [CrossRef]
- Wang, J.; Wen, X.; Liu, J.; Sun, H. Mitochondrial biogenesis Inhibitors for Anticancer Therapy: A Review of Recent Patents. Recent Pat. Anti-Cancer Drug Discov. 2016, 11, 332–341. [Google Scholar] [CrossRef]
- Whitaker, R.M.; Corum, D.; Beeson, C.C.; Schnellmann, R.G. Mitochondrial Biogenesis as a Pharmacological Target: A New Approach to Acute and Chronic Diseases. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 229–249. [Google Scholar] [CrossRef]
- Wideman, J.G.; Muñoz-Gómez, S.A. The evolution of ERMIONE in mitochondrial biogenesis and lipid homeostasis: An evolutionary view from comparative cell biology. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2016, 1861, 900–912. [Google Scholar] [CrossRef]
- Williams, J.A.; Zhao, K.; Jin, S.; Ding, W.-X. New methods for monitoring mitochondrial biogenesis and mitophagy in vitro and in vivo. Exp. Biol. Med. 2017, 242, 781–787. [Google Scholar] [CrossRef]
- Dos Santos, T.W.; Pereira, Q.C.; Teixeira, L.; Gambero, A.; Villena, J.A.; Ribeiro, M.L. Effects of Polyphenols on Thermogenesis and Mitochondrial Biogenesis. Int. J. Mol. Sci. 2018, 19, 2757. [Google Scholar] [CrossRef]
- Yambire, K.F.; Fernandez-Mosquera, L.; Steinfeld, R.; Mühle, C.; Ikonen, E.; Milosevic, I.; Raimundo, N. Mitochondrial biogenesis is transcriptionally repressed in lysosomal lipid storage diseases. eLife 2019, 8. [Google Scholar] [CrossRef]
- Yuan, Y.; Cruzat, V.F.; Newsholme, P.; Cheng, J.; Chen, Y.; Lu, Y. Regulation of SIRT1 in aging: Roles in mitochondrial function and biogenesis. Mech. Ageing Dev. 2016, 155, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Zamora, M.; Pardo, R.; Villena, J.A. Pharmacological induction of mitochondrial biogenesis as a therapeutic strategy for the treatment of type 2 diabetes. Biochem. Pharmacol. 2015, 98, 16–28. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, H. Translational regulation of mitochondrial biogenesis. Biochem. Soc. Trans. 2016, 44, 1717–1724. [Google Scholar] [CrossRef] [PubMed]
- Andhavarapu, S.; Katuri, A.; Bryant, J.; Patel, V.; Gupta, U.; Asemu, G.; Makar, T.K. Intersecting roles of ER stress, mitochondrial dysfunction, autophagy, and calcium homeostasis in HIV-associated neurocognitive disorder. J. NeuroVirol. 2020, 26, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Baquero, P.; Dawson, A.; Helgason, G.V. Autophagy and mitochondrial metabolism: Insights into their role and therapeutic potential in chronic myeloid leukaemia. FEBS J. 2019, 286, 1271–1283. [Google Scholar] [CrossRef]
- Chen, S.Y.; Gao, Y.; Sun, J.Y.; Meng, X.L.; Yang, D.; Fan, L.H.; Xiang, L.; Wang, P. Traditional Chinese Medicine: Role in Reducing Beta-Amyloid, Apoptosis, Autophagy, Neuroinflammation, Oxidative Stress, and Mitochondrial Dysfunction of Alzheimer’s Disease. Front. Pharmacol. 2020, 11, 497. [Google Scholar] [CrossRef]
- Cheng, J.; Wei, L.; Li, M. Progress in Regulation of Mitochondrial Dynamics and Mitochondrial Autophagy. Sheng Li Xue Bao 2020, 72, 475–487. [Google Scholar]
- Furukawa, K.; Innokentev, A.; Kanki, T. Regulatory Mechanisms of Mitochondrial Autophagy: Lessons from Yeast. Front. Plant. Sci. 2019, 10, 1479. [Google Scholar] [CrossRef]
- Gafar, A.A.; Draz, H.M.; Goldberg, A.A.; Bashandy, M.A.; Bakry, S.; Khalifa, M.A.; AbuShair, W.; Titorenko, V.I.; Sanderson, J.T. Lithocholic acid induces endoplasmic reticulum stress, autophagy and mitochondrial dysfunction in human prostate cancer cells. PeerJ 2016, 4, e2445. [Google Scholar] [CrossRef] [PubMed]
