
Critical Molecular and Cellular Contributors to Tau Pathology



Abstract
1. Introduction
2. Factors Involved in the Formation of Tau Seeds
2.1. Site-Specific Phosphorylation-Mediated Tau Self-Aggregation
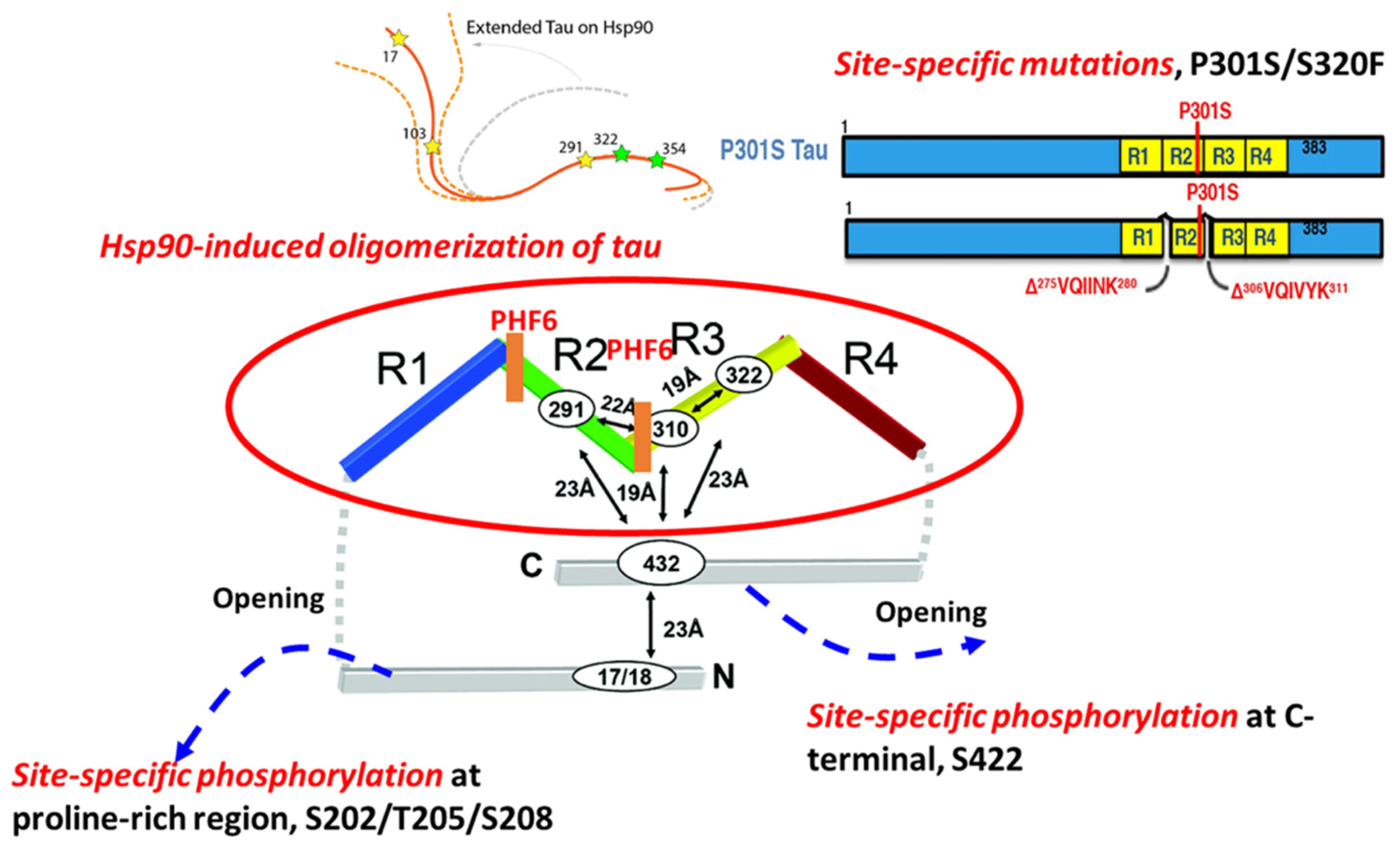
2.2. Hsp90-Mediated Tau Aggregation
2.3. Site-Specific Mutations and Tau Aggregation
3. Molecular Mechanisms of Tau Cellular Uptake
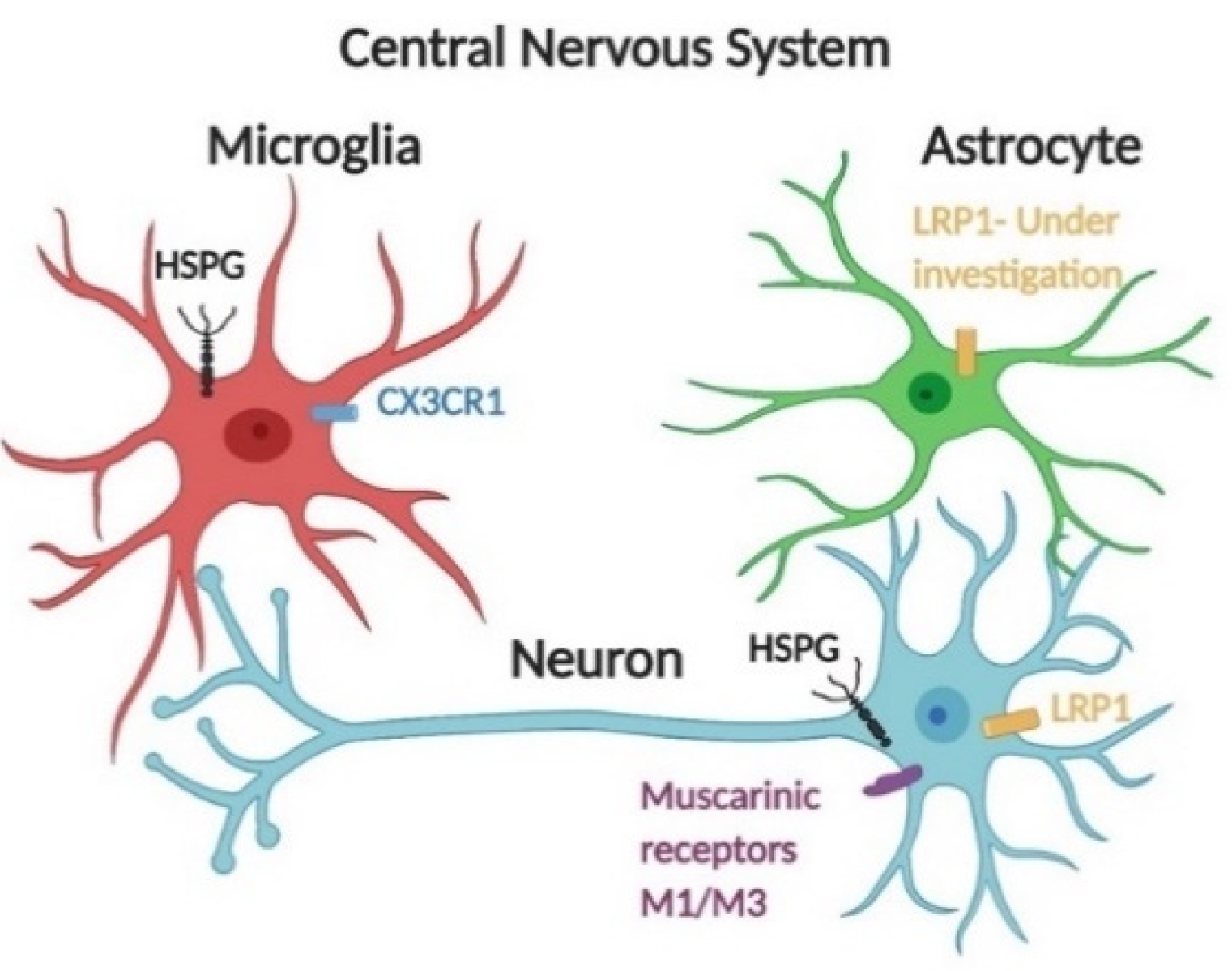
3.1. The Effects of HSPGs on the Cellular Uptake of Tau Seeds

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Forms of Tau | Animal/Cell Model | Conformation/Characteristics | ‘Prion-Like’ Transmission | Reference |
|---|---|---|---|---|
| 2N4R tau monomer | Mouse primary hippocampal and cortical neurons Non-neuronal cells including HEK293, Hela, MC17, and MLECs | Wild-type tau-paperclip like | Internalized by non-neuronal cells, MLECs 3-O-sulfation is required for HSPG-mediated tau monomer internalization | [41,42] |
| 2N4R tau oligomers | Spherical structure Diameters ranging from 10 nm to 30 nm | Dynamin-dependent endocytosis Bulk endocytosis Tau species in endosome transported in neurons anterogradely and retrogradely | ||
| Heparin-induced tau short fibrils (2N4R) | Helical twist Varied length, from 40 nm to 250 nm | |||
| Tau filaments purified from rTg4510 mouse brain | Primary neurons Hela cells P301L tau expressing mice | Fibrillary structure | Internalized by neuronal cells, but not non-neuronal cells 1-week post-injection, NFTs were detected by immunostaining against MC1at the tau aggregates injection site | |
| Heparin-induced tau long fibrils (2N4R) | Primary neurons Hela cells | Helical twist Varied length, from 200 nm to 1600 nm | No internalization detected in both neuronal cells and non-neuronal cells | |
| 2N4R tau monomer | Human neuroglioma, H4 cells Human SH-SY5Y hiPSC-derived neurons | Size averaged at 5.7 nm | Internalized by neuronal cells rapidly HSPG-dependent internalization Tau monomer interacts with 6-O-sulfation on the HSPGs, but not N-sulfation | [36] |
| Heparin-induced oligomer (2N4R) | Helical filaments/19 nm | |||
| Short fibrils (2N4R) | Helical fragments/33nm | |||
| Heparin-induced fibrils (2N4R) | Helical filaments/80 nm | Less internalization detected in both cell lines | ||
| Tau RD monomers | Mouse NPCs, C17.2 Primary neurons | Wild-type tau repeat domain | No internalization detected | [8,40] |
| Heparin-induced tau fibrils (both RD and WT 2N4R) | Fibrillary structure | Tau fibrils require HSPGs for neurons 6-O- and N-sulfation are required for tau aggregates internalization HSPG mediated intracellular tau seeding Actin-dependent macropinocytosis Clathrin- or dynamin-independent endocytosis | ||
| Tau P301S monomer | hiPSCs-derived cortical neurons | Seeding-prone form | Actin-dependent macropinocytosis Dynamin-dependent endocytosis | [35] |
| Heparin-induced tau fibrils (Tau P301S) | Fibrillary structure | Dynamin-dependent endocytosis Actin-independent endocytosis | ||
| Tau oligomers | Mouse primary neuronal cells HEK293 CHO cells | Spherical structure | Muscarinic receptor-mediated endocytosis of tau by neuronal and HEK293 cells, but not by CHO cells Muscarinic receptor highly expressed by neuronal cells | [43] |
| Soluble tau oligomers derived from AD patient | Mouse primary neuronal cells | Spherical structure | Muscarinic receptor-mediated endocytosis of tau | |
