
Therapeutic Status and Available Strategies in Pancreatic Ductal Adenocarcinoma

 , ,
, ,
Abstract
1. Introduction
2. Treatment Hurdles
3. Stem Cells
3.1. Cancer Stem Cells
3.2. Potential Approaches Targeting PCSCs
4. Signaling Pathways Involved in PCSCs
4.1. Notch Signaling Pathway
4.2. Hedgehog Signaling Pathway
4.3. WNT Signaling Pathway
5. Epithelial to Mesenchymal Transition
6. Role of Epigenetics
7. Role of G Protein-Coupled Receptor in Pancreatic Cancer
8. Role of Cysteine in Pancreatic Cancer
9. Role of Pyruvate Kinase M2
10. Repurposed Drugs for CSCs
11. Immunotherapy for Pancreatic Cancer
- Monotherapy includes the administration of several PD-1(MEDI4736, MPDL3280A, and pembrolizumab,) and CTL-4 (tremelimumab and ipilimumab) inhibitors and inhibition of double checkpoints: either by a combination of the above mentioned inhibitors or with other agents, such as anti-CCR-5 (mogamulizumab) [184].
- Combination of chemotherapeutic agents and immune checkpoint inhibitors: PD-1/CTL4 inhibitors leads to the activation of T cell that is efficient for immunotherapy. When PD-1/CTL4 inhibitors combined with commonly used chemotherapeutic agents such as Nab-paclitaxel, gemcitabine, carboplatin, and FOLFOX improved overall survival [47]. Remarkably, therapeutic procedures using a combination of immune checkpoint inhibitors with radiotherapy or chemotherapy have shown significant outcomes [185,186].
- Vaccination therapy is founded on the basis of the distribution of tumor antigens to antigen presenting cells (APCs), followed by induction of an organized immune response. Cancer specific DNA mutations produce new antigens, which, in turn, results in a unique sequence of the peptide. Variety of vaccines for pancreatic cancer treatment includes whole-cell vaccines, dendritic-cell based vaccines, peptide and DNA vaccines, telomerase peptide vaccines, Ras peptide vaccines, and survivin-targeted vaccines [187]; however, regardless of the enhanced immune system, showed poor clinical results. GVAX is an allogeneic irradiated whole-cell tumor vaccine genetically modified for the secretion of granulocyte macrophage colony stimulating factor and promotes cytolytic action against tumors, the most widely studied vaccine for pancreatic cancer [188]. Furthermore, the clinical studies when GVAX is applied in combination with 5-Fluorouracil/cyclophosphamide based chemotherapy have shown the same results regarding disease-free and median survival as that of GVAX applied alone [189]. On the other hand, when the above mentioned ipilimumab (immune checkpoint inhibitor) is applied in combination with GVAX, it leads to better survival [190].
- Adoptive T cell immunotherapy is based on the modification of autologous T cells, which stimulates the immune response against the tumor. The patients receiving mesothelin-targeting chimeric antigen receptor-T (CAR) cells have shown overexpression of a membrane antigen in pancreatic cancer, exhibited adequate patience but unsuccessful in showing good results [191]. Along with mesothelin, other cancer-associated antigens are being studied alone or in combination with chemotherapy as potential targets of CAR-T cells based therapy [191].
- Immune modulating agents that target the microenvironment of the pancreas can also exert extensive antitumor activity. Anti-CD40 agonistic antibodies used in combination with gemcitabine in PDAC patients showed significant results [192]. PDAC patients treated with a CCR2 inhibitor (PF-04136309) exhibited fractional response and constant tumor when used in combination with FOLFIRINOX [193]. Several chemokine receptor molecules are under examination in clinical trials against PDAC.
12. Natural Killer Cell Therapy
13. Discussion
14. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CSCs | Cancer stem cells |
| EMT | Epithelial mesenchymal transition |
| ESCs | Embryonic stem cells |
| GPCR | G protein-coupled receptor |
| GPER | G protein-coupled estrogen receptor |
| MSCs | Mesenchymal stem cells |
| NK cell | Natural killer cells |
| PCSCs | Pancreatic cancer stem cells |
| PDAC | Pancreatic ductal adenocarcinoma |
| PKM2 | Pyruvate kinase M2 |
| PMCA | Plasma membrane calcium pump |
| ROS | Reactive oxygen species |
| SHH | Sonic Hedgehog |
| Xc- | Cystine/glutamate transporter |
| ZEB | Zinc-finger E-box binding homeobox |
References
- Hong, S.; Won, Y.J.; Park, Y.R.; Jung, K.W.; Kong, H.J.; Lee, E.S. Cancer Statistics in Korea: Incidence, Mortality, Survival, and Prevalence in 2017. Cancer Res. Treat. Off. J. Kor. Cancer Assoc. 2020, 52, 335–350. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Mathur, P.; Sathishkumar, K.; Chaturvedi, M.; Das, P.; Sudarshan, K.L.; Santhappan, S.; Nallasamy, V.; John, A.; Narasimhan, S.; Roselind, F.S. ICMR-NCDIR-NCRP Investigator Group. Cancer Statistics, 2020: Report From National Cancer Registry Programme, India. JCO Glob. Oncol. 2020, 6, 1063–1075. [Google Scholar] [CrossRef]
- Ilic, M.; Ilic, I. Epidemiology of pancreatic cancer. World J. Gastroenterol. 2016, 22, 9694–9705. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Sarantis, P.; Koustas, E.; Papadimitropoulou, A.; Papavassiliou, A.G.; Karamouzis, M.V. Pancreatic ductal adenocarcinoma: Treatment hurdles, tumor microenvironment and immunotherapy. World J. Gastrointest. Oncol. 2020, 12, 173–181. [Google Scholar] [CrossRef]
- Ying, H.; Dey, P.; Yao, W.; Kimmelman, A.C.; Draetta, G.F.; Maitra, A.; DePinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2016, 30, 355–385. [Google Scholar] [CrossRef]
- Hidalgo, M.; Cascinu, S.; Kleeff, J.; Labianca, R.; Löhr, J.M.; Neoptolemos, J.; Real, F.X.; Van Laethem, J.L.; Heinemann, V. Addressing the challenges of pancreatic cancer: Future directions for improving outcomes. Pancreatology 2015, 15, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef]
- Zhou, J.; Enewold, L.; Stojadinovic, A.; Clifton, G.T.; Potter, J.F.; Peoples, G.E.; Zhu, K. Incidence rates of exocrine and endocrine pancreatic cancers in the United States. Cancer Causes Control. 2010, 21, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Bond-Smith, G.; Banga, N.; Hammond, T.M.; Imber, C.J. Pancreatic adenocarcinoma. BMJ 2012, 344, e2476. [Google Scholar] [CrossRef]
- Orth, M.; Metzger, P.; Gerum, S.; Mayerle, J.; Schneider, G.; Belka, C.; Schnurr, M.; Lauber, K. Pancreatic ductal adenocarcinoma: Biological hallmarks, current status, and future perspectives of combined modality treatment approaches. Radiat. Oncol. 2019, 14, 1–20. [Google Scholar] [CrossRef]
- Corbo, V.; Tortora, G.; Scarpa, A. Molecular pathology of pancreatic cancer: From bench-to-bedside translation. Curr. Drug Targ. 2012, 13, 744–752. [Google Scholar] [CrossRef] [PubMed]
- Ghaneh, P.; Costello, E.; Neoptolemos, J.P. Biology and management of pancreatic cancer. Postgrad. Med. J. 2008, 84, 478–497. [Google Scholar] [CrossRef] [PubMed]
- Porta, M.; Fabregat, X.; Malats, N.; Guarner, L.; Carrato, A.; de Miguel, A.; Ruiz, L.; Jariod, M.; Costafreda, S.; Coll, S.; et al. Exocrine pancreatic cancer: Symptoms at presentation and their relation to tumour site and stage. Clin. Transl. Oncol. 2005, 7, 189–197. [Google Scholar] [CrossRef] [PubMed]
- De Souza, A.; Khawaja, K.I.; Masud, F.; Saif, M.W. Metformin and pancreatic cancer: Is there a role? Cancer Chemother. Pharmacol. 2016, 77, 235–242. [Google Scholar] [CrossRef]
- Ben, Q.; Xu, M.; Ning, X.; Liu, J.; Hong, S.; Huang, W.; Zhang, H.; Li, Z. Diabetes mellitus and risk of pancreatic cancer: A meta-analysis of cohort studies. Eur. J. Cancer 2011, 47, 1928–1937. [Google Scholar] [CrossRef] [PubMed]
