The Structure, Activation and Signaling of IRE1 and Its Role in Determining Cell Fate

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
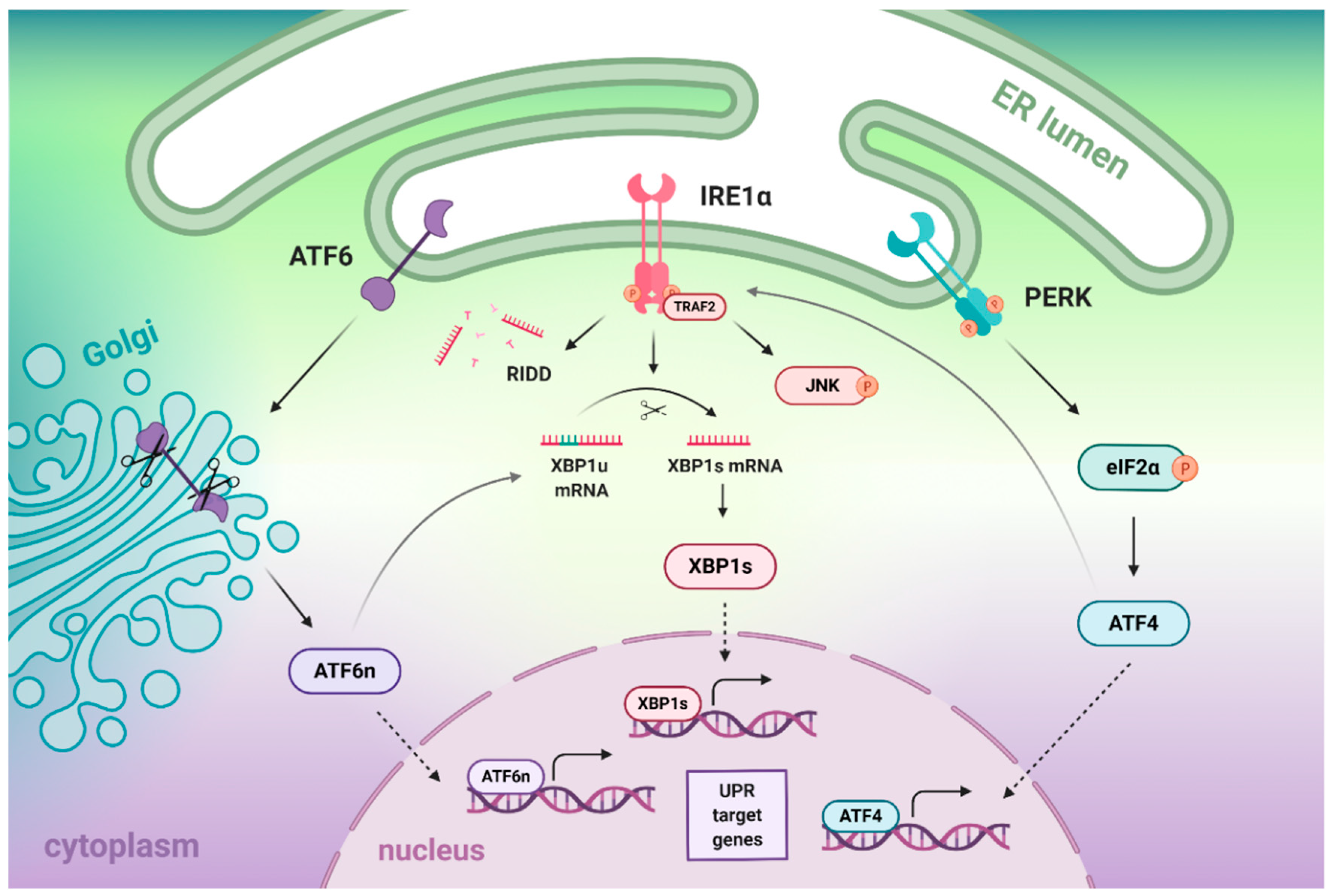
2. The Crosstalk between IRE1α, ATF6, and PERK Branches of the UPR
2.1. Crosstalk between IRE1- and ATF6-Dependent Signaling Branches
2.2. Crosstalk between IRE1- and PERK-Dependent Signaling Pathways
2.3. Coronavirus-Mediated Activation of the UPR Signaling Pathways and Their Possible Crosstalk
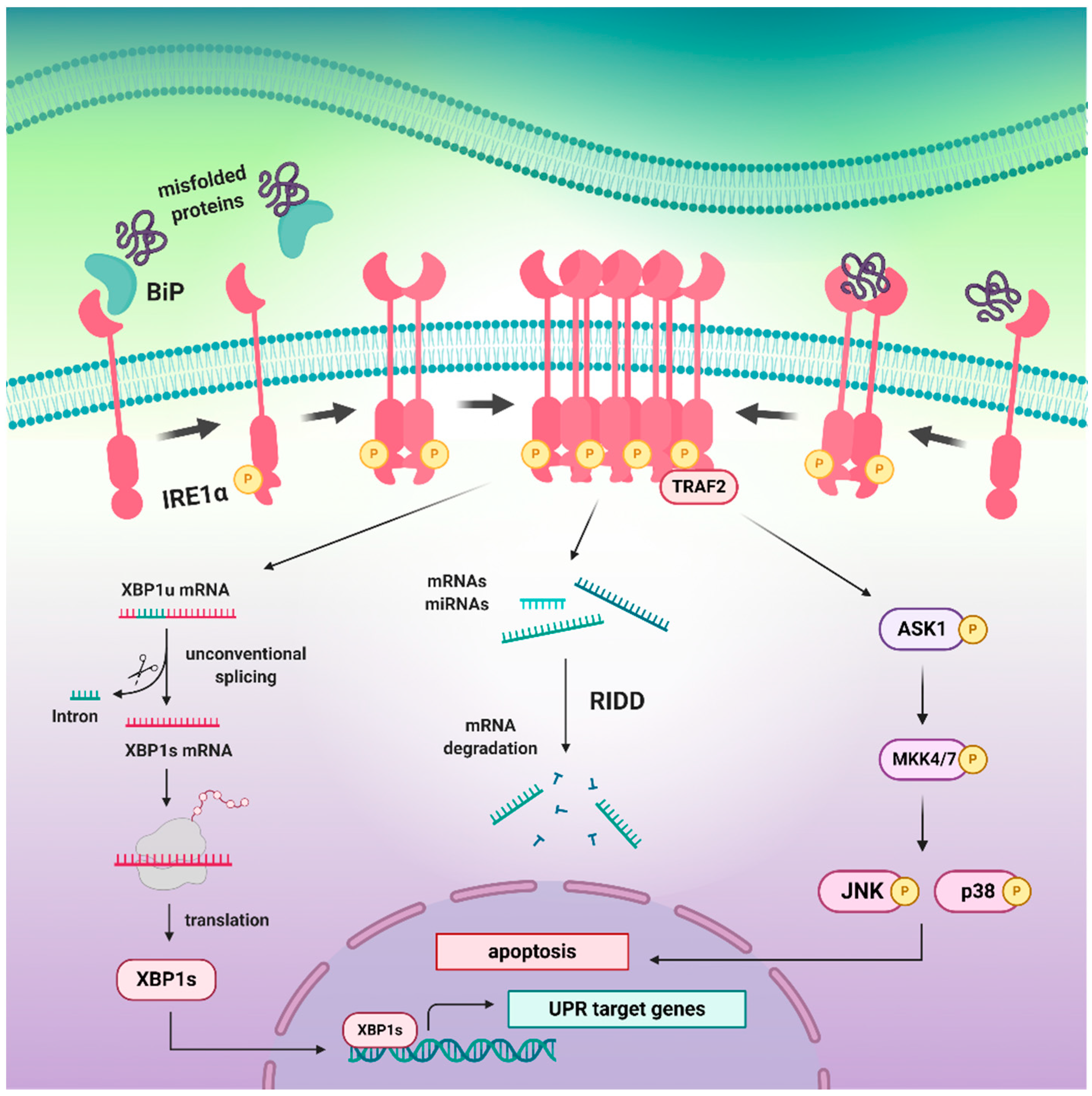
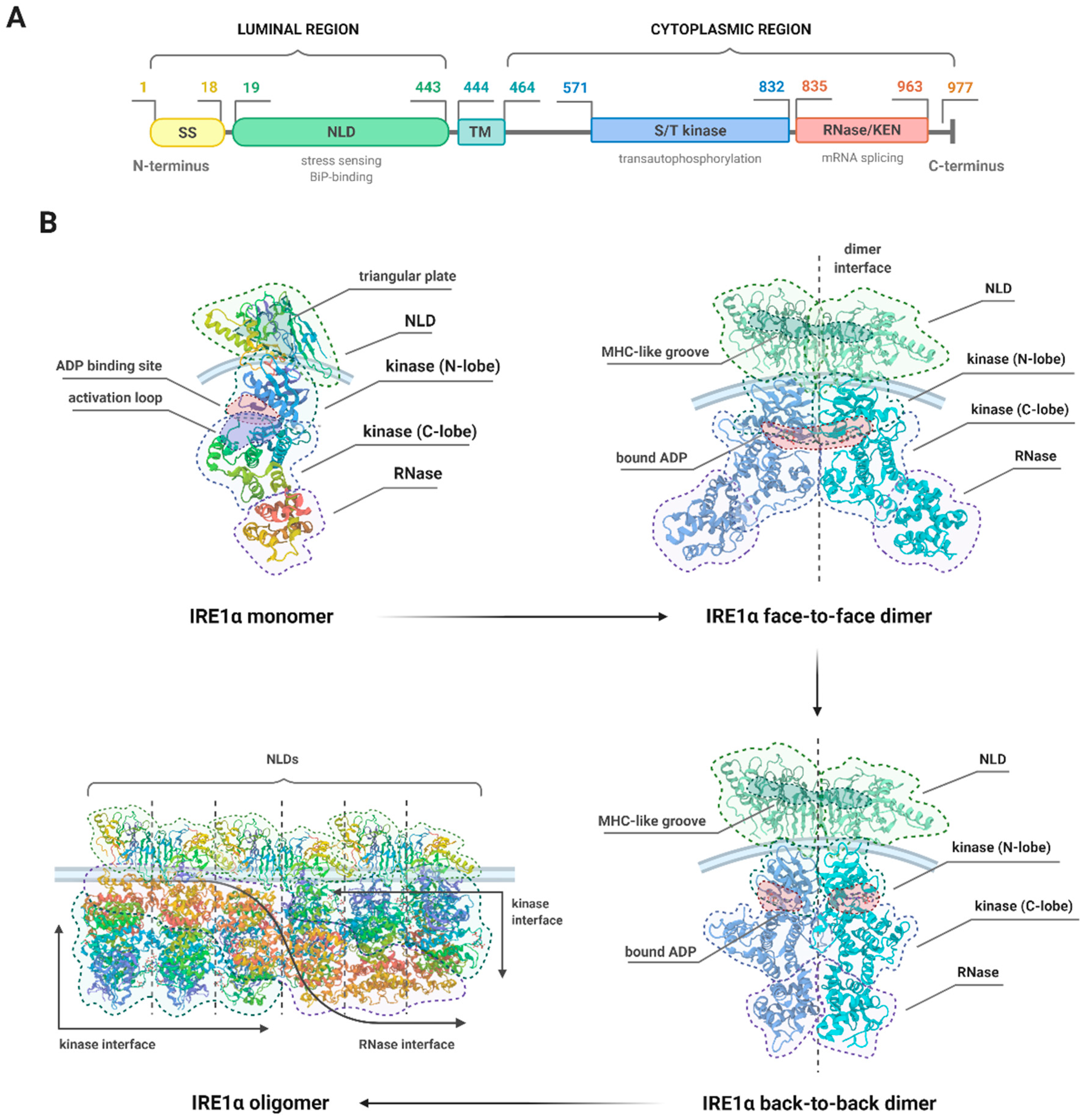
3. The Structure of IRE1α and Molecular Events Associated with Its Activation
3.1. The Inactive and Active States of the IRE1 upon Distinct Molecular Events
3.2. Details of the Structure of IRE1
3.3. Structural Changes and the Different Activities of IRE1
3.4. The Pro-Survival XBP1 Pathway Restores Cellular Proteostasis
3.5. Dual Role of RIDD Pathway in Cell Fate Regulation
3.6. The IRE1α/TRAF2/JNK Arm Initiates the Pro-Apoptotic Signals
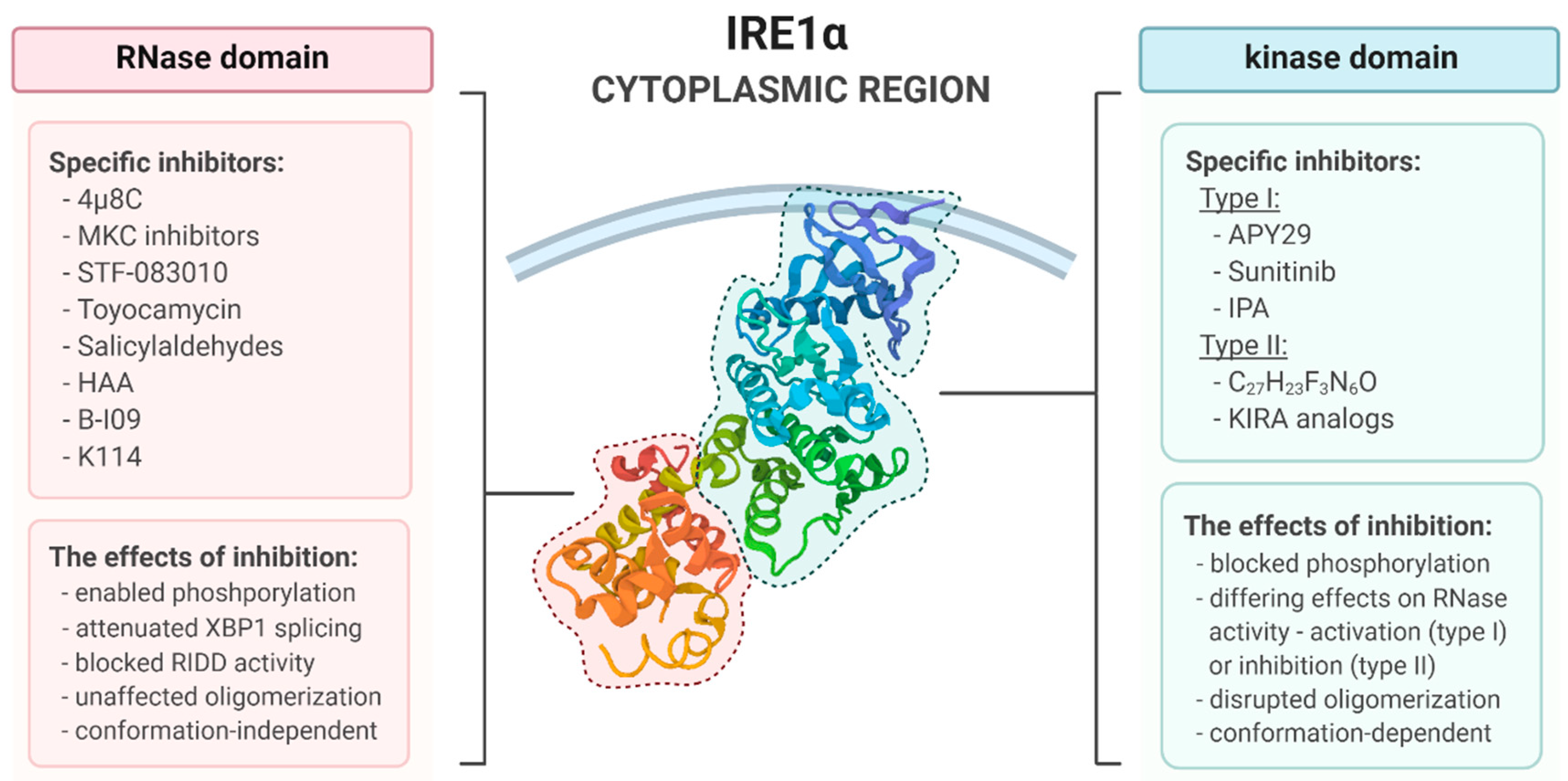
4. Pharmacological Modulation of IRE1 Activity
4.1. The Kinase Type I Inhbitors
4.1.1. APY29
4.1.2. Sunitinib
4.1.3. IPA
4.2. The Kinase Type II Inhbitors
4.2.1. C27H23F3N6O
4.2.2. KIRA Analogs
4.3. RNase Inhbitors
4.3.1. Salicylaldehydes
4.3.2. 4μ8C
4.3.3. STF-083010
4.3.4. Toyocamycin
4.3.5. MKC Inhibitors
4.3.6. HAA
4.3.7. B-I09
4.3.8. K114
4.4. Other Inhibitors Specific for IRE1
4.5. IRE1 Activators
4.5.1. Quercetin
4.5.2. NM-PP1
4.5.3. Other Activators of IRE1 Function
5. Summary and Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Sun, Z.; Brodsky, J.L. Protein quality control in the secretory pathway. J. Cell Biol. 2019, 218, 3171–3187. [Google Scholar] [CrossRef] [PubMed]
- Chino, H.; Mizushima, N. ER-Phagy: Quality Control and Turnover of Endoplasmic Reticulum. Trends Cell Biol. 2020, 30, 384–398. [Google Scholar] [CrossRef] [PubMed]
- Oakes, S.A.; Papa, F.R. The role of endoplasmic reticulum stress in human pathology. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 173–194. [Google Scholar] [CrossRef] [PubMed]
- Hiramatsu, N.; Chiang, W.C.; Kurt, T.D.; Sigurdson, C.J.; Lin, J.H. Multiple Mechanisms of Unfolded Protein Response-Induced Cell Death. Am. J. Pathol. 2015, 185, 1800–1808. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Li, H.; Zhang, Y.; Ron, D.; Walter, P. Divergent effects of PERK and IRE1 signaling on cell viability. PLoS ONE 2009, 4, e4170. [Google Scholar] [CrossRef] [PubMed]
- Tavernier, Q.; Bennana, E.; Poindessous, V.; Schaeffer, C.; Rampoldi, L.; Pietrancosta, N.; Pallet, N. Regulation of IRE1 RNase activity by the Ribonuclease inhibitor 1 (RNH1). Cell Cycle 2018, 17, 1901–1916. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Alvear, D.; Karagöz, G.E.; Fröhlich, F.; Li, H.; Walther, T.C.; Walter, P. The unfolded protein response and endoplasmic reticulum protein targeting machineries converge on the stress sensor IRE1. Elife 2018, 7. [Google Scholar] [CrossRef]
- Poothong, J.; Sopha, P.; Kaufman, R.J.; Tirasophon, W. Domain compatibility in Ire1 kinase is critical for the unfolded protein response. FEBS Lett. 2010, 584, 3203–3208. [Google Scholar] [CrossRef] [PubMed]
- Belyy, V.; Tran, N.H.; Walter, P. Quantitative microscopy reveals dynamics and fate of clustered IRE1α. Proc. Natl. Acad. Sci. USA 2020, 117, 1533–1542. [Google Scholar] [CrossRef]
- Tavernier, S.J.; Osorio, F.; Vandersarren, L.; Vetters, J.; Vanlangenakker, N.; Van Isterdael, G.; Vergote, K.; De Rycke, R.; Parthoens, E.; Van De Laar, L.; et al. Regulated IRE1-dependent mRNA decay sets the threshold for dendritic cell survival. Nat. Cell Biol. 2017, 19, 698–710. [Google Scholar] [CrossRef]