| 2N4R tau monomer, oligomers, and fibrils | H4 neuroglioma hiPSCs-derived neuronal cells | Spherical structure Filaments | Receptor-mediated tau endocytosis Knockdown of LRP1 blocks soluble tau uptake including tau monomers/oligomers, but only reduce tau fibrils uptake | [36] |
3.2. Receptor-Mediated Endocytosis of Tau
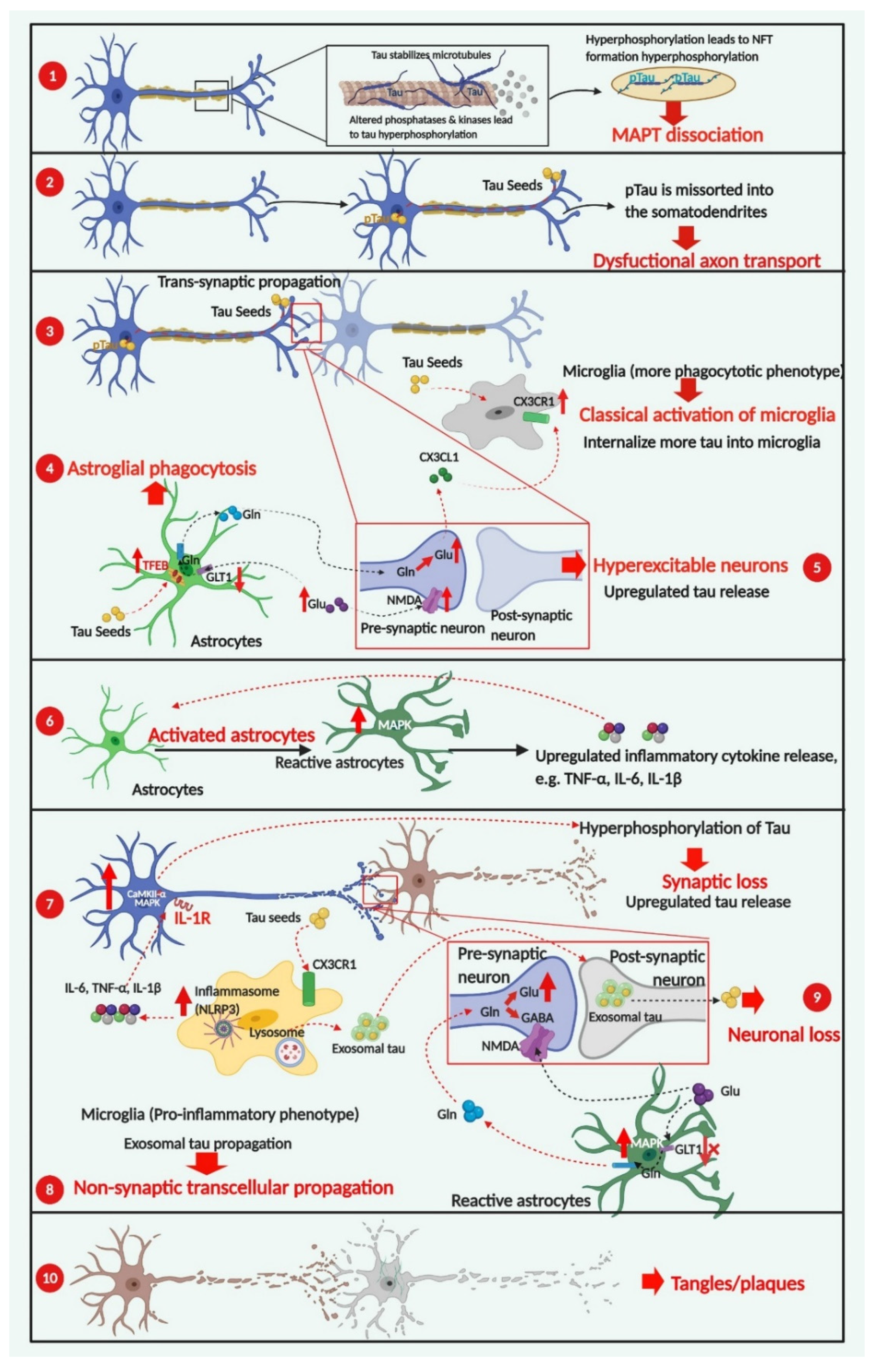
4. Cellular Contributors to Tau Pathology
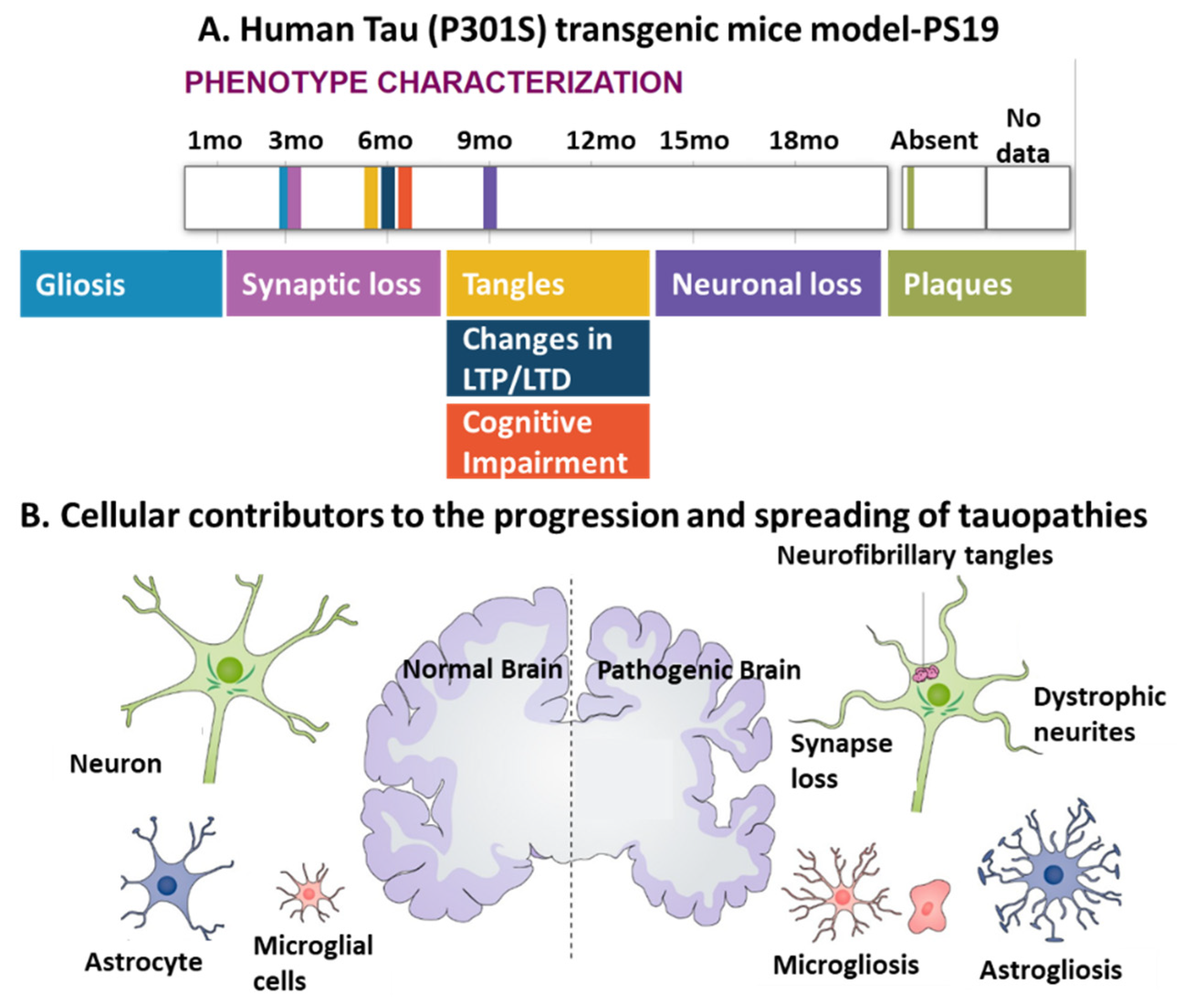
4.1. The Involvement of Neuronal Activity in the Spreading of Pathogenic Tau
4.2. Glial Cells Are Involved in the Progression of Tau Pathology
4.2.1. Microgliosis in Tauopathies
Neuroprotective Effects of CX3CL1/CX3CR1 Signaling
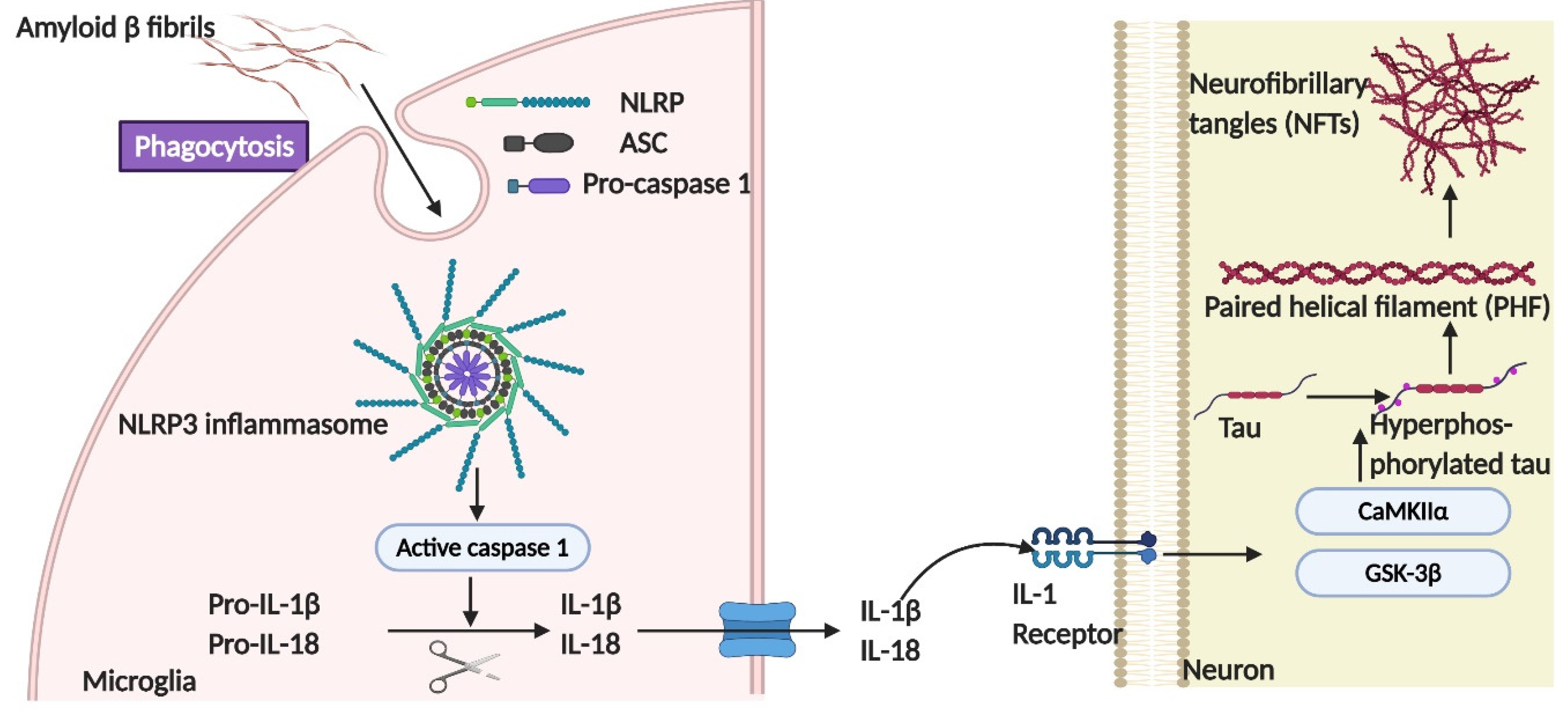
The Role of NLRP3-ASC Inflammasome Activation in Tau Phosphorylation
Non-Transsynaptic Tau Propagation-Microglial and Exosomal Spreading of Tau Species as an Alternative Pathway
4.2.2. Astrogliosis in Tauopathies
Reactive Astrocyte Phagocytosis Has a Neuroprotective Effect
Neuroinflammatory Microenvironment Induced by Reactive Astrocytes
Dysfunctional Neuronal-Astroglial Communication
4.3. ApoE4 Plays a Cell-Type Dependent Role in Tau Pathology
5. Therapeutic Approaches Targeting Molecular/Cellular Signaling Pathways
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Hock, E.M.; Polymenidou, M. Prion-like propagation as a pathogenic principle in frontotemporal dementia. J. Neurochem. 2016, 138 (Suppl. 1), 163–183. [Google Scholar] [CrossRef]
- Mirbaha, H.; Chen, D.; Morazova, O.A.; Ruff, K.M.; Sharma, A.M.; Liu, X.; Goodarzi, M.; Pappu, R.V.; Colby, D.W.; Mirzaei, H.; et al. Inert and seed-competent tau monomers suggest structural origins of aggregation. eLife 2018. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Li, B.; Tung, E.J.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Site-specific effects of tau phosphorylation on its microtubule assembly activity and self-aggregation. Eur. J. Neurosci. 2007, 26, 3429–3436. [Google Scholar] [CrossRef]
- Wang, Y.; Balaji, V.; Kaniyappan, S.; Kruger, L.; Irsen, S.; Tepper, K.; Chandupatla, R.; Maetzler, W.; Schneider, A.; Mandelkow, E.; et al. The release and trans-synaptic transmission of Tau via exosomes. Mol. Neurodegenerat. 2017, 12, 5. [Google Scholar] [CrossRef] [PubMed]
- Pooler, A.M.; Phillips, E.C.; Lau, D.H.W.; Noble, W.; Hanger, D.P. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 2013, 14, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Dal Pra, I.; Chiarini, A.; Gui, L.; Chakravarthy, B.; Pacchiana, R.; Gardenal, E.; Whitfield, J.F.; Armato, U. Do astrocytes collaborate with neurons in spreading the “infectious” abeta and Tau drivers of Alzheimer’s disease? Neuroscientist 2015, 21, 9–29. [Google Scholar] [CrossRef]
- Yamada, K.; Cirrito, J.R.; Stewart, F.R.; Jiang, H.; Finn, M.B.; Holmes, B.B.; Binder, L.I.; Mandelkow, E.M.; Diamond, M.I.; Lee, V.M.Y.; et al. In Vivo Microdialysis Reveals Age-Dependent Decrease of Brain Interstitial Fluid Tau Levels in P301S Human Tau Transgenic Mice. J. Neurosci. 2011, 31, 13110–13117. [Google Scholar] [CrossRef] [PubMed]
- Holmes, B.B.; DeVos, S.L.; Kfoury, N.; Li, M.; Jacks, R.; Yanamandra, K.; Ouidja, M.O.; Brodsky, F.M.; Marasa, J.; Bagchi, D.P.; et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. USA 2013, 110, E3138–E3147. [Google Scholar] [CrossRef] [PubMed]