- Wormann, S.M.; Algul, H. Risk factors and therapeutic targets in pancreatic cancer. Front. Oncol. 2013, 3, 282. [Google Scholar] [CrossRef]
- Mayo, S.C.; Nathan, H.; Cameron, J.L.; Olino, K.; Edil, B.H.; Herman, J.M.; Hirose, K.; Schulick, R.D.; Choti, M.A.; Wolfgang, C.L.; et al. Conditional survival in patients with pancreatic ductal adenocarcinoma resected with curative intent. Cancer 2012, 118, 2674–2681. [Google Scholar] [CrossRef]
- Kanojia, D.; Balyasnikova, I.V.; Morshed, R.A.; Frank, R.T.; Yu, D.; Zhang, L.; Spencer, D.A.; Kim, J.W.; Han, Y.; Yu, D.; et al. Neural stem cells secreting anti-her2 antibody improve survival in a preclinical model of her2 overexpressing breast cancer brain metastases. Stem Cells 2015, 33, 2985–2994. [Google Scholar] [CrossRef][Green Version]
- Wang, J.P.; Wu, C.Y.; Yeh, Y.C.; Shyr, Y.M.; Wu, Y.Y.; Kuo, C.Y.; Hung, Y.P.; Chen, M.H.; Lee, W.P.; Luo, J.C.; et al. Erlotinib is effective in pancreatic cancer with epidermal growth factor receptor mutations: A randomized, open-label, prospective trial. Oncotarget 2005, 6, 18162–18173. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ng, J.; Allendorf, J.; Saif, M.W. Locally advanced pancreatic adenocarcinoma: Are we making progress? In Proceedings of the 2011 ASCO Annual Meeting, Chicago, IL, USA, 3–7 June 2011; Volume 12, pp. 347–350. [Google Scholar] [CrossRef]
- Carpelan-Holmstrom, M.; Nordling, S.; Pukkala, E.; Sankila, R.; Lüttges, J.; Klöppel, G.; Haglund, C. Does anyone survive pancreatic ductal adenocarcinoma? A nationwide study re-evaluating the data of the Finnish Cancer Registry. Gut 2005, 54, 385–387. [Google Scholar] [CrossRef] [PubMed]
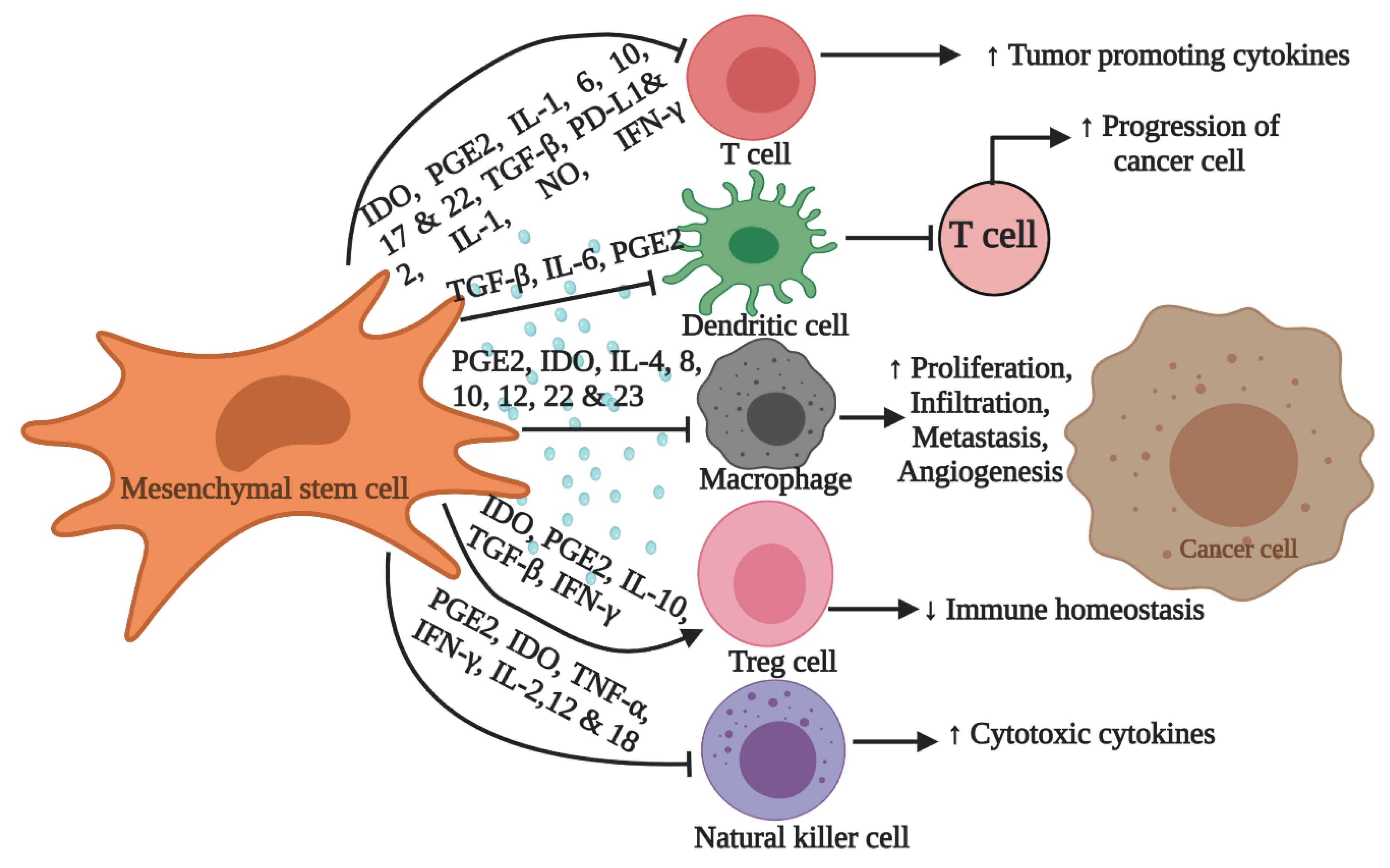
- Chopra, N.; Choudhury, S.; Bhargava, S.; Wajid, S.; Ganguly, N.K. Potentials of “stem cell-therapy” in pancreatic cancer: An update. Pancreatology 2019, 19, 1034–1042. [Google Scholar] [CrossRef]
- Momin, E.N.; Vela, G.; Zaidi, H.A.; Qui~nones-Hinojosa, A. The oncogenic potential of mesenchymal stem cells in the treatment of cancer: Directions for future research. Curr. Immunol. Rev. 2010, 6, 137–148. [Google Scholar] [CrossRef]
- Ullah, I.; Subbarao, R.B.; Rho, G.J. Human mesenchymal stem cells-current trends and future prospective. Biosci. Rep. 2015, 35. [Google Scholar] [CrossRef] [PubMed]
- Zachar, L.; Bacenkov, D.; Soltys, J.; Rosocha, J. Bioactive mediators associated with mesenchymal stem cells-mediated immunomodulation. J. Bone Stem Res. 2015, 1, 6. [Google Scholar]
- Weiss, A.R.R.; Dahlke, M.H. Immunomodulation by mesenchymal stem cells (MSCs): Mechanisms of action of living, apoptotic, and dead MSCs. Front. Immunol. 2019, 10, 1191. [Google Scholar] [CrossRef]
- Cousin, B.; Ravet, E.; Poglio, S.; De Toni, F.; Bertuzzi, M.; Lulka, H.; Touil, I.; André, M.; Grolleau, J.L.; Péron, J.M.; et al. Adult stromal cells derived from human adipose tissue provoke pancreatic cancer cell death both in vitro and in vivo. PLoS ONE 2009, 17, e6278. [Google Scholar] [CrossRef]
- Doi, C.; Maurya, D.K.; Pyle, M.M.; Troyer, D.; Tamura, M. Cytotherapy with naive rat umbilical cord matrix stem cells significantly attenuates growth of murine pancreatic cancer cells and increases survival in syngeneic mice. Cytotherapy 2010, 12, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Brini, A.T.; Coccè, V.; Ferreira, L.M.J.; Giannasi, C.; Cossellu, G.; Giannì, A.B.; Angiero, F.; Bonomi, A.; Pascucci, L.; Falchetti, M.L.; et al. Cell-mediated drug delivery by gingival interdental papilla mesenchymal stromal cells (GinPa-MSCs) loaded with paclitaxel. Exp. Opin. Drug Deliv. 2016, 13, 789–798. [Google Scholar] [CrossRef]
- Sun, Z.; Wang, S.; Zhao, R.C. The roles of mesenchymal stem cells in tumor inflammatory microenvironment. J. Hematol. Oncol. 2014, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Chulpanova, D.S.; Kitaeva, K.V.; Tazetdinova, L.G.; James, V.; Rizvanov, A.A.; Solovyeva, V.V. Application of mesenchymal stem cells for therapeutic agent delivery in anti-tumor treatment. Front. Pharmacol. 2018, 9, 259. [Google Scholar] [CrossRef] [PubMed]
- Valle, S.; Martin-Hijano, L.; Alcalá, S.; Alonso-Nocelo, M.; Sainz, B., Jr. The Ever-Evolving Concept of the Cancer Stem Cell in Pancreatic Cancer. Cancers 2018, 10, 33. [Google Scholar] [CrossRef]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [PubMed]
- O’Brien-Ball, C.; Biddle, A. Reprogramming to developmental plasticity in cancer stem cells. Dev. Biol. 2017, 430, 266–274. [Google Scholar] [CrossRef]
- Rao, C.V.; Mohammed, A. New insights into pancreatic cancer stem cells. World J. Stem Cells 2015, 7, 547–555. [Google Scholar] [CrossRef]
- Ercan, G.; Karlitepe, A.; Ozpolat, B. Pancreatic Cancer Stem Cells and Therapeutic Approaches. Anticancer Res. 2017, 37, 2761–2775. [Google Scholar] [CrossRef]
- Dawood, S.; Austin, L.; Cristofanilli, M. Cancer stem cells: Implications for cancer therapy. Oncology 2014, 28, 1101–1107. [Google Scholar]
- Subramaniam, D.; Kaushik, G.; Dandawate, P.; Anant, S. Targeting Cancer Stem Cells for Chemoprevention of Pancreatic Cancer. Curr. Med. Chem. 2018, 25, 2585–2594. [Google Scholar] [CrossRef] [PubMed]
- Najumudeen, A.K.; Jaiswal, A.; Lectez, B.; Oetken-Lindholm, C.; Guzmán, C.; Siljamäki, E.; Posada, I.M.; Lacey, E.; Aittokallio, T.; Abankwa, D. Cancer stem cell drugs target K-ras signaling in a stemness context. Oncogene 2016, 35, 5248–5262. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.M.; Dong, T.T.; Wang, L.L.; Feng, B.; Zhao, H.C.; Fan, X.K.; Zheng, M.H. Suppression of colorectal cancer metastasis by nigericin through inhibition of epithelial-mesenchymal transition. World J. Gastroenterol. 2012, 18, 2640–2648. [Google Scholar] [CrossRef]
- Lamb, R.; Ozsvari, B.; Lisanti, C.L.; Tanowitz, H.B.; Howell, A.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: Treating cancer like an infectious disease. Oncotarget 2015, 6, 4569–4584. [Google Scholar] [CrossRef]
- Balic, A.; Sørensen, M.D.; Trabulo, S.M.; Sainz, B., Jr.; Cioffi, M.; Vieira, C.R.; Miranda-Lorenzo, I.; Hidalgo, M.; Kleeff, J.; Erkan, M.; et al. Chloroquine targets pancreatic cancer stem cells via inhibition of CXCR4 and hedgehog signaling. Mol. Cancer Ther. 2014, 13, 1758–1771. [Google Scholar] [CrossRef]
- Coyle, C.; Cafferty, F.H.; Rowley, S.; MacKenzie, M.; Berkman, L.; Gupta, S.; Pramesh, C.S.; Gilbert, D.; Kynaston, H.; Cameron, D.; et al. Add-Aspirin investigators. ADD-ASPIRIN: A phase III, double-blind, placebo controlled, randomised trial assessing the effects of aspirin on disease recurrence and survival after primary therapy in common non-metastatic solid tumours. Contemp. Clin. Trials 2016, 51, 56–64. [Google Scholar] [CrossRef]
- Cong, J.; Wang, Y.; Zhang, X.; Zhang, N.; Liu, L.; Soukup, K.; Michelakos, T.; Hong, T.; DeLeo, A.; Cai, L.; et al. A novel chemoradiation targeting stem and nonstem pancreatic cancer cells by repurposing disulfiram. Cancer Lett. 2017, 409, 9–19. [Google Scholar] [CrossRef]