- Go, K.L.; Lee, S.; Zendejas, I.; Behrns, K.E.; Kim, J.S. Mitochondrial Dysfunction and Autophagy in Hepatic Ischemia/Reperfusion Injury. Biomed. Res. Int. 2015, 2015, 183469. [Google Scholar] [CrossRef]
- Hamacher-Brady, A.; Brady, N.R. Mitophagy programs: Mechanisms and physiological implications of mitochondrial targeting by autophagy. Cell. Mol. Life Sci. 2016, 73, 775–795. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.; Shi, Y. Regulation of autophagy by mitochondrial phospholipids in health and diseases. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2017, 1862, 114–129. [Google Scholar] [CrossRef]
- Huang, M.L.-H.; Chiang, S.; Kalinowski, D.S.; Bae, D.-H.; Sahni, S.; Richardson, D.R. The Role of the Antioxidant Response in Mitochondrial Dysfunction in Degenerative Diseases: Cross-Talk between Antioxidant Defense, Autophagy, and Apoptosis. Oxidative Med. Cell. Longev. 2019, 2019, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Katuri, A.; Bryant, J.L.; Patel, D.; Patel, V.; Andhavarapu, S.; Asemu, G.; Davis, H.; Makar, T.K. HIVAN associated tubular pathology with reference to ER stress, mitochondrial changes, and autophagy. Exp. Mol. Pathol. 2019, 106, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Kubli, D.A.; Gustafsson, Å.B. Unbreak My Heart: Targeting Mitochondrial Autophagy in Diabetic Cardiomyopathy. Antioxid. Redox Signal. 2015, 22, 1527–1544. [Google Scholar] [CrossRef]
- Madhavi, Y.V.; Gaikwad, N.; Yerra, V.G.; Kalvala, A.K.; Nanduri, S.; Kumar, A. Targeting AMPK in Diabetes and Diabetic Complications: Energy Homeostasis, Autophagy and Mitochondrial Health. Curr. Med. Chem. 2019, 26, 5207–5229. [Google Scholar] [CrossRef]
- Miyamoto, S. Hexokinase 2 mediated cellular protection: Interaction with Akt/mTORC1 to regulate mitochondrial protection and autophagy. Seikagaku. J. Jpn. Biochem. Soc. 2017, 89, 199–210. [Google Scholar]
- Ney, P.A. Mitochondrial Autophagy: Origins, Significance, and Role of Bnip3 and Nix. Biochim. Biophys. Acta 2015, 1853, 2775–2783. [Google Scholar] [CrossRef]
- Roca-Agujetas, V.; De Dios, C.; Lestón, L.; Marí, M.; Morales, A.; Colell, A. Recent Insights into the Mitochondrial Role in Autophagy and Its Regulation by Oxidative Stress. Oxidative Med. Cell. Longev. 2019, 2019, 1–16. [Google Scholar] [CrossRef]
- Saito, T.; Sadoshima, J. Molecular Mechanisms of Mitochondrial Autophagy/Mitophagy in the Heart. Circ. Res. 2015, 116, 1477–1490. [Google Scholar] [CrossRef]
- Shah, S.Z.A.; Zhao, D.; Hussain, T.; Sabir, N.; Mangi, M.H.; Yang, L. p62-Keap1-NRF2-ARE Pathway: A Contentious Player for Selective Targeting of Autophagy, Oxidative Stress and Mitochondrial Dysfunction in Prion Diseases. Front. Mol. Neurosci. 2018, 11, 310. [Google Scholar] [CrossRef]
- Sifuentes-Franco, S.; Pacheco-Moisés, F.P.; Rodríguez-Carrizalez, A.D.; Miranda-Díaz, A.G. The Role of Oxidative Stress, Mitochondrial Function, and Autophagy in Diabetic Polyneuropathy. J. Diabetes Res. 2017, 2017, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.A.; Yen, P.M. Thyroid hormone-mediated autophagy and mitochondrial turnover in NAFLD. Cell Biosci. 2016, 6, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Tagaya, M.; Arasaki, K. Regulation of Mitochondrial Dynamics and Autophagy by the Mitochondria-Associated Membrane. Adv. Exp. Med. Biol. 2017, 997, 33–47. [Google Scholar] [PubMed]