- Sozen, E.; Yazgan, B.; Tok, O.E.; Demirel, T.; Ercan, F.; Proto, J.D.; Ozer, N.K. Cholesterol induced autophagy via IRE1/JNK pathway promotes autophagic cell death in heart tissue. Metabolism 2020, 106, 154205. [Google Scholar] [CrossRef] [PubMed]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, R.B.; Bennett, B.S.; Cullinan, S.B.; Diehl, J.A. PERK and GCN2 contribute to eIF2α phosphorylation and cell cycle arrest after activation of the unfolded protein response pathway. Mol. Biol. Cell 2005, 16, 5493–5501. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, S.J.; Garcia-Bonilla, L.; Hu, J.; Harding, H.P.; Ron, D. Activation-dependent substrate recruitment by the eukaryotic translation initiation factor 2 kinase PERK. J. Cell Biol. 2006, 172, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Su, N.; Kilberg, M.S. C/EBP homology protein (CHOP) interacts with activating transcription factor 4 (ATF4) and negatively regulates the stress-dependent induction of the asparagine synthetase gene. J. Biol. Chem. 2008, 283, 35106–35117. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Davé, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Lee, A.-H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 Regulates a Subset of Endoplasmic Reticulum Resident Chaperone Genes in the Unfolded Protein Response. Mol. Cell. Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef]
- Huang, H.W.; Zeng, X.; Rhim, T.; Ron, D.; Ryoo, H.D. The requirement of IRE1 and XBP1 in resolving physiological stress during Drosophila development. J. Cell Sci. 2017, 130, 3040–3049. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Saito, M.; Kadokura, H.; Miyazaki, J.I.; Tashiro, F.; Imagawa, Y.; Iwawaki, T.; Kohno, K. IRE1-XBP1 pathway regulates oxidative proinsulin folding in pancreatic β cells. J. Cell Biol. 2018, 217, 1287–1301. [Google Scholar] [CrossRef]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef]
- Chopra, S.; Giovanelli, P.; Alvarado-Vazquez, P.A.; Alonso, S.; Song, M.; Sandoval, T.A.; Chae, C.S.; Tan, C.; Fonseca, M.M.; Gutierrez, S.; et al. IRE1α-XBP1 signaling in leukocytes controls prostaglandin biosynthesis and pain. Science 2019, 365. [Google Scholar] [CrossRef]
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491. [Google Scholar] [CrossRef]
- Zhao, N.; Cao, J.; Xu, L.; Tang, Q.; Dobrolecki, L.E.; Lv, X.; Talukdar, M.; Lu, Y.; Wang, X.; Hu, D.Z.; et al. Pharmacological targeting of MYC-regulated IRE1/XBP1 pathway suppresses MYC-driven breast cancer. J. Clin. Investig. 2018, 128, 1283–1299. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Jiang, M.; Chen, W.; Zhao, T.; Wei, Y. Cancer and ER stress: Mutual crosstalk between autophagy, oxidative stress and inflammatory response. Biomed. Pharmacother. 2019, 118, 109249. [Google Scholar] [CrossRef] [PubMed]
- Rozpędek-Kamińska, W.; Siwecka, N.; Wawrzynkiewicz, A.; Wojtczak, R.; Pytel, D.; Diehl, J.A.; Majsterek, I. The PERK-Dependent Molecular Mechanisms as a Novel Therapeutic Target for Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 2108. [Google Scholar] [CrossRef] [PubMed]
- Xiang, C.; Wang, Y.; Zhang, H.; Han, F. The role of endoplasmic reticulum stress in neurodegenerative disease. Apoptosis 2017, 22, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Szpigel, A.; Hainault, I.; Carlier, A.; Venteclef, N.; Batto, A.F.; Hajduch, E.; Bernard, C.; Ktorza, A.; Gautier, J.F.; Ferré, P.; et al. Lipid environment induces ER stress, TXNIP expression and inflammation in immune cells of individuals with type 2 diabetes. Diabetologia 2018, 61, 399–412. [Google Scholar] [CrossRef]
- Jin, B.; Ishikawa, T.; Taniguchi, M.; Ninagawa, S.; Okada, T.; Kagaya, S.; Mori, K. Development of a Rapid in vivo Assay to Evaluate the Efficacy of IRE1-specific Inhibitors of the Unfolded Protein Response Using Medaka Fish. Cell Struct. Funct. 2019, 45, 23–31. [Google Scholar] [CrossRef]
- Rosen, D.A.; Seki, S.M.; Fernández-Castañeda, A.; Beiter, R.M.; Eccles, J.D.; Woodfolk, J.A.; Gaultier, A. Modulation of the sigma-1 receptor-IRE1 pathway is beneficial in preclinical models of inflammation and sepsis. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Tufanli, O.; Akillilar, P.T.; Acosta-Alvear, D.; Kocaturk, B.; Onat, U.I.; Hamid, S.M.; Çimen, I.; Walter, P.; Weber, C.; Erbay, E. Targeting IRE1 with small molecules counteracts progression of atherosclerosis. Proc. Natl. Acad. Sci. USA 2017, 114, E1395–E1404. [Google Scholar] [CrossRef]