- Garnier, A.; Vykoukal, J.; Hubertus, J.; Alt, E.; von Schweinitz, D.; Kappler, R.; Berger, M.; Ilmer, M. Targeting the neurokinin-1 receptor inhibits growth of human colon cancer cells. Int. J. Oncol. 2015, 47, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Fiorillo, M.; Lamb, R.; Tanowitz, H.B.; Mutti, L.; Krstic-Demonacos, M.; Cappello, A.R.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Repurposing atovaquone: Targeting mitochondrial complex III and OXPHOS to eradicate cancer stem cells. Oncotarget 2016, 7, 34084–34099. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.Y.; Sharma, S.; Zhou, Y.Q.; Yao, H.P.; Hu, X.; Zhang, R.; Wang, M.H. Synergistic activities of MET/RON inhibitor BMS-777607 and mTOR inhibitor AZD8055 to polyploid cells derived from pancreatic cancer and cancer stem cells. Mol. Cancer Ther. 2014, 13, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Rangarajan, P.; Subramaniam, D.; Paul, S.; Kwatra, D.; Palaniyandi, K.; Islam, S.; Harihar, S.; Ramalingam, S.; Gutheil, W.; Putty, S. Crocetinic acid inhibits hedgehog signaling to inhibit pancreatic cancer stem cells. Oncotarget 2015, 6, 27661–27673. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Matsubara, S.; Ding, Q.; Tsukasa, K.; Yoshimitsu, M.; Kosai, K.; Takao, S. Efficient elimination of pancreatic cancer stem cells by hedgehog/GLI inhibitor GANT61 in combination with mTOR inhibition. Mol. Cancer 2016, 15, 49. [Google Scholar] [CrossRef]
- Blaj, C.; Bringmann, A.; Schmidt, E.M.; Urbischek, M.; Lamprecht, S.; Fröhlich, T.; Arnold, G.J.; Krebs, S.; Blum, H.; Hermeking, H. ADNP Is a Therapeutically Inducible Repressor of WNT Signaling in Colorectal Cancer. Clin. Cancer Res. 2017, 23, 2769–2780. [Google Scholar] [CrossRef] [PubMed]
- Reni, M.; Dugnani, E.; Cereda, S.; Belli, C.; Balzano, G.; Nicoletti, R.; Liberati, D.; Pasquale, V.; Scavini, M.; Maggiora, P.; et al. Irrelevance of Metformin Treatment in Patients with Metastatic Pancreatic Cancer: An Open-Label, Randomized Phase II Trial. Clin. Cancer Res. 2016, 22, 1076–1085. [Google Scholar] [CrossRef]
- Ponnurangam, S.; Dandawate, P.R.; Dhar, A.; Tawfik, O.W.; Parab, R.R.; Mishra, P.D.; Ranadive, P.; Sharma, R.; Mahajan, G.; Umar, S. Quinomycin A targets Notch signaling pathway in pancreatic cancer stem cells. Oncotarget 2016, 7, 3217–3232. [Google Scholar] [CrossRef]
- Matsubara, S.; Ding, Q.; Miyazaki, Y.; Kuwahata, T.; Tsukasa, K.; Takao, S. mTOR plays critical roles in pancreatic cancer stem cells through specific and stemness-related functions. Sci. Rep. 2013, 3, 3230. [Google Scholar] [CrossRef] [PubMed]
- Abel, E.V.; Kim, E.J.; Wu, J.; Hynes, M.; Bednar, F.; Proctor, E.; Wang, L.; Dziubinski, M.L.; Simeone, D.M. The Notch pathway is important in maintaining the cancer stem cell population in pancreatic cancer. PLoS ONE 2014, 9, 91983. [Google Scholar] [CrossRef]
- Ma, Y.; Yu, W.; Shrivastava, A.; Alemi, F.; Lankachandra, K.; Srivastava, R.K.; Shankar, S. Sanguinarine inhibits pancreatic cancer stem cell characteristics by inducing oxidative stress and suppressing sonic hedgehog-Gli-Nanog pathway. Carcinogenesis 2017, 38, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Renz, B.W.; D’Haese, J.G.; Werner, J.; Westphalen, C.B.; Ilmer, M. Repurposing Established Compounds to Target Pancreatic Cancer Stem Cells (CSCs). Med. Sci. 2017, 5, 14. [Google Scholar] [CrossRef]
- Endo, Y.; Kitago, M.; Aiura, K.; Shinoda, M.; Yagi, H.; Abe, Y.; Oshima, G.; Hori, S.; Nakano, Y.; Itano, O.; et al. Efficacy and safety of preoperative 5-fluorouracil, cisplatin, and mitomycin C in combination with radiotherapy in patients with resectable and borderline resectable pancreatic cancer: A long-term follow-up study. World J. Surg. Oncol. 2019, 17, 1–8. [Google Scholar] [CrossRef]
- Vreeland, T.J.; McAllister, F.; Javadi, S.; Prakash, L.R.; Fogelman, D.R.; Ho, L.; Varadhachary, G.; Aloia, T.A.; Vauthey, J.N.; Lee, J.E.; et al. Benefit of Gemcitabine/Nab-Paclitaxel Rescue of Patients With Borderline Resectable or Locally Advanced Pancreatic Adenocarcinoma After Early Failure of FOLFIRINOX. Pancreas 2019, 48, 837–843. [Google Scholar] [CrossRef]
- Babiker, H.M.; Karass, M.; Recio-Boiles, A.; Chandana, S.R.; McBride, A.; Mahadevan, D. Everolimus for the treatment of advanced pancreatic ductal adenocarcinoma (PDAC). Exp. Opin. Investig. Drugs 2019, 28, 583–592. [Google Scholar] [CrossRef]
- Patel, G.K.; Perry, J.B.; Abdul-Rahim, O.; Frankel, A.E.; Cameron, D.; Taylor, W.; Rocconi, R.P.; Abushahin, L.; Nelson, C.; Singh, A.P. Epidermal growth factor receptor-activating mutation (E746_T751> VP) in pancreatic ductal adenocarcinoma responds to erlotinib, followed by epidermal growth factor receptor resistance-mediating mutation (A647T): A case report and literature review. J. Cancer Res. Ther. 2020, 16, 950. [Google Scholar] [CrossRef]
- Golan, T.; Varadhachary, G.R.; Sela, T.; Fogelman, D.R.; Halperin, N.; Shroff, R.T.; Halparin, S.; Xiao, L.; Aderka, D.; Maitra, A.; et al. Phase II study of olaparib for BRCAness phenotype in pancreatic cancer. J. Clin. Oncol. 2018, 36, 297. [Google Scholar] [CrossRef]
- Mercadé, T.M.; Chen, L.T.; Li, C.P.; Siveke, J.T.; Cunningham, D.; Bodoky, G.; Blanc, J.F.; Lee, K.H.; Dean, A.; Belanger, B.; et al. Liposomal irinotecan+ 5-FU/LV in metastatic pancreatic cancer: Subgroup analyses of patient, tumor, and previous treatment characteristics in the pivotal NAPOLI-1 trial. Pancreas 2020, 49, 62. [Google Scholar] [CrossRef]
- Gaustad, J.V.; Simonsen, T.G.; Wegner, C.S.; Rofstad, E.K. Vascularization, Oxygenation, and the Effect of Sunitinib Treatment in Pancreatic Ductal Adenocarcinoma Xenografts. Front. Oncol. 2019, 9, 845. [Google Scholar] [CrossRef] [PubMed]
- Hong, Z.; Wang, Z.; Zhou, B.; Wang, J.; Tong, H.; Liao, Y.; Zheng, P.; Jamshed, M.B.; Zhang, Q.; Chen, H. Effects of evodiamine on PI3K/Akt and MAPK/ERK signaling pathways in pancreatic cancer cells. Int. J. Oncol. 2020, 56, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Grassilli, S.; Brugnoli, F.; Lattanzio, R.; Buglioni, S.; Bertagnolo, V. Vav1 Down-Modulates Akt2 Expression in Cells from Pancreatic Ductal Adenocarcinoma: Nuclear Vav1 as a Potential Regulator of Akt Related Malignancy in Pancreatic Cancer. Biomedicines 2020, 8, 379. [Google Scholar] [CrossRef] [PubMed]
- Semba, T.; Sammons, R.; Wang, X.; Xie, X.; Dalby, K.N.; Ueno, N.T. JNK Signaling in Stem Cell Self-Renewal and Differentiation. Int. J. Mol. Sci. 2020, 21, 2613. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, P.; Shen, D.; Liu, H.; Xu, L.; Wang, J.; Shen, D.; Sun, H.; Wu, H. FAM172A inhibits EMT in pancreatic cancer via ERK-MAPK signaling. Biol. Open 2020, 9. [Google Scholar] [CrossRef]
- Kaltschmidt, C.; Banz-Jansen, C.; Benhidjeb, T.; Beshay, M.; Förster, C.; Greiner, J.; Hamelmann, E.; Jorch, N.; Mertzlufft, F.; Pfitzenmaier, J.; et al. A role for NF-κB in organ specific cancer and cancer stem cells. Cancers 2019, 11, 655. [Google Scholar] [CrossRef] [PubMed]
- Delle Cave, D.; Di Guida, M.; Costa, V.; Sevillano, M.; Ferrante, L.; Heeschen, C.; Corona, M.; Cucciardi, A.; Lonardo, E. TGF-β1 secreted by pancreatic stellate cells promotes stemness and tumourigenicity in pancreatic cancer cells through L1CAM downregulation. Oncogene 2020, 1–15. [Google Scholar] [CrossRef]
- Weekes, C.D.; Winn, R.A. The many faces of wnt and pancreatic ductal adenocarcinoma oncogenesis. Cancers 2011, 3, 3676–3686. [Google Scholar] [CrossRef] [PubMed]
- Ram Makena, M.; Gatla, H.; Verlekar, D.; Sukhavasi, S.K.; Pandey, M.C.; Pramanik, K. Wnt/β-Catenin Signaling: The Culprit in Pancreatic Carcinogenesis and Therapeutic Resistance. Int. J. Mol. Sci. 2019, 20, 4242. [Google Scholar] [CrossRef]
- Beachy, P.A.; Hymowitz, S.G.; Lazarus, R.A.; Leahy, D.J.; Siebold, C. Interactions between Hedgehog proteins and their binding partners come into view. Genes Dev. 2010, 24, 2001–2012. [Google Scholar] [CrossRef] [PubMed]
- Sari, I.N.; Phi, L.T.H.; Jun, N.; Wijaya, Y.T.; Lee, S.; Kwon, H.Y. Hedgehog signaling in cancer: A prospective therapeutic target for eradicating cancer stem cells. Cells 2018, 7, 208. [Google Scholar] [CrossRef]