- Mudher, A.; Colin, M.; Dujardin, S.; Medina, M.; Dewachter, I.; Alavi Naini, S.M.; Mandelkow, E.M.; Mandelkow, E.; Buee, L.; Goedert, M.; et al. What is the evidence that tau pathology spreads through prion-like propagation? Acta Neuropathol. Commun. 2017, 5, 99. [Google Scholar] [CrossRef]
- Ozcelik, S.; Sprenger, F.; Skachokova, Z.; Fraser, G.; Abramowski, D.; Clavaguera, F.; Probst, A.; Frank, S.; Muller, M.; Staufenbiel, M.; et al. Co-expression of truncated and full-length tau induces severe neurotoxicity. Mol. Psychiatry 2016, 21, 1790–1798. [Google Scholar] [CrossRef] [PubMed]
- Leyns, C.E.G.; Holtzman, D.M. Glial contributions to neurodegeneration in tauopathies. Mol. Neurodegenerat. 2017, 12, 50. [Google Scholar] [CrossRef]
- Kahlson, M.A.; Colodner, K.J. Glial Tau Pathology in Tauopathies: Functional Consequences. J. Exp. Neurosci. 2015, 9, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Parachikova, A.; Agadjanyan, M.G.; Cribbs, D.H.; Blurton-Jones, M.; Perreau, V.; Rogers, J.; Beach, T.G.; Cotman, C.W. Inflammatory changes parallel the early stages of Alzheimer disease. Neurobiol. Aging 2007, 28, 1821–1833. [Google Scholar] [CrossRef] [PubMed]
- Chun, H.; Marriott, I.; Lee, C.J.; Cho, H. Elucidating the Interactive Roles of Glia in Alzheimer’s Disease Using Established and Newly Developed Experimental Models. Front. Neurol. 2018, 9, 797. [Google Scholar] [CrossRef]
- Cagnin, A.; Brooks, D.J.; Kennedy, A.M.; Gunn, R.N.; Myers, R.; Turkheimer, F.E.; Jones, T.; Banati, R.B. In-vivo measurement of activated microglia in dementia. Lancet 2001, 358, 461–467. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Mirbaha, H.; Holmes, B.B.; Sanders, D.W.; Bieschke, J.; Diamond, M.I. Tau trimers are the minimal propagation unit spontaneously internalized to seed intracellular aggregation. J. Biol. Chem. 2015. [Google Scholar] [CrossRef]
- Jeganathan, S.; von Bergen, M.; Brutlach, H.; Steinhoff, H.J.; Mandelkow, E. Global hairpin folding of tau in solution. Biochemistry 2006, 45, 2283–2293. [Google Scholar] [CrossRef]
- Tenreiro, S.; Eckermann, K.; Outeiro, T.F. Protein phosphorylation in neurodegeneration: Friend or foe? Front. Mol. Neurosci. 2014, 7, 42. [Google Scholar] [CrossRef] [PubMed]
- Haase, C.; Stieler, J.; Arendt, T.; Holzer, M. Pseudophosphorylation of tau protein alters its ability for self-aggregation. J. Neurochem. 2004, 88, 1509–1520. [Google Scholar] [CrossRef] [PubMed]
- Šimić, G.; Babić Leko, M.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; De Silva, R.; Di Giovanni, G. Tau protein hyperphosphorylation and aggregation in Alzheimer’s disease and other tauopathies, and possible neuroprotective strategies. Biomolecules 2016, 6, 6. [Google Scholar] [CrossRef]
- Despres, C.; Byrne, C.; Qi, H.; Cantrelle, F.X.; Huvent, I.; Chambraud, B.; Baulieu, E.E.; Jacquot, Y.; Landrieu, I.; Lippens, G.; et al. Identification of the Tau phosphorylation pattern that drives its aggregation. Proc. Natl. Acad. Sci. USA 2017, 114, 9080–9085. [Google Scholar] [CrossRef] [PubMed]
- Ercan-Herbst, E.; Ehrig, J.; Schöndorf, D.C.; Behrendt, A.; Klaus, B.; Ramos, B.G.; Oriol, N.P.; Weber, C.; Ehrnhoefer, D.E. A post-translational modification signature defines changes in soluble tau correlating with oligomerization in early stage Alzheimer’s disease brain. Acta Neuropathol. Commun. 2019, 7, 1–19. [Google Scholar] [CrossRef]
- Glover, J.R.; Lindquist, S. Hsp104, Hsp70, and Hsp40: A novel chaperone system that rescues previously aggregated proteins. Cell 1998, 94, 73–82. [Google Scholar] [CrossRef]