- Doultsinos, D.; Carlesso, A.; Chintha, C.; Paton, J.C.; Paton, A.W.; Samali, A.; Chevet, E.; Eriksson, L.A. Peptidomimetic-based identification of FDA-approved compounds inhibiting IRE1 activity. FEBS J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Adams, C.J.; Kopp, M.C.; Larburu, N.; Nowak, P.R.; Ali, M.M.U. Structure and molecular mechanism of ER stress signaling by the unfolded protein response signal activator IRE1. Front. Mol. Biosci. 2019, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; He, G.T.; Zhang, W.J.; Xu, J.; Huang, Q.B. IRE1α signaling pathways involved in mammalian cell fate determination. Cell. Physiol. Biochem. 2016, 38, 847–858. [Google Scholar] [CrossRef] [PubMed]
- Shemorry, A.; Harnoss, J.M.; Guttman, O.; Marsters, S.A.; Kőműves, L.G.; Lawrence, D.A.; Ashkenazi, A. Caspase-mediated cleavage of IRE1 controls apoptotic cell commitment during endoplasmic reticulum stress. Elife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Tsuru, A.; Imai, Y.; Saito, M.; Kohno, K. Novel mechanism of enhancing IRE1α-XBP1 signalling via the PERK-ATF4 pathway. Sci. Rep. 2016, 6, 24217. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Tirasophon, W.; Shen, X.; Michalak, M.; Prywes, R.; Okada, T.; Yoshida, H.; Mori, K.; Kaufman, R.J. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 2002, 16, 452–466. [Google Scholar] [CrossRef]
- Hampton, R.Y. IRE1: A role in UPREgulation of ER degradation. Dev. Cell 2003, 4, 144–146. [Google Scholar] [CrossRef]
- Walter, F.; O’Brien, A.; Concannon, C.G.; Düssmann, H.; Prehn, J.H.M. ER stress signaling has an activating transcription factor 6 (ATF6)-dependent “off-switch”. J. Biol. Chem. 2018, 293, 18270–18284. [Google Scholar] [CrossRef]
- Saraswat Ohri, S.; Howard, R.M.; Liu, Y.; Andres, K.R.; Shepard, C.T.; Hetman, M.; Whittemore, S.R. Oligodendrocyte-specific deletion of Xbp1 exacerbates the endoplasmic reticulum stress response and restricts locomotor recovery after thoracic spinal cord injury. Glia 2020, 69. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Li, H.; Yasumura, D.; Cohen, H.R.; Zhang, C.; Panning, B.; Shokat, K.M.; LaVail, M.M.; Walter, P. IRE1 signaling affects cell fate during the unfolded protein response. Science 2007, 318, 944–949. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.K.; Lawrence, D.A.; Lu, M.; Tan, J.; Harnoss, J.M.; Marsters, S.A.; Liu, P.; Sandoval, W.; Martin, S.E.; Ashkenazi, A. Coordination between Two Branches of the Unfolded Protein Response Determines Apoptotic Cell Fate. Mol. Cell 2018, 71, 629–636.e5. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.; Hollien, J. Ire1-mediated decay in mammalian cells relies on mRNA sequence, structure, and translational status. Mol. Biol. Cell 2015, 26, 2873–2884. [Google Scholar] [CrossRef] [PubMed]
- Walter, F.; Schmid, J.; Dussmann, H.; Concannon, C.G.; Prehn, J.H.M. Imaging of single cell responses to ER stress indicates that the relative dynamics of IRE1/XBP1 and PERK/ATF4 signalling rather than a switch between signalling branches determine cell survival. Cell Death Differ. 2015, 22, 1502–1516. [Google Scholar] [CrossRef]
- Sureda, A.; Alizadeh, J.; Nabavi, S.F.; Berindan-Neagoe, I.; Cismaru, C.A.; Jeandet, P.; Łos, M.J.; Clementi, E.; Nabavi, S.M.; Ghavami, S. Endoplasmic reticulum as a potential therapeutic target for covid-19 infection management? Eur. J. Pharmacol. 2020, 882. [Google Scholar] [CrossRef]
- Versteeg, G.A.; van de Nes, P.S.; Bredenbeek, P.J.; Spaan, W.J.M. The Coronavirus Spike Protein Induces Endoplasmic Reticulum Stress and Upregulation of Intracellular Chemokine mRNA Concentrations. J. Virol. 2007, 81, 10981–10990. [Google Scholar] [CrossRef]
- Bechill, J.; Chen, Z.; Brewer, J.W.; Baker, S.C. Coronavirus Infection Modulates the Unfolded Protein Response and Mediates Sustained Translational Repression. J. Virol. 2008, 82, 4492–4501. [Google Scholar] [CrossRef]
- Chan, C.-P.; Siu, K.-L.; Chin, K.-T.; Yuen, K.-Y.; Zheng, B.; Jin, D.-Y. Modulation of the Unfolded Protein Response by the Severe Acute Respiratory Syndrome Coronavirus Spike Protein. J. Virol. 2006, 80, 9279–9287. [Google Scholar] [CrossRef]
- Sicari, D.; Chatziioannou, A.; Koutsandreas, T.; Sitia, R.; Chevet, E. Role of the early secretory pathway in SARS-CoV-2 Infection. J. Cell Biol. 2020, 219, e202006005. [Google Scholar] [CrossRef]