- Tricarico, P.M.; Crovella, S.; Celsi, F. Mevalonate Pathway Blockade, Mitochondrial Dysfunction and Autophagy: A Possible Link. Int. J. Mol. Sci. 2015, 16, 16067–16084. [Google Scholar] [CrossRef]
- Van Houten, B.; Hunter, S.E.; Meyer, J.N. Mitochondrial DNA Damage Induced Autophagy, Cell Death, and Disease. Front. Biosci. (Landmark Ed.) 2016, 21, 42–54. [Google Scholar] [CrossRef]
- Wang, B.; Abraham, N.; Gao, G.; Yang, Q. Dysregulation of Autophagy and Mitochondrial Function in Parkinson’s Disease. Transl. Neurodegener. 2016, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Wang, G. Autophagy in Mitochondrial Quality Control. In Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Swtizerland, 2019; Volume 1206, pp. 421–434. [Google Scholar]
- Yamano, K.; Matsuda, N.; Tanaka, K. The Ubiquitin Signal and Autophagy: An Orchestrated Dance Leading to Mitochondrial Degradation. EMBO Rep. 2016, 17, 300–316. [Google Scholar] [CrossRef]
- Yamashita, S.I.; Kanki, T. How Autophagy Eats Large Mitochondria: Autophagosome Formation Coupled with Mitochondrial Fragmentation. Autophagy 2017, 13, 980–981. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Gao, G.; Mao, Z.; Yang, Q. Chaperone-Mediated Autophagy and Mitochondrial Homeostasis in Parkinson’s Disease. Park. Dis. 2016, 2016, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Culp, M.L.; Craver, J.G.; Darley-Usmar, V. Mitochondrial Function and Autophagy: Integrating Proteotoxic, Redox, and Metabolic Stress in Parkinson’s Disease. J. Neurochem. 2018, 144, 691–709. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zheng, Y.; Chen, Z. Autophagy and Mitochondrial Encephalomyopathies. In Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2020; pp. 103–110. [Google Scholar]
- Kopinski, P.K.; Janssen, K.A.; Schaefer, P.M.; Trefely, S.; Perry, C.E.; Potluri, P.; Tintos-Hernandez, J.A.; Singh, L.N.; Karch, K.R.; Campbell, S.L.; et al. Regulation of nuclear epigenome by mitochondrial DNA heteroplasmy. Proc. Natl. Acad. Sci. USA 2019, 116, 16028–16035. [Google Scholar] [CrossRef]
- Matilainen, O.; Quirós, P.M.; Auwerx, J. Mitochondria and Epigenetics—Crosstalk in Homeostasis and Stress. Trends Cell Biol. 2017, 27, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Schell, J.C.; Rutter, J. Mitochondria link metabolism and epigenetics in haematopoiesis. Nat. Cell Biol. 2017, 19, 589–591. [Google Scholar] [CrossRef]
- Sharma, N.; Pasala, M.S.; Prakash, A. Mitochondrial DNA: Epigenetics and environment. Environ. Mol. Mutagen. 2019, 60, 668–682. [Google Scholar] [CrossRef]
- Danzeisen, R.; Schwalenstoecker, B.; Gillardon, F.; Buerger, E.; Krzykalla, V.; Klinder, K.; Schild, L.; Hengerer, B.; Ludolph, A.C.; Dorner-Ciossek, C.; et al. Targeted Antioxidative and Neuroprotective Properties of the Dopamine Agonist Pramipexole and Its Nondopaminergic Enantiomer SND919CL2x [(+)2-Amino-4,5,6,7-tetrahydro-6-lpropylamino-benzathiazole Dihydrochloride]. J. Pharmacol. Exp. Ther. 2005, 316, 189–199. [Google Scholar] [CrossRef]
- Ferrari-Toninelli, G.; Maccarinelli, G.; Uberti, D.L.; Buerger, E.; Memo, M. Mitochondria-targeted antioxidant effects of S(-) and R(+) pramipexole. BMC Pharmacol. 2010, 10, 2. [Google Scholar] [CrossRef]
- Akhmadishina, R.A.; Garifullin, R.; Petrova, N.V.; Kamalov, M.I.; Abdullin, T.I. Triphenylphosphonium Moiety Modulates Proteolytic Stability and Potentiates Neuroprotective Activity of Antioxidant Tetrapeptides in Vitro. Front. Pharmacol. 2018, 9, 115. [Google Scholar] [CrossRef]