- Takebe, N.; Miele, L.; Harris, P.J.; Jeong, W.; Bando, H.; Kahn, M.; Yang, S.X.; Ivy, S.P. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: Clinical update. Nat. Rev. Clin. Oncol. 2015, 12, 445–464. [Google Scholar] [CrossRef]
- Gao, J.; Long, B.; Wang, Z. Role of Notch signaling pathway in pancreatic cancer. Am. J. Cancer Res. 2017, 7, 173–186. [Google Scholar] [PubMed]
- Pelullo, M.; Zema, S.; Nardozza, F.; Checquolo, S.; Screpanti, I.; Bellavia, D. Wnt, Notch, and TGF-β Pathways Impinge on Hedgehog Signaling Complexity: An Open Window on Cancer. Front. Genet. 2019, 10, 711. [Google Scholar] [CrossRef]
- Bailey, J.M.; Alsina, J.; Rasheed, Z.A.; McAllister, F.M.; Fu, Y.Y.; Plentz, R.; Zhang, H.; Pasricha, P.J.; Bardeesy, N.; Matsui, W.; et al. DCLK1 marks a morphologically distinct subpopulation of cells with stem cell properties in preinvasive pancreatic cancer. Gastroenterology 2014, 146, 245–256. [Google Scholar] [CrossRef]
- Thakur, G.; Lee, H.J.; Jeon, R.H.; Lee, S.L.; Rho, G.J. Small Molecule-Induced Pancreatic β-Like Cell Development: Mechanistic Approaches and Available Strategies. Int. J. Mol. Sci. 2020, 21, 2388. [Google Scholar] [CrossRef]
- Castellanos, J.A.; Merchant, N.B.; Nagathihalli, N.S. Emerging targets in pancreatic cancer: Epithelial–mesenchymal transition and cancer stem cells. Onco. Targ. Ther. 2013, 6, 1261. [Google Scholar] [CrossRef]
- Yang, Y.; Li, X.; Wang, T.; Guo, Q.; Xi, T.; Zheng, L. Emerging agents that target signaling pathways in cancer stem cells. J. Hematol. Oncol. 2020, 13, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, I.; Miele, L. Notch inhibitors for cancer treatment. Pharmacol. Ther. 2013, 139, 95–110. [Google Scholar] [CrossRef]
- Wang, Z.; Banerjee, S.; Li, Y.; Rahman, K.M.; Zhang, Y.; Sarkar, F.H. Down-regulation of notch-1 inhibits invasion by inactivation of nuclear factor-kappaB, vascular endothelial growth factor, and matrix metalloproteinase-9 in pancreatic cancer cells. Cancer Res. 2006, 66, 2778–2784. [Google Scholar] [CrossRef] [PubMed]
- Blaumueller, C.M.; Qi, H.; Zagouras, P.; Artavanis-Tsakonas, S. Intracellular cleavage of Notch leads to a heterodimeric receptor on the plasma membrane. Cell 1997, 90, 281–291. [Google Scholar] [CrossRef]
- Xie, M.; Zhang, L.; He, C.S.; Xu, F.; Liu, J.L.; Hu, Z.H.; Zhao, L.P.; Tian, Y. Activation of Notch-1 enhances epithelial–mesenchymal transition in gefitinib-acquired resistant lung cancer cells. J. Cell. Biochem. 2012, 113, 1501–1513. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.W.; Rizzo, P.; Pannuti, A.; Golde, T.; Osborne, B.; Miele, L. Notch signals in the endothelium and cancer “stem-like” cells: Opportunities for cancer therapy. Vasc. Cell 2012, 4, 7. [Google Scholar] [CrossRef][Green Version]
- Güngör, C.; Zander, H.; Effenberger, K.E.; Vashist, Y.K.; Kalinina, T.; Izbicki, J.R.; Yekebas, E.; Bockhorn, M. Notch signaling activated by replication stressinduced expression of midkine drives epithelial–mesenchymal transition and chemoresistance in pancreatic cancer. Cancer Res. 2011, 71, 5009–5019. [Google Scholar] [CrossRef] [PubMed]
- Sethi, S.; Macoska, J.; Chen, W.; Sarkar, F.H. Molecular signature of epithelial– mesenchymal transition (EMT) in human prostate cancer bone metastasis. Am. J. Transl. Res. 2020, 3, 90–99. [Google Scholar]
- Hu, H.; Zhou, L.; Awadallah, A.; Xin, W. Significance of Notch1-signaling pathway in human pancreatic development and carcinogenesis. Appl. Immunohistochem. Mol. Morphol. 2013, 21, 242–247. [Google Scholar] [CrossRef]
- Vizio, B.; Mauri, F.A.; Prati, A.; Trivedi, P.; Giacobino, A.; Novarino, A.; Satolli, M.A.; Ciuffreda, L.; Camandona, M.; Gasparri, G.; et al. Comparative evaluation of cancer stem cell markers in normal pancreas and pancreatic ductal adenocarcinoma. Oncol. Rep. 2012, 27, 69–76. [Google Scholar] [CrossRef][Green Version]
- De La, O.J.; Emerson, L.L.; Goodman, J.L.; Froebe, S.C.; Illum, B.E.; Curtis, A.B.; Murtaugh, L.C. Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. Proc. Natl. Acad. Sci. USA 2008, 105, 18907–18912. [Google Scholar] [CrossRef]
- Mazur, P.K.; Einwächter, H.; Lee, M.; Sipos, B.; Nakhai, H.; Rad, R.; Zimber-Strobl, U.; Strobl, L.J.; Radtke, F.; Klöppel, G.; et al. Notch2 is required for progression of pancreatic intraepithelial neoplasia and development of pancreatic ductal adenocarcinoma. Proc. Natl. Acad. Sci. USA 2010, 107, 13438–13443. [Google Scholar] [CrossRef] [PubMed]
- Güngör, C.; Hofmann, B.T.; Wolters-Eisfeld, G.; Bockhorn, M. Pancreatic cancer. Br. J. Pharmacol. 2014, 171, 849–858. [Google Scholar] [CrossRef]
- Buscail, L.; Bournet, B.; Cordelier, P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 153–168. [Google Scholar] [CrossRef]
- Reedijk, M.; Odorcic, S.; Chang, L.; Zhang, H.; Miller, N.; McCready, D.R.; Lockwood, G.; Egan, S.E. High-level coexpression of JAG1 and NOTCH1 is observed in human breast cancer and is associated with poor overall survival. Cancer Res. 2005, 65, 8530–8537. [Google Scholar] [CrossRef] [PubMed]
- Bostad, M.; Berg, K.; Hogset, A.; Skarpen, E.; Stenmark, H.; Selbo, P.K. Photochemical internalization (PCI) of immunotoxins targeting CD133 is specific and highly potent at femtomolar levels in cells with cancer stem cell properties. J. Control. Release Off. J. Control. Release Soc. 2013, 168, 317–326. [Google Scholar] [CrossRef]
- Işeri, Ö.D.; Kars, M.D.; Arpaci, F.; Atalay, C.; Pak, I.; Gündüz, U. Drug resistant MCF- 7 cells exhibit epithelial–mesenchymal transition gene expression pattern. Biomed. Pharmacother. 2011, 65, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Ahmad, A.; Li, Y.; Azmi, A.S.; Miele, L.; Sarkar, F.H. Targeting notch to eradicate pancreatic cancer stem cells for cancer therapy. Anticancer Res. 2011, 31, 1105–1113. [Google Scholar] [PubMed]
- Song, H.Y.; Wang, Y.; Lan, H.; Zhang, Y.X. Expression of Notch receptors and their ligands in pancreatic ductal adenocarcinoma. Exp. Ther. Med. 2018, 16, 53–60. [Google Scholar] [CrossRef]
- Li, Y.; Ma, J.; Qian, X.; Wu, Q.; Xia, J.; Miele, L.H.; Sarkar, F.; Wang, Z. Regulation of EMT by Notch Signaling Pathway in Tumor Progression. Curr. Cancer Drug Targ. 2013, 13, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Yabuuchi, S.; Pai, S.G.; Campbell, N.R.; De Wilde, R.F.; De Oliveira, E.; Korangath, P.; Streppel, M.M.; Rasheed, Z.A.; Hidalgo, M.; Maitra, A.; et al. Notch signaling pathway targeted therapy suppresses tumor progression and metastatic spread in pancreatic cancer. Cancer Lett. 2013, 335, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Behnsawy, H.M.; Shigemura, K.; Meligy, F.Y.; Yamamichi, F.; Yamashita, M.; Haung, W.C.; Li, X.; Miyake, H.; Tanaka, K.; Kawabata, M.; et al. Possible role of sonic hedgehog and epithelial-mesenchymal transition in renal cell cancer progression. Kor. J. Urol. 2013, 54, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Merchant, A.A.; Matsui, W. Targeting Hedgehog—a cancer stem cell pathway. Clin. Cancer Res. 2010, 16, 3130–3140. [Google Scholar] [CrossRef]
- Lei, J.; Ma, J.; Ma, Q.; Li, X.; Liu, H.; Xu, Q.; Duan, W.; Sun, Q.; Xu, J.; Wu, Z.; et al. Hedgehog signaling regulates hypoxia induced epithelial to mesenchymal transition and invasion in pancreatic cancer cells via a ligand-independent manner. Mol. Cancer 2013, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Petrova, R.; Joyner, A.L. Roles for Hedgehog signaling in adult organ homeostasis and repair. Development 2014, 141, 3445–3457. [Google Scholar] [CrossRef]