- Fung, T.S.; Liu, D.X. The ER stress sensor IRE1 and MAP kinase ERK modulate autophagy induction in cells infected with coronavirus infectious bronchitis virus. Virology 2019, 533, 34–44. [Google Scholar] [CrossRef]
- Fung, T.S.; Liao, Y.; Liu, D.X. The Endoplasmic Reticulum Stress Sensor IRE1 Protects Cells from Apoptosis Induced by the Coronavirus Infectious Bronchitis Virus. J. Virol. 2014, 88, 12752–12764. [Google Scholar] [CrossRef] [PubMed]
- Amin-Wetzel, N.; Saunders, R.A.; Kamphuis, M.J.; Rato, C.; Preissler, S.; Harding, H.P.; Ron, D. A J-Protein Co-chaperone Recruits BiP to Monomerize IRE1 and Repress the Unfolded Protein Response. Cell 2017, 171, 1625–1637.e13. [Google Scholar] [CrossRef] [PubMed]
- Amin-Wetzel, N.; Neidhardt, L.; Yan, Y.; Mayer, M.P.; Ron, D. Unstructured regions in IRE1α specify BiP-mediated destabilisation of the luminal domain dimer and repression of the UPR. Elife 2019, 8, e50793. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation; American Association for the Advancement of Science: Washington, DC, USA, 2011; Volume 334, pp. 1081–1086. [Google Scholar]
- Kopp, M.C.; Nowak, P.R.; Larburu, N.; Adams, C.J.; Ali, M.M.U. In vitro FRET analysis of IRE1 and BiP association and dissociation upon endoplasmic reticulum stress. Elife 2018, 7, e30257. [Google Scholar] [CrossRef]
- Karagöz, G.E.; Acosta-Alvear, D.; Nguyen, H.T.; Lee, C.P.; Chu, F.; Walter, P. An unfolded protein-induced conformational switch activates mammalian IRE1. Elife 2017, 6, e30700. [Google Scholar] [CrossRef] [PubMed]
- Preissler, S.; Ron, D. Early events in the endoplasmic reticulum unfolded protein response. Cold Spring Harb. Perspect. Biol. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Li, J.; Tao, J.; Sha, B. The luminal domain of the ER stress sensor protein PERK binds misfolded proteins and thereby triggers PERK oligomerization. J. Biol. Chem. 2018, 293, 4110–4121. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, D.; Kimata, Y.; Kohno, K.; Iwawaki, T. Activation of mammalian IRE1α upon ER stress depends on dissociation of BiP rather than on direct interaction with unfolded proteins. Exp. Cell Res. 2009, 315, 2496–2504. [Google Scholar] [CrossRef] [PubMed]
- Halbleib, K.; Pesek, K.; Covino, R.; Hofbauer, H.F.; Wunnicke, D.; Hänelt, I.; Hummer, G.; Ernst, R. Activation of the Unfolded Protein Response by Lipid Bilayer Stress. Mol. Cell 2017, 67, 673–684.e8. [Google Scholar] [CrossRef] [PubMed]
- Bobrovnikova-Marjon, E.; Pytel, D.; Riese, M.J.; Vaites, L.P.; Singh, N.; Koretzky, G.A.; Witze, E.S.; Diehl, J.A. PERK Utilizes Intrinsic Lipid Kinase Activity To Generate Phosphatidic Acid, Mediate Akt Activation, and Promote Adipocyte Differentiation. Mol. Cell. Biol. 2012, 32, 2268–2278. [Google Scholar] [CrossRef] [PubMed]
- Nagai, A.; Kadowaki, H.; Maruyama, T.; Takeda, K.; Nishitoh, H.; Ichijo, H. USP14 inhibits ER-associated degradation via interaction with IRE1α. Biochem. Biophys. Res. Commun. 2009, 379, 995–1000. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.P.K.; Dey, M.; Neculai, D.; Cao, C.; Dever, T.E.; Sicheri, F. Structure of the Dual Enzyme Ire1 Reveals the Basis for Catalysis and Regulation in Nonconventional RNA Splicing. Cell 2008, 132, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, H.; Li, L.; Liu, H.; Shi, W.; Yuan, X.; Wu, L. Structural Insights into IRE1 Functions in the Unfolded Protein Response. Curr. Med. Chem. 2016, 23, 4706–4716. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.; Newbatt, Y.; McAndrew, P.C.; Stubbs, M.; Burke, R.; Richards, M.W.; Bhatia, C.; Caldwell, J.J.; McHardy, T.; Collins, I.; et al. Molecular mechanisms of human IRE1 activation through dimerization and ligand binding. Oncotarget 2015, 6, 13019–13035. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, C.Y.; Back, S.H.; Clark, R.L.; Peisach, D.; Xu, Z.; Kaufman, R.J. The crystal structure of human IRE1 luminal domain reveals a conserved dimerization interface required for activation of the unfolded protein response. Proc. Natl. Acad. Sci. USA 2006, 103, 14343–14348. [Google Scholar] [CrossRef]