- Kundel, F.; De, S.; Flagmeier, P.; Horrocks, M.H.; Kjaergaard, M.; Shammas, S.L.; Jackson, S.E.; Dobson, C.M.; Klenerman, D. Hsp70 inhibits the nucleation and elongation of tau and sequesters tau aggregates with high affinity. ACS Chem. Biol. 2018, 13, 636–646. [Google Scholar] [CrossRef]
- Patterson, K.R.; Ward, S.M.; Combs, B.; Voss, K.; Kanaan, N.M.; Morfini, G.; Brady, S.T.; Gamblin, T.C.; Binder, L.I. Heat shock protein 70 prevents both tau aggregation and the inhibitory effects of preexisting tau aggregates on fast axonal transport. Biochemistry 2011, 50, 10300–10310. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, S.; Zhang, L.; Lu, J.; Zhao, C.; Luo, F.; Li, D.; Li, X.; Liu, C. Heat shock protein 104 (HSP104) chaperones soluble Tau via a mechanism distinct from its disaggregase activity. J. Biol. Chem. 2019, 294, 4956–4965. [Google Scholar] [CrossRef] [PubMed]
- Tortosa, E.; Santa-Maria, I.; Moreno, F.; Lim, F.; Perez, M.; Avila, J. Binding of Hsp90 to tau promotes a conformational change and aggregation of tau protein. J. Alzheimers Dis. 2009, 17, 319–325. [Google Scholar] [CrossRef]
- Weickert, S.; Wawrzyniuk, M.; John, L.H.; Rudiger, S.G.D.; Drescher, M. The mechanism of Hsp90-induced oligomerizaton of Tau. Sci. Adv. 2020, 6, eaax6999. [Google Scholar] [CrossRef]
- Kaufman, S.K.; Sanders, D.W.; Thomas, T.L.; Ruchinskas, A.J.; Vaquer-Alicea, J.; Sharma, A.M.; Miller, T.M.; Diamond, M.I. Tau Prion Strains Dictate Patterns of Cell Pathology, Progression Rate, and Regional Vulnerability In Vivo. Neuron 2016, 92, 796–812. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.; Bastone, A.; Piccoli, E.; Mazzoleni, G.; Morbin, M.; Uggetti, A.; Giaccone, G.; Sperber, S.; Beeg, M.; Salmona, M.; et al. New mutations in MAPT gene causing frontotemporal lobar degeneration: Biochemical and structural characterization. Neurobiol. Aging 2012, 33, 834.e1–834.e6. [Google Scholar] [CrossRef] [PubMed]
- Stein, K.C.; True, H.L. Extensive diversity of prion strains is defined by differential chaperone interactions and distinct amyloidogenic regions. PLoS Genet. 2014, 10, e1004337. [Google Scholar] [CrossRef]
- Strang, K.H.; Croft, C.L.; Sorrentino, Z.A.; Chakrabarty, P.; Golde, T.E.; Giasson, B.I. Distinct differences in prion-like seeding and aggregation between Tau protein variants provide mechanistic insights into tauopathies. J. Biol. Chem. 2018, 293, 4579. [Google Scholar] [CrossRef]
- Holmes, B.B.; Diamond, M.I. Prion-like properties of Tau protein: The importance of extracellular Tau as a therapeutic target. J. Biol. Chem. 2014, 289, 19855–19861. [Google Scholar] [CrossRef] [PubMed]
- Evans, L.D.; Wassmer, T.; Fraser, G.; Smith, J.; Perkinton, M.; Billinton, A.; Livesey, F.J. Extracellular Monomeric and Aggregated Tau Efficiently Enter Human Neurons through Overlapping but Distinct Pathways. Cell Rep. 2018, 22, 3612–3624. [Google Scholar] [CrossRef]
- Rauch, J.N.; Chen, J.J.; Sorum, A.W.; Miller, G.M.; Sharf, T.; See, S.K.; Hsieh-Wilson, L.C.; Kampmann, M.; Kosik, K.S. Tau Internalization is Regulated by 6-O Sulfation on Heparan Sulfate Proteoglycans (HSPGs). Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Jacks, R.L. Spreading the Seeds of Neurodegeneration: Tau Fibrils Enter Cells by Macroendocytosis. Ph.D. Thesis, University of California, San Francisco, CA, USA, 2010. [Google Scholar]