- Wong, S.Y.; Reiter, J.F. The primary cilium at the crossroads of mammalian hedgehog signaling. Curr. Top. Dev. Biol. 2008, 85, 225–260. [Google Scholar] [CrossRef] [PubMed]
- Odoux, C.; Fohrer, H.; Hoppo, T.; Guzik, L.; Stolz, D.B.; Lewis, D.W.; Gollin, S.M.; Gamblin, T.C.; Geller, D.A.; Lagasse, E. A stochastic model for cancer stem cell origin in metastatic colon cancer. Cancer Res. 2008, 68, 6932–6941. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kong, D.; Ahmad, A.; Bao, B.; Sarkar, F.H. Pancreatic cancer stem cells: Emerging target for designing novel therapy. Cancer Lett. 2013, 338, 94–100. [Google Scholar] [CrossRef]
- Hao, K.; Tian, X.D.; Qin, C.F.; Xie, X.H.; Yang, Y.M. Hedgehog signaling pathway regulates human pancreatic cancer cell proliferation and metastasis. Oncol. Rep. 2013, 29, 1124–1132. [Google Scholar] [CrossRef]
- Barakat, M.T.; Humke, E.W.; Scott, M.P. Learning from Jekyll to control Hyde: Hedgehog signaling in development and cancer. Trends Mol. Med. 2010, 16, 337–348. [Google Scholar] [CrossRef]
- Subramani, R.; Gonzalez, E.; Nandy, S.B.; Arumugam, A.; Camacho, F.; Medel, J.; Alabi, D.; Lakshmanaswamy, R. Gedunin inhibits pancreatic cancer by altering sonic hedgehog signaling pathway. Oncotarget 2017, 8, 10891. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.; Lim, K.S.; Eberhart, C.G. Arsenic trioxide inhibits Hedgehog, Notch and stem cell properties in glioblastoma neurospheres. Acta Neuropathol. Comm. 2014, 2, 31. [Google Scholar] [CrossRef]
- Schreck, K.C.; Taylor, P.; Marchionni, L.; Gopalakrishnan, V.; Bar, E.E.; Gaiano, N.; Eberhart, C.G. The Notch target Hes1 directly modulates Gli1 expression and Hedgehog signaling: A potential mechanism of therapeutic resistance. Clin. Cancer Res. 2010, 16, 6060–6070. [Google Scholar] [CrossRef] [PubMed]
- Wall, D.S.; Mears, A.J.; McNeill, B.; Mazerolle, C.; Thurig, S.; Wang, Y.; Kageyama, R.; Wallace, V.A. Progenitor cell proliferation in the retina is dependent on Notch-independent sonic hedgehog/Hes1 activity. J. Cell Biol. 2009, 184, 101–112. [Google Scholar] [CrossRef]
- Ristorcelli, E.; Lombardo, D. Targeting Notch signaling in pancreatic cancer. Exp. Opin. Ther. Targ. 2010, 14, 541–552. [Google Scholar] [CrossRef]
- Mohelnikova-Duchonova, B.; Kocik, M.; Duchonova, B.; Brynychova, V.; Oliverius, M.; Hlavsa, J.; Honsova, E.; Mazanec, J.; Kala, Z.; Ojima, I.; et al. Hedgehog pathway overexpression in pancreatic cancer is abrogated by new-generation taxoid SB-T-1216. Pharmacogenom. J. 2017, 17, 452–460. [Google Scholar] [CrossRef]
- Collins, M.A.; Bednar, F.; Zhang, Y.; Brisset, J.C.; Galban, S.; Galban, C.J.; Rakshit, S.; Flannagan, K.S.; Adsay, N.V.; di Magliano, M.P. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J. Clin. Investig. 2012, 122, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. Wnt/beta-catenin signaling in development and disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef]
- Chien, A.J.; Conrad, W.H.; Moon, R.T. A Wnt survival guide: From flies to human disease. J. Investig. Dermatol. 2009, 129, 1614–1627. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Jiang, W.; Wang, S.; Wang, L.; Xie, K. Role of Wnt/β-catenin signaling in drug resistance of pancreatic cancer. Curr. Pharm. Des. 2012, 18, 2464–2471. [Google Scholar] [CrossRef]
- Liu, S.; Dontu, G.; Wicha, M.S. Mammary stem cells, self-renewal pathways, and carcinogenesis. Breast Cancer Res. 2005, 7, 86–95. [Google Scholar] [CrossRef]
- Tiemann, K.; Heitling, U.; Kosmahl, M.; Kloppel, G. Solid pseudopapillary neoplasms of the pancreas show an interruption of the Wnt-signaling pathway and express gene products of 11q. Modern Pathol. 2007, 20, 955–960. [Google Scholar] [CrossRef]
- Sano, M.; Driscoll, D.R.; DeJesus-Monge, W.E.; Quattrochi, B.; Appleman, V.A.; Ou, J.; Zhu, L.J.; Yoshida, N.; Yamazaki, S.; Takayama, T.; et al. Activation of WNT/β-Catenin Signaling Enhances Pancreatic Cancer Development and the Malignant Potential Via Up-regulation of Cyr61. Neoplasia 2016, 18, 785–794. [Google Scholar] [CrossRef]
- White, B.D.; Chien, A.J.; Dawson, D.W. Dysregulation of Wnt/beta-catenin signaling in gastrointestinal cancers. Gastroenterology 2012, 142, 219–232. [Google Scholar] [CrossRef]
- Pires-daSilva, A.; Sommer, R.J. The evolution of signalling pathways in animal development. Nat. Rev. Genet. 2003, 4, 39–49. [Google Scholar] [CrossRef]
- Zeng, Y.A.; Nusse, R. Wnt proteins are self-renewal factors for mammary stem cells and promote their long-term expansion in culture. Cell Stem Cell 2010, 6, 568–577. [Google Scholar] [CrossRef]
- Li, X.Y.; Zhai, W.J.; Teng, C.B. Notch Signaling in Pancreatic Development. Int. J. Mol. Sci. 2016, 17, 48. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial–mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P. Epithelial–mesenchymal transitions in cancer onset and progression. Bull. Acad. Nat. Med. 2009, 193, 1969–1978. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, I.; Miele, L. Deadly crosstalk: Notch signaling at the intersection of EMT and cancer stem cells. Cancer Lett. 2013, 341, 41–45. [Google Scholar] [CrossRef]
- Potenta, S.; Zeisberg, E.; Kalluri, R. The role of endothelial-to-mesenchymal transition in cancer progression. Br. J. Cancer 2008, 99, 1375–1379. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, B.P. Epithelial-mesenchymal transition in breast cancer progression and metastasis. Chin. J. Cancer 2011, 30, 603–611. [Google Scholar] [CrossRef]
- Dudás, J.; Ladányi, A.; Ingruber, J.; Steinbichler, T.B.; Riechelmann, H. Epithelial to Mesenchymal Transition: A Mechanism that Fuels Cancer Radio/Chemoresistance. Cells 2020, 9, 428. [Google Scholar] [CrossRef]
- Ciernikova, S.; Earl, J.; García Bermejo, M.L.; Stevurkova, V.; Carrato, A.; Smolkova, B. Epigenetic Landscape in Pancreatic Ductal Adenocarcinoma: On the Way to Overcoming Drug Resistance? Int. J. Mol. Sci. 2020, 21, 4091. [Google Scholar] [CrossRef]
- Thompson, M.J.; Rubbi, L.; Dawson, D.W.; Donahue, T.R.; Pellegrini, M. Pancreatic cancer patient survival correlates with DNA methylation of pancreas development genes. PLoS ONE 2015, 10, e0128814. [Google Scholar] [CrossRef]
- Liu, Z.; Gao, Y.; Li, X. Cancer epigenetics and the potential of epigenetic drugs for treating solid tumors. Exp. Rev. Anticancer Ther. 2019, 19, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Burk, U.; Schubert, J.; Wellner, U.; Schmalhofer, O.; Vincan, E.; Spaderna, S.; Brabletz, T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008, 9, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Schmalhofer, O.; Brabletz, S.; Brabletz, T. E-cadherin, β-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. 2009, 28, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Aigner, K.; Dampier, B.; Descovich, L.; Mikula, M.; Sultan, A.; Schreiber, M.; Mikulits, W.; Brabletz, T.; Strand, D.; Obrist, P.; et al. The transcription factor ZEB1 (δ EF1) promotes tumour cell dedifferentiation by repressing master regulators of epithelial polarity. Oncogene 2007, 26, 6979–6988. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, L.; Li, A.; Han, X. The roles of ZEB1 in tumorigenic progression and epigenetic modifications. Biomed. Pharmacother. 2019, 110, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; Zur Hausen, A.; et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Chen, B.; Zhu, Z.; Ye, W.; Zeng, J.; Liu, G.; Wang, S.; Gao, J.; Xu, G.; Huang, Z. Prognostic value of ZEB-1 in solid tumors: A meta-analysis. BMC Cancer 2019, 19, 635. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.; Salmerón, C.; Wiley, S.Z.; Insel, P.A. GPCRs in pancreatic adenocarcinoma: Contributors to tumour biology and novel therapeutic targets. Br. J. Pharmacol. 2020, 177, 2434–2455. [Google Scholar] [CrossRef]