- Afzal, A.J.; Wood, A.J.; Lightfoot, D.A. Plant receptor-like serine threonine kinases: Roles in signaling and plant defense. Mol. Plant Microbe Interact. 2008, 21, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Wong, H.N.; Schauerte, J.A.; Kaufman, R.J. The protein kinase/endoribonuclease IRE1α that signals the unfolded protein response has a luminal N-terminal ligand-independent dimerization domain. J. Biol. Chem. 2002, 277, 18346–18356. [Google Scholar] [CrossRef]
- Cho, H.; Stanzione, F.; Oak, A.; Kim, G.H.; Yerneni, S.; Qi, L.; Sum, A.K.; Chan, C. Intrinsic Structural Features of the Human IRE1α Transmembrane Domain Sense Membrane Lipid Saturation. Cell Rep. 2019, 27, 307–320.e5. [Google Scholar] [CrossRef] [PubMed]
- Carlesso, A.; Chintha, C.; Gorman, A.M.; Samali, A.; Eriksson, L.A. Effect of Kinase Inhibiting RNase Attenuator (KIRA) Compounds on the Formation of Face-to-Face Dimers of Inositol-Requiring Enzyme 1: Insights from Computational Modeling. Int. J. Mol. Sci. 2019, 20, 5538. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.M.U.U.; Bagratuni, T.; Davenport, E.L.; Nowak, P.R.; Silva-Santisteban, M.C.; Hardcastle, A.; McAndrews, C.; Rowlands, M.G.; Morgan, G.J.; Aherne, W.; et al. Structure of the Ire1 autophosphorylation complex and implications for the unfolded protein response. EMBO J. 2011, 30, 894–905. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Xu, Z.; Kaufman, R.J. Structure and intermolecular interactions of the luminal dimerization domain of human IRE1α. J. Biol. Chem. 2003, 278, 17680–17687. [Google Scholar] [CrossRef]
- Feldman, H.C.; Tong, M.; Wang, L.; Meza-Acevedo, R.; Gobillot, T.A.; Lebedev, I.; Gliedt, M.J.; Hari, S.B.; Mitra, A.K.; Backes, B.J.; et al. Structural and Functional Analysis of the Allosteric Inhibition of IRE1α with ATP-Competitive Ligands. ACS Chem. Biol. 2016, 11, 2195–2205. [Google Scholar] [CrossRef] [PubMed]
- Prischi, F.; Nowak, P.R.; Carrara, M.; Ali, M.M.U. Phosphoregulation of Ire1 RNase splicing activity. Nat. Commun. 2014, 5, 1–11. [Google Scholar] [CrossRef]
- Tirasophon, W.; Lee, K.; Callaghan, B.; Welihinda, A.; Kaufman, R.J. The endoribonuclease activity of mammalian IRE1 autoregulates its mRNA and is required for the unfolded protein response. Genes Dev. 2000, 14, 2725–2736. [Google Scholar] [CrossRef] [PubMed]
- Korennykh, A.V.; Egea, P.F.; Korostelev, A.A.; Finer-Moore, J.; Zhang, C.; Shokat, K.M.; Stroud, R.M.; Walter, P. The unfolded protein response signals through high-order assembly of Ire1. Nature 2009, 457, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Itzhak, D.; Bright, M.; McAndrew, P.; Mirza, A.; Newbatt, Y.; Strover, J.; Widya, M.; Thompson, A.; Morgan, G.; Collins, I.; et al. Multiple autophosphorylations significantly enhance the endoribonuclease activity of human inositol requiring enzyme 1α. BMC Biochem. 2014, 15, 3. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Korennykh, A.V.; Behrman, S.L.; Walter, P. Mammalian endoplasmic reticulum stress sensor IRE1 signals by dynamic clustering. Proc. Natl. Acad. Sci. USA 2010, 107, 16113–16118. [Google Scholar] [CrossRef]
- Tam, A.B.; Koong, A.C.; Niwa, M. Ire1 has distinct catalytic mechanisms for XBP1/HAC1 splicing and RIDD. Cell Rep. 2014, 9, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Ricci, D.; Marrocco, I.; Blumenthal, D.; Dibos, M.; Eletto, D.; Vargas, J.; Boyle, S.; Iwamoto, Y.; Chomistek, S.; Paton, J.C.; et al. Clustering of IRE1α depends on sensing ER stress but not on its RNase activity. FASEB J. 2019, 33, 9811–9827. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, C.; Wang, A. Divergence and Conservation of the Major UPR Branch IRE1-bZIP Signaling Pathway across Eukaryotes. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Oku, M.; Suzuki, M.; Mori, K. pXBP1(U) encoded in XBP1 pre-mRNA negatively regulates unfolded protein response activator pXBP1(S) in mammalian ER stress response. J. Cell Biol. 2006, 172, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Kanemoto, S.; Kondo, S.; Ogata, M.; Murakami, T.; Urano, F.; Imaizumi, K. XBP1 activates the transcription of its target genes via an ACGT core sequence under ER stress. Biochem. Biophys. Res. Commun. 2005, 331, 1146–1153. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H. Unconventional splicing of XBP-1 mRNA in the unfolded protein response. Antioxid. Redox Signal. 2007, 9, 2323–2333. [Google Scholar] [CrossRef]