- Fichou, Y.; Vigers, M.; Goring, A.K.; Eschmann, N.A.; Han, S. Correction: Heparin-induced tau filaments are structurally heterogeneous and differ from Alzheimer’s disease filaments. Chem. Commun. 2018, 54, 8653. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Huvent, I.; Lippens, G.; Eliezer, D.; Zhang, A.; Li, Q.; Tessier, P.; Linhardt, R.J.; Zhang, F.; Wang, C. Glycan Determinants of Heparin-Tau Interaction. Biophys. J. 2017, 112, 921–932. [Google Scholar] [CrossRef] [PubMed]
- Stopschinski, B.E.; Holmes, B.B.; Miller, G.M.; Manon, V.A.; Vaquer-Alicea, J.; Prueitt, W.L.; Hsieh-Wilson, L.C.; Diamond, M.I. Specific glycosaminoglycan chain length and sulfation patterns are required for cell uptake of tau versus alpha-synuclein and beta-amyloid aggregates. J. Biol. Chem. 2018, 293, 10826–10840. [Google Scholar] [CrossRef]
- Zhao, J.; Zhu, Y.; Song, X.; Xiao, Y.; Su, G.; Liu, X.; Wang, Z.; Xu, Y.; Liu, J.; Eliezer, D.; et al. 3-O-Sulfation of Heparan Sulfate Enhances Tau Interaction and Cellular Uptake. Angew. Chem. Int. Ed. Engl. 2020, 59, 1818–1827. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.W.; Herman, M.; Liu, L.; Simoes, S.; Acker, C.M.; Figueroa, H.; Steinberg, J.I.; Margittai, M.; Kayed, R.; Zurzolo, C.; et al. Small misfolded Tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J. Biol. Chem. 2013, 288, 1856–1870. [Google Scholar] [CrossRef] [PubMed]
- Morozova, V.; Cohen, L.S.; Makki, A.E.; Shur, A.; Pilar, G.; El Idrissi, A.; Alonso, A.D. Normal and Pathological Tau Uptake Mediated by M1/M3 Muscarinic Receptors Promotes Opposite Neuronal Changes. Front. Cell Neurosci. 2019, 13, 403. [Google Scholar] [CrossRef]
- Rauch, J.N.; Luna, G.; Guzman, E.; Audouard, M.; Challis, C.; Sibih, Y.E.; Leshuk, C.; Hernandez, I.; Wegmann, S.; Hyman, B.T.; et al. LRP1 is a master regulator of tau uptake and spread. Nature 2020, 580, 381–385. [Google Scholar] [CrossRef]
- Bolos, M.; Llorens-Martin, M.; Jurado-Arjona, J.; Hernandez, F.; Rabano, A.; Avila, J. Direct Evidence of Internalization of Tau by Microglia In Vitro and In Vivo. J. Alzheimers Dis. 2016, 50, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Perea, J.R.; López, E.; Díez-Ballesteros, J.C.; Ávila, J.; Hernández, F.; Bolós, M. Extracellular Monomeric Tau Is Internalized by Astrocytes. Front. Neurosci. 2019, 13, 442. [Google Scholar] [CrossRef]
- Yang, L.; Liu, C.C.; Zheng, H.; Kanekiyo, T.; Atagi, Y.; Jia, L.; Wang, D.; N’Songo, A.; Can, D.; Xu, H.; et al. LRP1 modulates the microglial immune response via regulation of JNK and NF-kappaB signaling pathways. J. Neuroinflam. 2016, 13, 304. [Google Scholar] [CrossRef]
- Liu, C.C.; Hu, J.; Zhao, N.; Wang, J.; Wang, N.; Cirrito, J.R.; Kanekiyo, T.; Holtzman, D.M.; Bu, G. Astrocytic LRP1 Mediates Brain Abeta Clearance and Impacts Amyloid Deposition. J. Soc. Neurosci. 2017, 37, 4023–4031. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Ghosh, A.; Torraville, S.E.; Mukherjee, B.; Walling, S.G.; Martin, G.M.; Harley, C.W.; Yuan, Q. An experimental model of Braak’s pretangle proposal for the origin of Alzheimer’s disease: The role of locus coeruleus in early symptom development. Alzheimer’s Res. Ther. 2019, 11, 59. [Google Scholar] [CrossRef]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Sasner, M. Tau P301S (Line PS19). Available online: https://www.alzforum.org/research-models/tau-p301s-line-ps19 (accessed on 4 January 2021).