- Hauser, A.S.; Chavali, S.; Masuho, I.; Jahn, L.J.; Martemyanov, K.A.; Gloriam, D.E.; Babu, M.M. Pharmacogenomics of GPCR Drug Targets. Cell 2018, 172, 41–54. [Google Scholar] [CrossRef]
- Lappano, R.; Maggiolini, M. G protein-coupled receptors: Novel targets for drug discovery in cancer. Nat. Rev. Drug Discov. 2011, 10, 47–60. [Google Scholar] [CrossRef]
- Hothersall, J.D.; Brown, A.J.; Dale, I.; Rawlins, P. Can residence time offer a useful strategy to target agonist drugs for sustained GPCR responses? Drug Discov. Today 2016, 21, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Natale, C.A.; Li, J.; Pitarresi, J.R.; Norgard, R.J.; Dentchev, T.; Capell, B.C.; Seykora, J.T.; Stanger, B.Z.; Ridky, T.W. Pharmacologic Activation of the G Protein-Coupled Estrogen Receptor Inhibits Pancreatic Ductal Adenocarcinoma. Cell. Mol. Gastroenterol. Hepatol. 2020, 10, 868–880. [Google Scholar] [CrossRef] [PubMed]
- Chimento, A.; Sirianni, R.; Casaburi, I.; Zolea, F.; Rizza, P.; Avena, P.; Malivindi, R.; De Luca, A.; Campana, C.; Martire, E.; et al. GPER agonist G-1 decreases adrenocortical carcinoma (ACC) cell growth in vitro and in vivo. Oncotarget 2015, 6, 19190–19203. [Google Scholar] [CrossRef]
- Koppula, P.; Zhuang, L.; Gan, B. Cystine transporter SLC7A11/xCT in cancer: Ferroptosis, nutrient dependency, and cancer therapy. Protein Cell 2020, 1–22. [Google Scholar] [CrossRef]
- Daher, B.; Parks, S.K.; Durivault, J.; Cormerais, Y.; Baidarjad, H.; Tambutte, E.; Pouysségur, J.; Vučetić, M. Genetic ablation of the cystine transporter xCT in PDAC cells inhibits mTORC1, growth, survival, and tumor formation via nutrient and oxidative stresses. Cancer Res. 2019, 79, 3877–3890. [Google Scholar] [CrossRef]
- Badgley, M.A.; Kremer, D.M.; Maurer, H.C.; DelGiorno, K.E.; Lee, H.J.; Purohit, V.; Sagalovskiy, I.R.; Ma, A.; Kapilian, J.; Firl, C.E.; et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 2020, 368, 85–89. [Google Scholar] [CrossRef]
- Bannai, S.; Tsukeda, H.; Okumura, H. Effect of antioxidants on cultured human diploid fibroblasts exposed to cystine-free medium. Biochem. Biophys. Res. Commun. 1977, 74, 1582–1588. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Mazurek, S. Pyruvate kinase type M2: A key regulator of the metabolic budget system in tumor cells. Int. J. Biochem. Cell Biol. 2011, 43, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Marie, J.; Levin, M.J.; Simon, M.P.; Kahn, A. Genetic and epigenetic control of the pyruvate kinase isozymes in mammals. Isozymes 1983, 7, 221–240. [Google Scholar] [PubMed]
- Dong, G.; Mao, Q.; Xia, W.; Xu, Y.; Wang, J.; Xu, L.; Jiang, F. PKM2 and cancer: The function of PKM2 beyond glycolysis. Oncology Lett. 2016, 11, 1980–1986. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhao, Z.; Zhou, Z.; Liu, R. PKM2 promotes cell survival and invasion under metabolic stress by enhancing Warburg effect in pancreatic ductal adenocarcinoma. Digest. Dis. Sci. 2016, 61, 767–773. [Google Scholar] [CrossRef]
- James, A.D.; Richardson, D.A.; Oh, I.W.; Sritangos, P.; Attard, T.; Barrett, L.; Bruce, J.I. Cutting off the fuel supply to calcium pumps in pancreatic cancer cells: Role of pyruvate kinase-M2 (PKM2). Br. J. Cancer 2020, 122, 266–278. [Google Scholar] [CrossRef]
- James, A.D.; Patel, W.; Butt, Z.; Adiamah, M.; Dakhel, R.; Latif, A.; Uggenti, C.; Swanton, E.; Imamura, H.; Siriwardena, A.K.; et al. The plasma membrane calcium pump in pancreatic cancer cells exhibiting the Warburg effect relies on glycolytic ATP. J. Biol. Chem. 2015, 290, 24760–24771. [Google Scholar] [CrossRef]
- Yokoyama, M.; Tanuma, N.; Shibuya, R.; Shiroki, T.; Abue, M.; Yamamoto, K.; Miura, K.; Yamaguchi, K.; Sato, I.; Tamai, K.; et al. Pyruvate kinase type M2 contributes to the development of pancreatic ductal adenocarcinoma by regulating the production of metabolites and reactive oxygen species. Int. J. Oncol. 2018, 52, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.Y.; Yang, Y.C.; Wang, H.P.; Tien, Y.W.; Shun, C.T.; Huang, H.Y.; Hsiao, M.; Hua, K.T. Pyruvate kinase M2 promotes pancreatic ductal adenocarcinoma invasion and metastasis through phosphorylation and stabilization of PAK2 protein. Oncogene 2018, 37, 1730–1742. [Google Scholar] [CrossRef]
- Chen, J.; Xie, J.; Jiang, Z.; Wang, B.; Wang, Y.; Hu, X. Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2. Oncogene 2011, 30, 4297–4306. [Google Scholar] [CrossRef]
- Schenk, M.; Aykut, B.; Teske, C.; Giese, N.A.; Weitz, J.; Welsch, T. Salinomycin inhibits growth of pancreatic cancer and cancer cell migration by disruption of actin stress fiber integrity. Cancer Lett. 2015, 358, 161–169. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, Y.; Yao, S.; Cui, X.; Pan, W.; Huang, W.; Gao, J.; Dong, T.; Zhang, S. Nigericin inhibits epithelial ovarian cancer metastasis by suppressing the cell cycle and epithelial−mesenchymal transition. Biochem. Moscow 2017, 82, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, A.; Janakiram, N.B.; Brewer, M.; Ritchie, R.L.; Marya, A.; Lightfoot, S.; Steele, V.E.; Rao, C.V. Antidiabetic Drug Metformin Prevents Progression of Pancreatic Cancer by Targeting in Part Cancer Stem Cells and mTOR Signaling. Transl. Oncol. 2013, 6, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Nechushtan, H.; Hamamreh, Y.; Nidal, S.; Gotfried, M.; Baron, A.; Shalev, Y.I.; Nisman, B.; Peretz, T.; Peylan-Ramu, N. A phase IIb trial assessing the addition of disulfiram to chemotherapy for the treatment of metastatic non-small cell lung cancer. Oncologist 2015, 20, 366–367. [Google Scholar] [CrossRef]
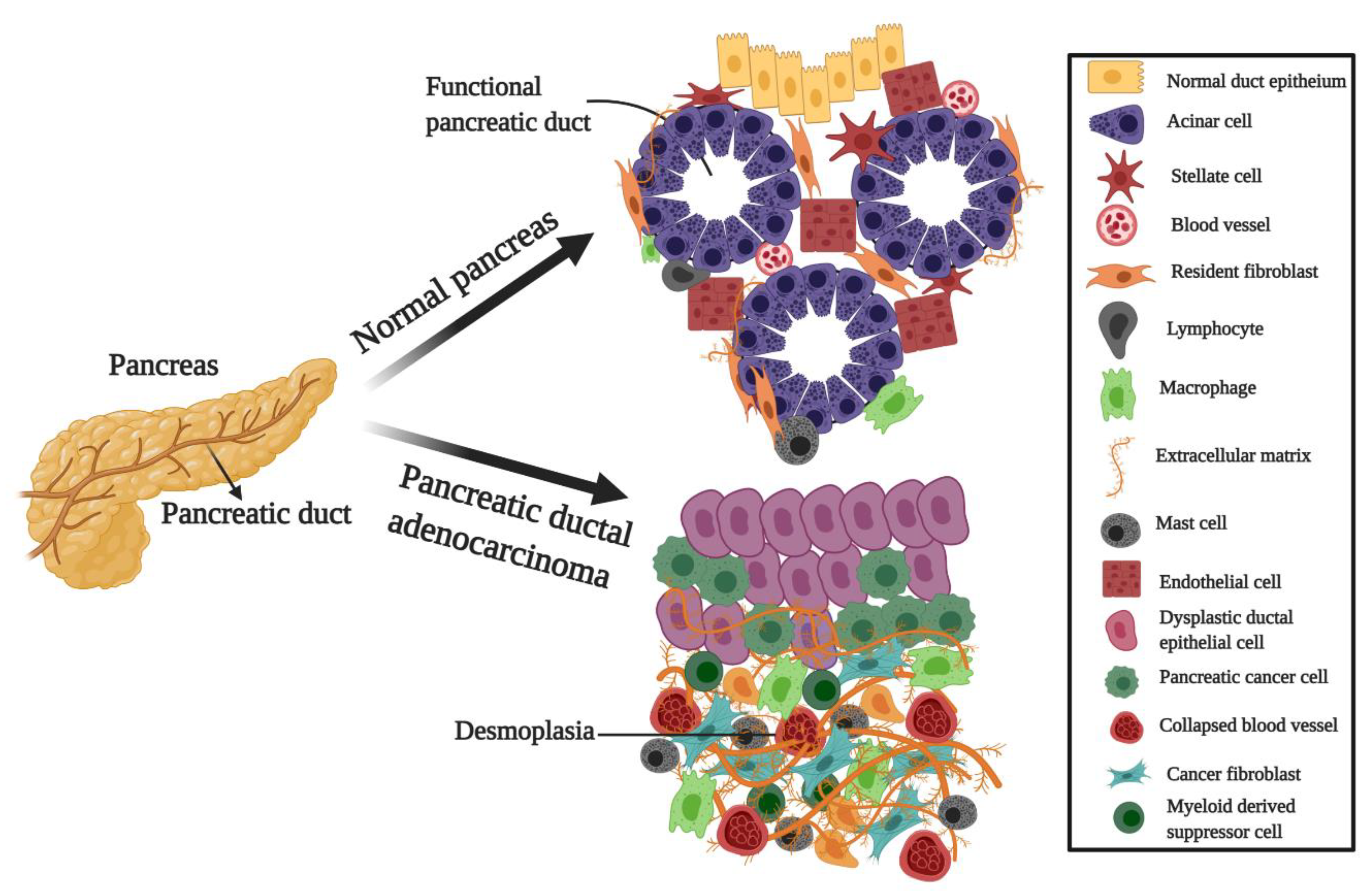
- Neesse, A.; Michl, P.; Frese, K.K.; Feig, C.; Cook, N.; Jacobetz, M.A.; Lolkema, M.P.; Buchholz, M.; Olive, K.P.; Gress, T.M.; et al. Stromal biology and therapy in pancreatic cancer. Gut 2011, 60, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, E.; Frias-Aldeguer, J.; Hermann, P.C.; Heeschen, C. Pancreatic stellate cells form a niche for cancer stem cells and promote their self-renewal and invasiveness. Cell Cycle 2012, 11, 1282–1290. [Google Scholar] [CrossRef] [PubMed]
- Wörmann, S.M.; Diakopoulos, K.N.; Lesina, M.; Algül, H. The immune network in pancreatic cancer development and progression. Oncogene 2014, 33, 2956–2967. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Washington, M.K.; Chaturvedi, R.; Drosos, Y.; Revetta, F.L.; Weaver, C.J.; Buzhardt, E.; Yull, F.E.; Blackwell, T.S.; Sosa-Pineda, B.; et al. Fibrogenesis in pancreatic cancer is a dynamic process regulated by macrophage–stellate cell interaction. Lab. Investig. 2014, 94, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Ene–Obong, A.; Clear, A.J.; Watt, J.; Wang, J.; Fatah, R.; Riches, J.C.; Marshall, J.F.; Chin–Aleong, J.; Chelala, C.; Gribben, J.G.; et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology 2013, 145, 1121–1132. [Google Scholar] [CrossRef]
- Tang, D.; Gao, J.; Wang, S.; Yuan, Z.; Ye, N.; Chong, Y.; Xu, C.; Jiang, X.; Li, B.; Yin, W.; et al. Apoptosis and anergy of T cell induced by pancreatic stellate cells-derived galectin-1 in pancreatic cancer. Tumor Biol. 2015, 36, 5617–5626. [Google Scholar] [CrossRef] [PubMed]