- Coelho, D.S.; Gaspar, C.J.; Domingos, P.M. Ire1 mediated mRNA splicing in a C-terminus deletion mutant of drosophila Xbp1. PLoS ONE 2014, 9, e105588. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.B.; Lee, K.; Vink, E.; Kaufman, R.J. Cytoplasmic IRE1α-mediated XBP1 mRNA splicing in the absence of nuclear processing and endoplasmic reticulum stress. J. Biol. Chem. 2006, 281, 18691–18706. [Google Scholar] [CrossRef]
- Iwawaki, T.; Akai, R. Analysis of the XBP1 splicing mechanism using endoplasmic reticulum stress-indicators. Biochem. Biophys. Res. Commun. 2006, 350, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xing, P.; Cui, W.; Wang, W.; Cui, Y.; Ying, G.; Wang, X.; Li, B. Acute endoplasmic reticulum stress-independent unconventional splicing of XBP1 mRNA in the nucleus of mammalian cells. Int. J. Mol. Sci. 2015, 16, 13302–13321. [Google Scholar] [CrossRef] [PubMed]
- Hollien, J.; Lin, J.H.; Li, H.; Stevens, N.; Walter, P.; Weissman, J.S. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 2009, 186, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Maurel, M.; Chevet, E.; Tavernier, J.; Gerlo, S. Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem. Sci. 2014, 39, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, D.; Tokuda, M.; Hosoda, A.; Iwawaki, T. Identification of a consensus element recognized and cleaved by IRE1α. Nucleic Acids Res. 2010, 38, 6265–6273. [Google Scholar] [CrossRef] [PubMed]
- Coelho, D.S.; Domingos, P.M. Physiological roles of regulated Ire1 dependent decay. Front. Genet. 2014, 5. [Google Scholar] [CrossRef]
- Chen, Y.; Brandizzi, F. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 2013, 23, 547–555. [Google Scholar] [CrossRef]
- Iurlaro, R.; Muñoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.Z.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Saveljeva, S.; Mc Laughlin, S.L.; Vandenabeele, P.; Samali, A.; Bertrand, M.J.M. Endoplasmic reticulum stress induces ligand-independent TNFR1-mediated necroptosis in L929 cells. Cell Death Dis. 2015, 6, e1587. [Google Scholar] [CrossRef] [PubMed]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed]
- Kanekura, K.; Ma, X.; Murphy, J.T.; Zhu, L.J.; Diwan, A.; Urano, F. IRE1 prevents endoplasmic reticulum membrane permeabilization and cell death under pathological conditions. Sci. Signal. 2015, 8, ra62. [Google Scholar] [CrossRef]
- Brown, M.; Strudwick, N.; Suwara, M.; Sutcliffe, L.K.; Mihai, A.D.; Ali, A.A.; Watson, J.N.; Schröder, M. An initial phase of JNK activation inhibits cell death early in the endoplasmic reticulum stress response. J. Cell Sci. 2016, 129, 2317–2328. [Google Scholar] [CrossRef] [PubMed]
- Treiber, D.K.; Shah, N.P. Ins and outs of kinase DFG motifs. Chem. Biol. 2013, 20, 745–746. [Google Scholar] [CrossRef] [PubMed]
- Grandjean, J.M.D.; Wiseman, R.L. Small molecule strategies to harness the unfolded protein response: Where do we go from here? J. Biol. Chem. 2020, 295, 15692–15711. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Perera, B.G.K.; Hari, S.B.; Bhhatarai, B.; Backes, B.J.; Seeliger, M.A.; Schürer, S.C.; Oakes, S.A.; Papa, F.R.; Maly, D.J. Divergent allosteric control of the IRE1α endoribonuclease using kinase inhibitors. Nat. Chem. Biol. 2012, 8, 982–989. [Google Scholar] [CrossRef] [PubMed]
- Tomasio, S.M.; Harding, H.P.; Ron, D.; Cross, B.C.S.; Bond, P.J. Selective inhibition of the unfolded protein response: Targeting catalytic sites for Schiff base modification. Mol. Biosyst. 2013, 9, 2408–2416. [Google Scholar] [CrossRef] [PubMed]
- Medinas, D.B.; González, J.V.; Falcon, P.; Hetz, C. Fine-tuning ER stress signal transducers to treat amyotrophic lateral sclerosis. Front. Mol. Neurosci. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Thakur, P.C.; Miller-Ocuin, J.L.; Nguyen, K.; Matsuda, R.; Singhi, A.D.; Zeh, H.J.; Bahary, N. Inhibition of endoplasmic-reticulum-stress-mediated autophagy enhances the effectiveness of chemotherapeutics on pancreatic cancer. J. Transl. Med. 2018, 16, 190. [Google Scholar] [CrossRef] [PubMed]