- Congdon, E.E.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 399–415. [Google Scholar] [CrossRef]
- Liu, L.; Drouet, V.; Wu, J.W.; Witter, M.P.; Small, S.A.; Clelland, C.; Duff, K. Trans-synaptic spread of tau pathology in vivo. PLoS ONE 2012, 7, e31302. [Google Scholar] [CrossRef]
- Bolos, M.; Llorens-Martin, M.; Perea, J.R.; Jurado-Arjona, J.; Rabano, A.; Hernandez, F.; Avila, J. Absence of CX3CR1 impairs the internalization of Tau by microglia. Mol. Neurodegener. 2017, 12, 59. [Google Scholar] [CrossRef]
- Song, J.X.; Malampati, S.; Zeng, Y.; Durairajan, S.S.K.; Yang, C.B.; Tong, B.C.; Iyaswamy, A.; Shang, W.B.; Sreenivasmurthy, S.G.; Zhu, Z.; et al. A small molecule transcription factor EB activator ameliorates beta-amyloid precursor protein and Tau pathology in Alzheimer’s disease models. Aging Cell 2020, 19, e13069. [Google Scholar] [CrossRef]
- Sidoryk-Wegrzynowicz, M.; Struzynska, L. Astroglial contribution to tau-dependent neurodegeneration. Biochem. J. 2019, 476, 3493–3504. [Google Scholar] [CrossRef]
- Maphis, N.; Xu, G.; Kokiko-Cochran, O.N.; Jiang, S.; Cardona, A.; Ransohoff, R.M.; Lamb, B.T.; Bhaskar, K. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain 2015, 138, 1738–1755. [Google Scholar] [CrossRef]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kugler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef]
- Iwata, M.; Watanabe, S.; Yamane, A.; Miyasaka, T.; Misonou, H. Regulatory mechanisms for the axonal localization of tau protein in neurons. Mol. Biol. Cell 2019, 30, 2441–2457. [Google Scholar] [CrossRef]
- Zempel, H.; Mandelkow, E. Lost after translation: Missorting of Tau protein and consequences for Alzheimer disease. Trends Neurosci. 2014, 37, 721–732. [Google Scholar] [CrossRef]
- de Calignon, A.; Polydoro, M.; Suarez-Calvet, M.; William, C.; Adamowicz, D.H.; Kopeikina, K.J.; Pitstick, R.; Sahara, N.; Ashe, K.H.; Carlson, G.A.; et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 2012, 73, 685–697. [Google Scholar] [CrossRef]
- Maia, L.F.; Kaeser, S.A.; Reichwald, J.; Hruscha, M.; Martus, P.; Staufenbiel, M.; Jucker, M. Changes in amyloid-beta and Tau in the cerebrospinal fluid of transgenic mice overexpressing amyloid precursor protein. Sci. Transl. Med. 2013, 5, 194re192. [Google Scholar] [CrossRef]
- Siskova, Z.; Justus, D.; Kaneko, H.; Friedrichs, D.; Henneberg, N.; Beutel, T.; Pitsch, J.; Schoch, S.; Becker, A.; von der Kammer, H.; et al. Dendritic structural degeneration is functionally linked to cellular hyperexcitability in a mouse model of Alzheimer’s disease. Neuron 2014, 84, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.W.; Hussaini, S.A.; Bastille, I.M.; Rodriguez, G.A.; Mrejeru, A.; Rilett, K.; Sanders, D.W.; Cook, C.; Fu, H.; Boonen, R.A.; et al. Neuronal activity enhances tau propagation and tau pathology in vivo. Nat. Neurosci. 2016, 19, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
- Assefa, B.T.; Gebre, A.K.; Altaye, B.M. Reactive Astrocytes as Drug Target in Alzheimer’s Disease. Biomed. Res. Int. 2018, 2018, 4160247. [Google Scholar] [CrossRef]
- Amadoro, G.; Ciotti, M.T.; Costanzi, M.; Cestari, V.; Calissano, P.; Canu, N. NMDA receptor mediates tau-induced neurotoxicity by calpain and ERK/MAPK activation. Proc. Natl. Acad. Sci. USA 2006, 103, 2892–2897. [Google Scholar] [CrossRef] [PubMed]
- Hynd, M.R.; Scott, H.L.; Dodd, P.R. Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer’s disease. Neurochem. Int. 2004, 45, 583–595. [Google Scholar] [CrossRef] [PubMed]
- Greenhalgh, A.D.; David, S.; Bennett, F.C. Immune cell regulation of glia during CNS injury and disease. Nat. Rev. Neurosci. 2020, 21, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Hopp, S.C.; Lin, Y.; Oakley, D.; Roe, A.D.; DeVos, S.L.; Hanlon, D.; Hyman, B.T. The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer’s disease. J. Neuroinflam. 2018, 15, 269. [Google Scholar] [CrossRef]
- Lloyd, A.F.; Davies, C.L.; Miron, V.E. Microglia: Origins, homeostasis, and roles in myelin repair. Curr. Opin. Neurobiol. 2017, 47, 113–120. [Google Scholar] [CrossRef]
- Song, L.; Yuan, X.; Jones, Z.; Vied, C.; Miao, Y.; Marzano, M.; Hua, T.; Sang, Q.A.; Guan, J.; Ma, T.; et al. Functionalization of Brain Region-specific Spheroids with Isogenic Microglia-like Cells. Sci. Rep. 2019, 9, 11055. [Google Scholar] [CrossRef]
- Perea, J.R.; Llorens-Martin, M.; Avila, J.; Bolos, M. The Role of Microglia in the Spread of Tau: Relevance for Tauopathies. Front. Cell Neurosci. 2018, 12, 172. [Google Scholar] [CrossRef]
- Wolf, S.A.; Boddeke, H.W.; Kettenmann, H. Microglia in Physiology and Disease. Annu. Rev. Physiol. 2017, 79, 619–643. [Google Scholar] [CrossRef] [PubMed]
- Limatola, C.; Ransohoff, R.M. Modulating neurotoxicity through CX3CL1/CX3CR1 signaling. Front. Cell Neurosci. 2014, 8, 229. [Google Scholar] [CrossRef]
- Graeber, M.B. Changing face of microglia. Science 2010, 330, 783–788. [Google Scholar] [CrossRef]
- Cho, S.H.; Sun, B.; Zhou, Y.; Kauppinen, T.M.; Halabisky, B.; Wes, P.; Ransohoff, R.M.; Gan, L. CX3CR1 protein signaling modulates microglial activation and protects against plaque-independent cognitive deficits in a mouse model of Alzheimer disease. J. Biol. Chem. 2011, 286, 32713–32722. [Google Scholar] [CrossRef]
- Kim, T.S.; Lim, H.K.; Lee, J.Y.; Kim, D.J.; Park, S.; Lee, C.; Lee, C.U. Changes in the levels of plasma soluble fractalkine in patients with mild cognitive impairment and Alzheimer’s disease. Neurosci. Lett. 2008, 436, 196–200. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e1217. [Google Scholar] [CrossRef]