- Minici, C.; Rigamonti, E.; Lanzillotta, M.; Monno, A.; Rovati, L.; Maehara, T.; Kaneko, N.; Deshpande, V.; Protti, M.P.; De Monte, L.; et al. B lymphocytes contribute to stromal reaction in pancreatic ductal adenocarcinoma. Oncoimmunology 2020, 9, 1794359. [Google Scholar] [CrossRef]
- Yuen, G.J.; Demissie, E.; Pillai, S. B lymphocytes and cancer: A love–hate relationship. Trends Cancer 2016, 2, 747–757. [Google Scholar] [CrossRef]
- Le, D.T.; Jaffee, E.M. Next-generation cancer vaccine approaches: Integrating lessons learned from current successes with promising biotechnologic advances. J. Natl. Compr. Cancer Network 2013, 11, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Gibney, G.T.; Weiner, L.M.; Atkins, M.B. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016, 17, 542–551. [Google Scholar] [CrossRef]
- Zhang, J.; Wolfgang, C.L.; Zheng, L. Precision Immuno-Oncology: Prospects of Individualized Immunotherapy for Pancreatic Cancer. Cancers 2018, 10, 39. [Google Scholar] [CrossRef] [PubMed]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef] [PubMed]
- AstraZeneca. Study of Tremelimumab in Patients with Advanced Solid Tumors. 2015. Available online: https://clinicaltrials.gov/show/NCT02527434 (accessed on 20 December 2020).
- Blank, C.U.; Enk, A. Therapeutic use of anti-CTLA-4 antibodies. Int. Immunol. 2015, 27, 3–10. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Oh, D.Y.; Dhani, N.; Renouf, D.J.; Lee, M.A.; Sun, W.; Fisher, G.; Hezel, A.; Chang, S.C.; Vlahovic, G.; et al. Durvalumab With or Without Tremelimumab for Patients With Metastatic Pancreatic Ductal Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 1431–1438. [Google Scholar] [CrossRef]
- Kamath, S.D.; Kalyan, A.; Kircher, S.; Nimeiri, H.; Fought, A.J.; Benson, A.; Mulcahy, M. Ipilimumab and Gemcitabine for Advanced Pancreatic Cancer: A Phase Ib Study. Oncologist 2020, 25, 808–815. [Google Scholar] [CrossRef]
- Weiss, G.J.; Blaydorn, L.; Beck, J.; Bornemann-Kolatzki, K.; Urnovitz, H.; Schutz, E.; Khemka, V. Correction to: Phase Ib/II study of gemcitabine, nab-paclitaxel, and pembrolizumab in metastatic pancreatic adenocarcinoma. Investig. New Drugs 2019, 37, 797. [Google Scholar] [CrossRef]
- Schizas, D.; Charalampakis, N.; Kole, C.; Economopoulou, P.; Koustas, E.; Gkotsis, E.; Ziogas, D.; Psyrri, A.; Karamouzis, M.V. Immunotherapy for pancreatic cancer: A 2020 update. Cancer Treat. Rev. 2020, 86, 102016. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.M.; Santarsiero, L.M.; Lutz, E.R.; Armstrong, T.D.; Chen, Y.C.; Huang, L.Q.; Laheru, D.A.; Goggins, M.; Hruban, R.H.; Jaffee, E.M. Mesothelin-specific CD8(+) T cell responses provide evidence of in vivo cross-priming by antigen-presenting cells in vaccinated pancreatic cancer patients. J. Exp. Med. 2004, 200, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Picozzi, V.J.; Ko, A.H.; Wainberg, Z.A.; Kindler, H.; Wang-Gillam, A.; Oberstein, P.; Morse, M.A.; Zeh, H.J.; Weekes, C.; et al. Results from a Phase IIb, Randomized, Multicenter Study of GVAX Pancreas and CRS-207 Compared with Chemotherapy in Adults with Previously Treated Metastatic Pancreatic Adenocarcinoma (ECLIPSE Study). Clin. Cancer Res. 2019, 25, 5493–5502. [Google Scholar] [CrossRef]
- Le, D.T.; Lutz, E.; Uram, J.N.; Sugar, E.A.; Onners, B.; Solt, S.; Zheng, L.; Diaz, L.A.; Donehower, R.C.; Jaffee, E.M.; et al. Evaluation of ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer. J. Immunother. 2013, 36, 382–389. [Google Scholar] [CrossRef]
- Thind, K.; Padrnos, L.J.; Ramanathan, R.K.; Borad, M.J. Immunotherapy in pancreatic cancer treatment: A new frontier. Ther. Adv. Gastroenterol. 2017, 10, 168–194. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Torigian, D.A.; Chiorean, E.G.; Saboury, B.; Brothers, A.; Alavi, A.; Troxel, A.B.; Sun, W.; Teitelbaum, U.R.; Vonderheide, R.H.; et al. A phase I study of an agonist CD40 monoclonal antibody (CP- 870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2013, 19, 6286–6295. [Google Scholar] [CrossRef]
- Nywening, T.M.; Wang-Gillam, A.; Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Cusworth, B.M.; Toriola, A.T.; Nieman, R.K.; Worley, L.A.; Yano, M.; et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: A single-centre, open-label, dose-finding, nonrandomised, phase 1b trial. Lancet Oncol. 2016, 17, 651–662. [Google Scholar] [CrossRef]
- Chiossone, L.; Dumas, P.Y.; Vienne, M.; Vivier, E. Natural killer cells and other innate lymphoid cells in cancer. Nat. Rev. Immunol. 2018, 18, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Van Audenaerde, J.; Roeyen, G.; Darcy, P.K.; Kershaw, M.H.; Peeters, M.; Smits, E. Natural killer cells and their therapeutic role in pancreatic cancer: A systematic review. Pharmacol. Ther. 2018, 189, 31–44. [Google Scholar] [CrossRef]
- Lee, J.; Kang, T.H.; Yoo, W.; Choi, H.; Jo, S.; Kong, K.; Lee, S.R.; Kim, S.U.; Kim, J.S.; Cho, D.; et al. An antibody designed to improve adoptive NK-cell therapy inhibits pancreatic cancer progression in a murine model. Cancer Immunol. Res. 2019, 7, 219–229. [Google Scholar] [CrossRef]
- Goedegebuure, P.; Mitchem, J.B.; Porembka, M.R.; Tan, M.C.; Belt, B.A.; WangGillam, A.E.; Gillanders, W.G.; Hawkins, W.C.; Linehan, D. Myeloid-derived suppressor cells: General characteristics and relevance to clinical management of pancreatic cancer. Curr. Cancer Drug Targ. 2011, 11, 734–751. [Google Scholar] [CrossRef] [PubMed]
- D’Alterio, C.; Scala, S.; Sozzi, G.; Roz, L.; Bertolini, G. Paradoxical effects of chemotherapy on tumor relapse and metastasis promotion. Semin. Cancer Biol. 2020, 60, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Won, M.; Kim, J.H.; Jung, E.; Min, K.; Jangili, P.; Kim, J.S. Cancer stem cell-targeted bio-imaging and chemotherapeutic perspective. Chem. Soc. Rev. 2020, 49, 7856–7878. [Google Scholar] [CrossRef] [PubMed]
- Aponte, P.M.; Caicedo, A. Stemness in cancer: Stem cells, cancer stem cells, and their microenvironment. Stem Cells Int. 2017. [Google Scholar] [CrossRef]
- Razidlo, G.L.; Magnine, C.; Sletten, A.C.; Hurley, R.M.; Almada, L.L.; Fernandez-Zapico, M.E.; Ji, B.; McNiven, M.A. Targeting pancreatic cancer metastasis by inhibition of Vav1, a driver of tumor cell invasion. Cancer Res. 2015, 75, 2907–2915. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Drug | Structure | Pathway inVolved | Mechanism of Action | Accession Number | References | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Salinomycin |  | WNT/EMT | Inhibits the growth of CSCs | DB11544 | [42] | |||||
| Nigericin |  | EMT | Inhibit the cell viability | DB14056 | [43] | |||||
| Azithromycin |  | Mitochondria | Inhibiting protein synthesis and translation | DB00207 | [44] | |||||
| Chloroquine |  | OXPHOS | Inhibits the autophagy pathway | DB00608 | [45] | |||||
| Aspirin |  | ALDH1, NF-κB | Blocks prostaglandin synthesis | DB00945 | [46] | |||||
| Disulfiram |  | WNT, β-catenin and NF-κB | Induce apoptosis in cancer stem cells | DB00822 | [47] | |||||
| Aprepitant |  | WNT | Inhibit emesis induced by cytotoxic chemotherapeutic agents | DB00673 | [48] | |||||
| Atovaquone |  | HER2/β-catenin | Antiprotozoal, antimicrobial and antipneumocystis activity | DB01117 | [49] | |||||
| AZD8055 |  | mTOR | Activation of epidermal growth factor receptor | DB12774 | [50] | |||||
| Crocetinic acid |  | Hedgehog | Inhibited epidermal growth factor receptor and Akt phosphorylation | NA | [51] | |||||
| GANT-61 |  | Hedgehog | Inhibited cell viability. protects autophagy | NA | [52] | |||||
| Ketamine |  | WNT | Inhibiting proliferation, invasion, and migration | DB01221 | [53] | |||||