- Mendez, A.S.; Alfaro, J.; Morales-Soto, M.A.; Dar, A.C.; McCullagh, E.; Gotthardt, K.; Li, H.; Acosta-Alvear, D.; Sidrauski, C.; Korennykh, A.V.; et al. Endoplasmic reticulum stress-independent activation of unfolded protein response kinases by a small molecule ATP-mimic. Elife 2015, 4, e05434. [Google Scholar] [CrossRef] [PubMed]
- Feldman, H.C.; Vidadala, V.N.; Potter, Z.E.; Papa, F.R.; Backes, B.J.; Maly, D.J. Development of a Chemical Toolset for Studying the Paralog-Specific Function of IRE1. ACS Chem. Biol. 2019, 14, 2595–2605. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Wang, L.; Wang, E.S.; Perera, B.G.K.; Igbaria, A.; Morita, S.; Prado, K.; Thamsen, M.; Caswell, D.; Macias, H.; et al. Allosteric inhibition of the IRE1α RNase preserves cell viability and function during endoplasmic reticulum stress. Cell 2014, 158, 534–548. [Google Scholar] [CrossRef] [PubMed]
- Kolpikova, E.P.; Tronco, A.R.; Den Hartigh, A.B.; Jackson, K.J.; Iwawaki, T.; Fink, S.L. IRE1α promotes zika virus infection via XBP1. Viruses 2020, 12, 278. [Google Scholar] [CrossRef] [PubMed]
- Anthony Tang, C.H.; Chang, S.; Paton, A.W.; Paton, J.C.; Gabrilovich, D.I.; Ploegh, H.L.; Del Valle, J.R.; Andrew Hu, C.C. Phosphorylation of IRE1 at S729 regulates RIDD in B cells and antibody production after immunization. J. Cell Biol. 2018, 217, 1739–1755. [Google Scholar] [CrossRef]
- Mahameed, M.; Wilhelm, T.; Darawshi, O.; Obiedat, A.; Tommy, W.S.; Chintha, C.; Schubert, T.; Samali, A.; Chevet, E.; Eriksson, L.A.; et al. The unfolded protein response modulators GSK2606414 and KIRA6 are potent KIT inhibitors. Cell Death Dis. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Thamsen, M.; Ghosh, R.; Auyeung, V.C.; Brumwell, A.; Chapman, H.A.; Backes, B.J.; Perara, G.; Maly, D.J.; Sheppard, D.; Papa, F.R. Small molecule inhibition of IRE1α kinase/ RNase has anti-fibrotic effects in the lung. PLoS ONE 2019, 14, e0209824. [Google Scholar] [CrossRef] [PubMed]
- Harrington, P.E.; Biswas, K.; Malwitz, D.; Tasker, A.S.; Mohr, C.; Andrews, K.L.; Dellamaggiore, K.; Kendall, R.; Beckmann, H.; Jaeckel, P.; et al. Unfolded protein response in cancer: IRE1α inhibition by selective kinase ligands does not impair tumor cell viability. ACS Med. Chem. Lett. 2015, 6, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Harnoss, J.M.; Le Thomas, A.; Shemorry, A.; Marsters, S.A.; Lawrence, D.A.; Lu, M.; Chen, Y.C.A.; Qing, J.; Totpal, K.; Kan, D.; et al. Disruption of IRE1α through its kinase domain attenuates multiple myeloma. Proc. Natl. Acad. Sci. USA 2019, 116, 16420–16429. [Google Scholar] [CrossRef] [PubMed]
- Volkmann, K.; Lucas, J.L.; Vuga, D.; Wang, X.; Brumm, D.; Stiles, C.; Kriebel, D.; Der-Sarkissian, A.; Krishnan, K.; Schweitzer, C.; et al. Potent and selective inhibitors of the inositol-requiring enzyme 1 endoribonuclease. J. Biol. Chem. 2011, 286, 12743–12755. [Google Scholar] [CrossRef]
- Chien, W.; Ding, L.W.; Sun, Q.Y.; Torres-Fernandez, L.A.; Tan, S.Z.; Xiao, J.; Lim, S.L.; Garg, M.; Lee, K.L.; Kitajima, S.; et al. Selective inhibition of unfolded protein response induces apoptosis in pancreatic cancer cells. Oncotarget 2014, 5, 4881–4894. [Google Scholar] [CrossRef] [PubMed]
- Cross, B.C.S.; Bond, P.J.; Sadowski, P.G.; Jha, B.K.; Zak, J.; Goodman, J.M.; Silverman, R.H.; Neubert, T.A.; Baxendale, I.R.; Ron, D.; et al. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc. Natl. Acad. Sci. USA 2012, 109. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.; Estrada, A.; Kim, P.; Wang, D.; Wei, Y.; Gentile, C.; Pagliassotti, M. Regulation of IRE1α by the small molecule inhibitor 4μ8c in hepatoma cells. Endoplasmic Reticulum Stress Dis. 2017, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Liu, H.; Song, Z.; Jiang, Y.; Kim, H.; Samavati, L.; Nguyen, H.M.; Yang, Z.Q. The UPR Transducer IRE1 Promotes Breast Cancer Malignancy by Degrading Tumor Suppressor microRNAs. iScience 2020, 23, 101503. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Nosak, C.; Sollazzo, P.; Odisho, T.; Volchuk, A. IRE1 inhibition perturbs the unfolded protein response in a pancreatic β-cell line expressing mutant proinsulin, but does not sensitize the cells to apoptosis. BMC Cell Biol. 2014, 15, 29. [Google Scholar] [CrossRef]