- Bhaskar, K.; Konerth, M.; Kokiko-Cochran, O.N.; Cardona, A.; Ransohoff, R.M.; Lamb, B.T. Regulation of tau pathology by the microglial fractalkine receptor. Neuron 2010, 68, 19–31. [Google Scholar] [CrossRef]
- Chidambaram, H.; Das, R.; Chinnathambi, S. Interaction of Tau with the chemokine receptor, CX3CR1 and its effect on microglial activation, migration and proliferation. Cell Biosci. 2020, 10, 1–9. [Google Scholar] [CrossRef]
- Nash, K.R.; Lee, D.C.; Hunt, J.B., Jr.; Morganti, J.M.; Selenica, M.L.; Moran, P.; Reid, P.; Brownlow, M.; Guang-Yu Yang, C.; Savalia, M.; et al. Fractalkine overexpression suppresses tau pathology in a mouse model of tauopathy. Neurobiol. Aging 2013, 34, 1540–1548. [Google Scholar] [CrossRef]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Stancu, I.C.; Cremers, N.; Vanrusselt, H.; Couturier, J.; Vanoosthuyse, A.; Kessels, S.; Lodder, C.; Brone, B.; Huaux, F.; Octave, J.N.; et al. Aggregated Tau activates NLRP3-ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded Tau pathology in vivo. Acta Neuropathol. 2019, 137, 599–617. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Dong, Z.; Song, W. NLRP3 inflammasome as a novel therapeutic target for Alzheimer’s disease. Signal Transduct. Target Ther. 2020, 5, 37. [Google Scholar] [CrossRef]
- Bahrini, I.; Song, J.H.; Diez, D.; Hanayama, R. Neuronal exosomes facilitate synaptic pruning by up-regulating complement factors in microglia. Sci. Rep. 2015, 5, 7989. [Google Scholar] [CrossRef] [PubMed]
- Pascual, M.; Ibanez, F.; Guerri, C. Exosomes as mediators of neuron-glia communication in neuroinflammation. Neural Regen. Res. 2020, 15, 796–801. [Google Scholar] [CrossRef]
- Calafate, S.; Buist, A.; Miskiewicz, K.; Vijayan, V.; Daneels, G.; de Strooper, B.; de Wit, J.; Verstreken, P.; Moechars, D. Synaptic Contacts Enhance Cell-to-Cell Tau Pathology Propagation. Cell Rep. 2015, 11, 1176–1183. [Google Scholar] [CrossRef]
- Kalus, P.; Slotboom, J.; Gallinat, J.; Mahlberg, R.; Cattapan-Ludewig, K.; Wiest, R.; Nyffeler, T.; Buri, C.; Federspiel, A.; Kunz, D.; et al. Examining the gateway to the limbic system with diffusion tensor imaging: The perforant pathway in dementia. Neuroimage 2006, 30, 713–720. [Google Scholar] [CrossRef]
- Perea, G.; Navarrete, M.; Araque, A. Tripartite synapses: Astrocytes process and control synaptic information. Trends Neurosci. 2009, 32, 421–431. [Google Scholar] [CrossRef]
- Cahoy, J.D.; Emery, B.; Kaushal, A.; Foo, L.C.; Zamanian, J.L.; Christopherson, K.S.; Xing, Y.; Lubischer, J.L.; Krieg, P.A.; Krupenko, S.A.; et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. J. Neurosci. 2008, 28, 264–278. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Park, J.; Choi, Y.K. The Role of Astrocytes in the Central Nervous System Focused on BK Channel and Heme Oxygenase Metabolites: A Review. Antioxidants 2019, 8, 121. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cheng, D.; Cheng, R.; Zhu, X.; Wan, T.; Liu, J.; Zhang, R. Mechanisms of U87 astrocytoma cell uptake and trafficking of monomeric versus protofibril Alzheimer’s disease amyloid-beta proteins. PLoS ONE 2014, 9, e99939. [Google Scholar] [CrossRef]
- Martini-Stoica, H.; Cole, A.L.; Swartzlander, D.B.; Chen, F.; Wan, Y.W.; Bajaj, L.; Bader, D.A.; Lee, V.M.Y.; Trojanowski, J.Q.; Liu, Z.; et al. TFEB enhances astroglial uptake of extracellular tau species and reduces tau spreading. J. Exp. Med. 2018, 215, 2355–2377. [Google Scholar] [CrossRef]
- Piacentini, R.; Li Puma, D.D.; Mainardi, M.; Lazzarino, G.; Tavazzi, B.; Arancio, O.; Grassi, C. Reduced gliotransmitter release from astrocytes mediates tau-induced synaptic dysfunction in cultured hippocampal neurons. Glia 2017, 65, 1302–1316. [Google Scholar] [CrossRef] [PubMed]
- Scuderi, C.; Facchinetti, R.; Steardo, L.; Valenza, M. Neuroinflammation in Alzheimer’s Disease: Friend or Foe? FASEB J. 2020, 34. [Google Scholar] [CrossRef]
- Nilsen, L.H.; Rae, C.; Ittner, L.M.; Gotz, J.; Sonnewald, U. Glutamate metabolism is impaired in transgenic mice with tau hyperphosphorylation. J. Cereb. Blood Flow Metab. 2013, 33, 684–691. [Google Scholar] [CrossRef]
- Sidoryk-Wegrzynowicz, M.; Gerber, Y.N.; Ries, M.; Sastre, M.; Tolkovsky, A.M.; Spillantini, M.G. Astrocytes in mouse models of tauopathies acquire early deficits and lose neurosupportive functions. Acta Neuropathol. Commun. 2017, 5, 89. [Google Scholar] [CrossRef] [PubMed]
- Sonnewald, U.; Westergaard, N.; Schousboe, A. Glutamate transport and metabolism in astrocytes. Glia 1997, 21, 56–63. [Google Scholar] [CrossRef]
- Corrêa, S.A.; Eales, K.L. The role of p38 MAPK and its substrates in neuronal plasticity and neurodegenerative disease. J. Signal Transduct. 2012, 2012. [Google Scholar] [CrossRef]
- Tesseur, I.; Van Dorpe, J.; Spittaels, K.; Van den Haute, C.; Moechars, D.; Van Leuven, F. Expression of human apolipoprotein E4 in neurons causes hyperphosphorylation of protein tau in the brains of transgenic mice. Am. J. Pathol. 2000, 156, 951–964. [Google Scholar] [CrossRef]
- Shi, Y.; Yamada, K.; Liddelow, S.A.; Smith, S.T.; Zhao, L.; Luo, W.; Tsai, R.M.; Spina, S.; Grinberg, L.T.; Rojas, J.C.; et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 2017, 549, 523–527. [Google Scholar] [CrossRef]
- Lin, Y.T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 2018, 98, 1294. [Google Scholar] [CrossRef]
- Wang, C.; Najm, R.; Xu, Q.; Jeong, D.E.; Walker, D.; Balestra, M.E.; Yoon, S.Y.; Yuan, H.; Li, G.; Miller, Z.A.; et al. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat. Med. 2018, 24, 647–657. [Google Scholar] [CrossRef]