| Metformin |  | mTOR, PI3K/Akt | Antineoplastic activity | DB00331 | [54] | |||||
| Quinomycin A |  | Notch | Suppresses CSCs growth | DB15582 | [55] | |||||
| Rapamycin |  | mTOR | Reduced the viability of CSCs | DB00877 | [56] | |||||
| RO-4929097 |  | Notch | Suppresses the tumor initiating potential of cancer cells | DB11870 | [57] | |||||
| Sanguinarine |  | Hedgehog | Inhibits the growth of CSCs | NA | [58] | |||||
| Tigecycline |  | OXPHOS | Inhibits cell proliferation, migration and invasion | DB00560 | [59] | |||||
| 5-FU (Fluorouracil Injection) |  | Antineoplastic antimetabolite | Inhibition of the formation of thymidylate from uracil | DB00544 | [60] | |||||
| Mitomycin |  | Antineoplastic antibiotic | Alkylating agent which inhibits DNA synthesis | DB00305 | [60] | |||||
| Abraxane/Paclitaxel |  | Microtubule associated protein | Stabilizes microtubules by preventing depolymerization | DB01229 | [61] | |||||
| Gemcitabine Hydrochloride/Gemzar |  | Antineoplastic anti-metabolite | Inhibits thymidylate synthetase | DB00441 | [61] | |||||
| Afinitor/Everolimus |  | mTOR inhibition | Immunosuppressant | DB01590 | [62] | |||||
| Erlotinib Hydrochloride/Tarceva |  | Epidermal growth factor receptor (EGFR) | Inhibits the intracellular phosphorylation of tyrosine kinase | DB00530 | [63] | |||||
| Lynparza/Olaparib |  | Poly (ADP-ribose) polymerase (PARP) inhibitor | Inhibit growth of tumor cells | DB09074 | [64] | |||||
| Onivyde/Irinotecan |  | Antineoplastic enzyme inhibitor | Inhibits the action of topoisomerase I | DB00762 | [65] | |||||
| Sunitinib Malate/Sutent |  | Multi-targeted receptor tyrosine kinase (RTK) inhibitor | Inhibits cellular signaling by targeting multiple RTKs | DB01268 | [66] | |||||
| Drug | Accession Number | Function | Mechanism of Action |
|---|---|---|---|
| FOLFIRINOX | |||
| FOL: Folinic acid/Leucovorin | DB00650 | Antidote | Enhances the effects of 5-fluorouracil |
| F: Fluorouracil | DB00544 | Pyrimidine analog and antimetabolite | Inhibit DNA synthesis |
| IRIN: Irinotecan/Camptosar | DB00762 | Topoisomerase inhibitor | Prevents DNA from uncoiling and duplicating |
| OX: Oxaliplatin/Eloxatin | DB00526 | Platinum-based antineoplastic agent | Inhibits DNA repair and synthesis |
| GEMCITABINE-OXALIPLATIN | |||
| Gemcitabine | DB00441 | Antineoplastic anti-metabolite | Inhibits thymidylate synthetase |
| Oxaliplatin | DB00526 | Platinum-based antineoplastic agent | Inhibits DNA repair and synthesis |
| GEMCITABINE-CISPLATIN | |||
| Gemcitabine | DB00441 | Antineoplastic anti-metabolite | Inhibits thymidylate Synthetase |
| Cisplatin | DB00515 | Antineoplastic | Alkylating agents |
| OFF | |||
| O: Oxaliplatin | DB00526 | Platinum-based antineoplastic agent | Inhibits DNA repair and synthesis |
| F: Fluorouracil | DB00544 | Antineoplastic antimetabolite | Inhibition of the formation of thymidylate from uracil |
| F: Folinic Acid/Leucovorin | DB00650 | Antidote | Enhances the effects of 5-fluorouracil |
| Pathological Condition | Enrolled Patients | Intervention | National Clinical Trial Number | Outcome Measures | Phase | Status | Result |
|---|---|---|---|---|---|---|---|
| Neoplasms, Pancreas | 40 | Cancer stem cell vaccine | NCT02074046 | Determine the safety of immunization | Phase 1/2 | Completed | CTLs harvested from CSC-vaccinated hosts were capable of killing CSCs in vitro |
| Metastatic pancreatic cancer | 98 | Gemcitabine, Nab-Paclitaxel, GDC-0449 | NCT01088815 | Progression free survival, safety of combination therapy | Phase 2 | Completed | Median progression-free survival and overall survival were 5.42 months and 9.79 months, respectively |
| Metastatic pancreatic adenocarcinoma | 139 | BBI608 either in combination with Gemcitabine and nab-Paclitaxel, mFOLFIRINOX, FOLFIRI, or MM-398 with 5-FU and Leucovorin | NCT02231723 | Safety, Adverse effects | Phase 1 | Completed | Inhibit cancer stemness pathways, including Nanog, by targeting stemness kinases. |
| Metastatic Pancreatic Ductal Adenocarcinoma | 65 | MEDI4736 Monotherapy, Tremelimumab + MEDI4736 | NCT02558894 | Response Rate, Overall survival, progression free survival, | Phase 2 | Completed | Monotherapy reflected a population of patients with mPDAC who had poor prognoses and rapidly progressing disease |
| PDAC, Pancreatic Cancer | 21 | Ipilimumab, Gemcitabine hydrochloride | NCT01473940 | Overall survival, progression free survival, recovery of tumor immune surveillance | Phase 1 | Completed | Median progression-free and overall survival were 2.78 months and 6.90 months, respectively. |
| Second-line, third-line and Greater Metastatic Pancreatic Cancer | 303 | GVAX Pancreas Vaccine, CRS-207, Chemotherapy, Cyclophosphamide | NCT02004262 | Overall survival and adverse effects | Phase 2 | Completed | Median overall survival in the primary cohort was 3.7, 5.4, and 4.6 months in arms A, B, and C, respectively (*) |
| Pancreatic Neoplasm | 22 | Monoclonal antibody, chemotherapy | NCT00711191 | Overall survival, progression free survival, and time to Progression | Phase 1 | Completed | Well tolerated and associated with antitumor activity in patients with PDAC and improved overall survival |
| Pancreatic Adenocarcinoma metastatic | 10 | Melphalan, BCNU, Vitamin B12, Vitamin C, and autologous hematopoietic stem cell | NCT04150042 | Response rate in metastatic lesions, overall survival, progression free survival | Phase 1 | Ongoing | NA |
| Resectable pancreatic adenocarcinoma | 42 | HIPEC-Gemcitabine | NCT03251365 | Morbidity, survival | Phase 2/3 | Ongoing | NA |
| PDAC, pancreatic cancer, metastasis | 36 | Ascorbic acid, Paclitaxel, Cisplatin, Gemcitabine | NCT03410030 | Determination of preliminary efficacy | Phase 1/2 | Ongoing | NA |
| Pancreatic Cancer | 81 | Pembrolizumab, Gemcitabine, Docetaxel, Nab-paclitaxel, Vinorelbine, Irinotecan, Liposomal Doxorubicin | NCT02331251 | Determine the recommended phase 2 dose | Phase 1/2 | Terminated | The median progression-free survival and overall survival was 9.1 and 15.0 months, respectively |
| Pancreatic Cancer | 15 | Fludarabine, Anti-mesothelin chimeric T cell receptor (CAR) transduced peripheral blood lymphocytes (PBL), Cyclophosphamide, Aldesleukin | NCT01583686 | Tumor regression response and adverse effects | Phase 1/2 | Terminated | MORAb-009 (chimeric monoclonal antibody) is well tolerated |
| Pancreatic adenocarcinoma | 10 | Allogeneic hematopoietic stem cell transplantation | NCT02207985 | Disease free survival | Phase 1/2 | Unknown | Patients are tumor-free for 9 years after diagnosis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thakur, G.; Kumar, R.; Kim, S.-B.; Lee, S.-Y.; Lee, S.-L.; Rho, G.-J. Therapeutic Status and Available Strategies in Pancreatic Ductal Adenocarcinoma. Biomedicines 2021, 9, 178. https://doi.org/10.3390/biomedicines9020178
Thakur G, Kumar R, Kim S-B, Lee S-Y, Lee S-L, Rho G-J. Therapeutic Status and Available Strategies in Pancreatic Ductal Adenocarcinoma. Biomedicines. 2021; 9(2):178. https://doi.org/10.3390/biomedicines9020178
Chicago/Turabian StyleThakur, Gitika, Raj Kumar, Saet-Byul Kim, Sang-Yeob Lee, Sung-Lim Lee, and Gyu-Jin Rho. 2021. "Therapeutic Status and Available Strategies in Pancreatic Ductal Adenocarcinoma" Biomedicines 9, no. 2: 178. https://doi.org/10.3390/biomedicines9020178
APA StyleThakur, G., Kumar, R., Kim, S.-B., Lee, S.-Y., Lee, S.-L., & Rho, G.-J. (2021). Therapeutic Status and Available Strategies in Pancreatic Ductal Adenocarcinoma. Biomedicines, 9(2), 178. https://doi.org/10.3390/biomedicines9020178