- Battogtokh, G.; Choi, Y.S.; Kang, D.S.; Park, S.J.; Shim, M.S.; Huh, K.M.; Cho, Y.-Y.; Lee, J.Y.; Lee, H.S.; Kang, H.C. Mitochondria-targeting drug conjugates for cytotoxic, anti-oxidizing and sensing purposes: Current strategies and future perspectives. Acta Pharm. Sin. B 2018, 8, 862–880. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Liaw, K.; Sharma, R.; Zhang, Z.; Kannan, S.; Kannan, R.M. Targeting Mitochondrial Dysfunction and Oxidative Stress in Activated Microglia Using Dendrimer-Based Therapeutics. Theranostics 2018, 8, 5529–5547. [Google Scholar] [CrossRef]
- Titova, E.; Shagieva, G.; Ivanova, O.; Domnina, L.; Domninskaya, M.; Strelkova, O.; Khromova, N.; Kopnin, P.; Chernyak, B.; Skulachev, V.; et al. Mitochondria-targeted antioxidant SkQ1 suppresses fibrosarcoma and rhabdomyosarcoma tumour cell growth. Cell Cycle 2018, 17, 1797–1811. [Google Scholar] [CrossRef]
- Apostolova, N.; Victor, V.M. Molecular Strategies for Targeting Antioxidants to Mitochondria: Therapeutic Implications. Antioxid. Redox Signal. 2015, 22, 686–729. [Google Scholar] [CrossRef]
- Fetisova, E.K.; Antoschina, M.M.; Cherepanynets, V.D.; Izumov, D.S.; Kireev, I.I.; Kireev, R.I.; Lyamzaev, K.G.; Riabchenko, N.I.; Chernyak, B.V.; Skulachev, V.P. Radioprotective Effects of Mitochondria-Targeted Antioxidant SkQR. Radiat. Res. 2015, 183, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Genrikhs, E.E.; Stelmashook, E.V.; Popova, O.V.; Kapay, N.A.; Korshunova, G.A.; Sumbatyan, N.V.; Skrebitsky, V.G.; Skulachev, V.P.; Isaev, N.K. Mitochondria-Targeted Antioxidant Skqt1 Decreases Trauma-Induced Neurological Deficit in Rat and Prevents Amyloid-Beta-Induced Impairment of Long-Term Potentiation in Rat Hippocampal Slices. J. Drug Target. 2015, 23, 347–352. [Google Scholar] [CrossRef]
- Silachev, D.N.; Plotnikov, E.Y.; Zorova, L.D.; Pevzner, I.B.; Sumbatyan, N.V.; Korshunova, G.A.; Gulyaev, M.V.; Pirogov, Y.A.; Skulachev, V.P.; Zorov, D.B. Neuroprotective Effects of Mitochondria-Targeted Plastoquinone and Thymoquinone in a Rat Model of Brain Ischemia/Reperfusion Injury. Molecules 2015, 20, 14487–14503. [Google Scholar] [CrossRef]
- Margulis, L. Symbiotic theory of the origin of eukaryotic organelles; criteria for proof. Symp. Soc. Exp. Biol. 1975, 29, 21–38. [Google Scholar]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bennett, J.P., Jr.; Onyango, I.G. Energy, Entropy and Quantum Tunneling of Protons and Electrons in Brain Mitochondria: Relation to Mitochondrial Impairment in Aging-Related Human Brain Diseases and Therapeutic Measures. Biomedicines 2021, 9, 225. https://doi.org/10.3390/biomedicines9020225
Bennett JP Jr., Onyango IG. Energy, Entropy and Quantum Tunneling of Protons and Electrons in Brain Mitochondria: Relation to Mitochondrial Impairment in Aging-Related Human Brain Diseases and Therapeutic Measures. Biomedicines. 2021; 9(2):225. https://doi.org/10.3390/biomedicines9020225
Chicago/Turabian StyleBennett, James P., Jr., and Isaac G. Onyango. 2021. "Energy, Entropy and Quantum Tunneling of Protons and Electrons in Brain Mitochondria: Relation to Mitochondrial Impairment in Aging-Related Human Brain Diseases and Therapeutic Measures" Biomedicines 9, no. 2: 225. https://doi.org/10.3390/biomedicines9020225
APA StyleBennett, J. P., Jr., & Onyango, I. G. (2021). Energy, Entropy and Quantum Tunneling of Protons and Electrons in Brain Mitochondria: Relation to Mitochondrial Impairment in Aging-Related Human Brain Diseases and Therapeutic Measures. Biomedicines, 9(2), 225. https://doi.org/10.3390/biomedicines9020225