- Friedberg, J.S.; Aytan, N.; Cherry, J.D.; Xia, W.; Standring, O.J.; Alvarez, V.E.; Nicks, R.; Svirsky, S.; Meng, G.; Jun, G.; et al. Associations between brain inflammatory profiles and human neuropathology are altered based on apolipoprotein E epsilon4 genotype. Sci. Rep. 2020, 10, 2924. [Google Scholar] [CrossRef]
- Alavi Naini, S.M.; Soussi-Yanicostas, N. Heparan Sulfate as a Therapeutic Target in Tauopathies: Insights from Zebrafish. Front. Cell Dev. Biol. 2018, 6, 163. [Google Scholar] [CrossRef]
- Zahid, A.; Li, B.; Kombe, A.J.K.; Jin, T.; Tao, J. Pharmacological Inhibitors of the NLRP3 Inflammasome. Front. Immunol. 2019, 10, 2538. [Google Scholar] [CrossRef]
- Thawkar, B.S.; Kaur, G. Inhibitors of NF-kappaB and P2X7/NLRP3/Caspase 1 pathway in microglia: Novel therapeutic opportunities in neuroinflammation induced early-stage Alzheimer’s disease. J. Neuroimmunol. 2019, 326, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Shelton, L.B.; Baker, J.D.; Zheng, D.; Sullivan, L.E.; Solanki, P.K.; Webster, J.M.; Sun, Z.; Sabbagh, J.J.; Nordhues, B.A.; Koren, J., 3rd; et al. Hsp90 activator Aha1 drives production of pathological tau aggregates. Proc. Natl. Acad. Sci. USA 2017, 114, 9707–9712. [Google Scholar] [CrossRef]
- Fan, Q.; He, W.; Gayen, M.; Benoit, M.R.; Luo, X.; Hu, X.; Yan, R. Activated CX3CL1/Smad2 Signals Prevent Neuronal Loss and Alzheimer’s Tau Pathology-Mediated Cognitive Dysfunction. J. Neurosci. 2020, 40, 1133–1144. [Google Scholar] [CrossRef]
- Vizi, E.S.; Kisfali, M.; Lorincz, T. Role of nonsynaptic GluN2B-containing NMDA receptors in excitotoxicity: Evidence that fluoxetine selectively inhibits these receptors and may have neuroprotective effects. Brain Res. Bull. 2013, 93, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Companys-Alemany, J.; Turcu, A.L.; Bellver-Sanchis, A.; Loza, M.I.; Brea, J.M.; Canudas, A.M.; Leiva, R.; Vazquez, S.; Pallas, M.; Grinan-Ferre, C. A Novel NMDA Receptor Antagonist Protects against Cognitive Decline Presented by Senescent Mice. Pharmaceutics 2020, 12, 284. [Google Scholar] [CrossRef]
- Perez-Nievas, B.G.; Serrano-Pozo, A. Deciphering the Astrocyte Reaction in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 114. [Google Scholar] [CrossRef] [PubMed]
- Kong, Q.; Chang, L.C.; Takahashi, K.; Liu, Q.; Schulte, D.A.; Lai, L.; Ibabao, B.; Lin, Y.; Stouffer, N.; Das Mukhopadhyay, C.; et al. Small-molecule activator of glutamate transporter EAAT2 translation provides neuroprotection. J. Clin. Invest. 2014, 124, 1255–1267. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.B.; Kaplitt, M.G.; De, B.P.; Chen, A.; Flagiello, T.; Salami, C.; Pey, E.; Zhao, L.; Ricart Arbona, R.J.; Monette, S.; et al. AAVrh.10-Mediated APOE2 Central Nervous System Gene Therapy for APOE4-Associated Alzheimer’s Disease. Hum. Gene Ther. Clin. Dev. 2018, 29, 24–47. [Google Scholar] [CrossRef]





| Abbreviation | Explanation |
|---|---|
| AD | Alzheimer’s disease |
| MAPT | Microtubule-associated protein tau |
| ApoE | Apolipoprotein E |
| NFT | Neurofibrillary tangles |
| PSP | Supranuclear palsy |
| CBD | Corticobasal degeneration |
| PiD | Pick’s disease |
| FTDP-17 | Frontotemporal dementia with Parkinsonism linked to chromosome 17 |
| CNS | Central nervous system |
| MBD | Microtubule-binding domain |
| Aβ | Amyloid β |
| PRR | Proline-rich region |
| PHF | Paired helical filaments |
| ISF | Interstitial fluid |
| CSF | Cerebrospinal fluid |
| HSPG | Heparan sulfate proteoglycans |
| ALP | Autophagy-lysosomal pathway |
| NMDA | N-methyl-D-aspartate |
| TFEB | Transcription factor EB |
| LTP | Long-term potentiation |
| LTD | Long-term depression |
| Signaling Targets | Therapeutics | Settings/Organisms | Outcomes/Affected Functional Effects | Affected Phosphorylated Sites | State of Process | Reference |
|---|---|---|---|---|---|---|
| Interfering HSPG-tau interaction | HS oligosaccharide containing two 3-O-sulfo group | Mouse lung endothelial cells (MLECs) |
| NA | Research stage | [41] |
| Intervening the formation of NLRP3 inflammasome | Cias1 and Pycard gene dysfunction | FTD mice model, ACS- or NLRP3 -deficient Tau22 mice |
| pS416 | Research stage | [59,87] |
| Hsp90 cochaperones, Aha1 | KU-177 | Inducible HEK-P301L cells transfected with Aha1 |
| pS396/404 | Research stage | [111] |
| Promoting the autophagy-lysosomal pathway (ALP) | Curcumin analog C1 | Transgenic mice models, 5×FAD, P301S, and 3×Tg |
| pS396/404 pS202/T205 | Research stage | [56] |
| Promoting CX3CL1 regulated signaling | CX3CL1 overexpression | P19 tau pathology mice model |
| NA | Research stage | [112] |
| Inhibiting extra-synaptic NMDA receptor | RL-208 | A mice model of late-onset AD (LOAD) |
| pS396 | Research stage | [114] |
| GLT1 upregulation | LDN/OSU-0212320 | Neuron and astrocyte coculture |
| NA | Research stage | [116] |
| ApoE4 gene therapy | AAV ApoE2-expressing vector targeting CNS/CSF ApoE4 | AD patients |
| NA | Phase I | [117] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, L.; Wells, E.A.; Robinson, A.S. Critical Molecular and Cellular Contributors to Tau Pathology. Biomedicines 2021, 9, 190. https://doi.org/10.3390/biomedicines9020190
Song L, Wells EA, Robinson AS. Critical Molecular and Cellular Contributors to Tau Pathology. Biomedicines. 2021; 9(2):190. https://doi.org/10.3390/biomedicines9020190
Chicago/Turabian StyleSong, Liqing, Evan A. Wells, and Anne Skaja Robinson. 2021. "Critical Molecular and Cellular Contributors to Tau Pathology" Biomedicines 9, no. 2: 190. https://doi.org/10.3390/biomedicines9020190
APA StyleSong, L., Wells, E. A., & Robinson, A. S. (2021). Critical Molecular and Cellular Contributors to Tau Pathology. Biomedicines, 9(2), 190. https://doi.org/10.3390/biomedicines9020190