- Chan, S.M.H.; Lowe, M.P.; Bernard, A.; Miller, A.A.; Herbert, T.P. The inositol-requiring enzyme 1 (IRE1α) RNAse inhibitor, 4m8C, is also a potent cellular antioxidant. Biochem. J. 2018, 475, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Masouleh, B.K.; Geng, H.; Hurtz, C.; Chan, L.N.; Logan, A.C.; Chang, M.S.; Huang, C.; Swaminathan, S.; Sun, H.; Paietta, E.; et al. Mechanistic rationale for targeting the unfolded protein response in pre-B acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA 2014, 111. [Google Scholar] [CrossRef]
- Sun, H.; Lin, D.C.; Guo, X.; Masouleh, B.K.; Gery, S.; Cao, Q.; Alkan, S.; Ikezoe, T.; Akiba, C.; Paquette, R.; et al. Inhibition of IRE1α-driven pro-survival pathways is a promising therapeutic application in acute myeloid leukemia. Oncotarget 2016, 7, 18736–18749. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, I.; Denko, N.C.; Olson, M.; Van Melckebeke, H.; Lust, S.; Tam, A.; Solow-Cordero, D.E.; Bouley, D.M.; Offner, F.; Niwa, M.; et al. Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood 2011, 117, 1311–1314. [Google Scholar] [CrossRef] [PubMed]
- Deck, K.; Abad, J.G.; Askew, D. Testing Inhibitory Drugs of the Human UPR Sensor IRE1 on Aspergillus fumigatus. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Ri, M.; Tashiro, E.; Oikawa, D.; Shinjo, S.; Tokuda, M.; Yokouchi, Y.; Narita, T.; Masaki, A.; Ito, A.; Ding, J.; et al. Identification of Toyocamycin, an agent cytotoxic for multiple myeloma cells, as a potent inhibitor of ER stress-induced XBP1 mRNA splicing. Blood Cancer, J. 2012, 2. [Google Scholar] [CrossRef]
- Park, S.G.; Kim, S.H.; Kim, K.Y.; Yu, S.N.; Choi, H.D.; Kim, Y.W.; Nam, H.W.; Seo, Y.K.; Ahn, S.C. Toyocamycin induces apoptosis via the crosstalk between reactive oxygen species and p38/ERK MAPKs signaling pathway in human prostate cancer PC-3 cells. Pharmacol. Rep. 2017, 69, 90–96. [Google Scholar] [CrossRef]
- Bodeau, S.; Sauzay, C.; Nyga, R.; Louandre, C.; Descamps, V.; François, C.; Godin, C.; Choukroun, G.; Galmiche, A. Targeting the unfolded protein response as a potential therapeutic strategy in renal carcinoma cells exposed to cyclosporine a. Anticancer Res. 2017, 37, 1049–1057. [Google Scholar] [CrossRef]
- Li, Y.; Tinoco, R.; Elmén, L.; Segota, I.; Xian, Y.; Fujita, Y.; Sahu, A.; Zarecki, R.; Marie, K.; Feng, Y.; et al. Gut microbiota dependent anti-tumor immunity restricts melanoma growth in Rnf5 −/− mice. Nat. Commun. 2019, 10, 1492. [Google Scholar] [CrossRef] [PubMed]
- Cawley, K.; Logue, S.E.; Gorman, A.M.; Zeng, Q.; Patterson, J.; Gupta, S.; Samali, A. Disruption of microRNA biogenesis confers resistance to ER stress-induced cell death upstream of the mitochondrion. PLoS ONE 2013, 8, e73870. [Google Scholar] [CrossRef]
- Le Reste, P.J.; Pineau, R.; Voutetakis, K.; Samal, J.; Jégou, G.; Lhomond, S.; Gorman, A.M.; Samali, A.; Patterson, J.B.; Zeng, Q.; et al. Local intracerebral inhibition of IRE1 by MKC8866 sensitizes glioblastoma to irradiation/chemotherapy in vivo. Cancer Lett. 2020, 494, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Logue, S.E.; McGrath, E.P.; Cleary, P.; Greene, S.; Mnich, K.; Almanza, A.; Chevet, E.; Dwyer, R.M.; Oommen, A.; Legembre, P.; et al. Inhibition of IRE1 RNase activity modulates the tumor cell secretome and enhances response to chemotherapy. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Vieri, M.; Preisinger, C.; Schemionek, M.; Salimi, A.; Patterson, J.B.; Samali, A.; Brümmendorf, T.H.; Appelmann, I.; Kharabi Masouleh, B. Synergistic Dual Inhibition of BCR-ABL1 and the Unfolded Protein Response Causes p38 MAPK-Mediated Cell Death and Sensitizes BCR-ABL1+ Acute Lymphoblastic Leukemia to Dexamethasone. Blood 2018, 132, 4674. [Google Scholar] [CrossRef]
- Sheng, X.; Nenseth, H.Z.; Qu, S.; Kuzu, O.F.; Frahnow, T.; Simon, L.; Greene, S.; Zeng, Q.; Fazli, L.; Rennie, P.S.; et al. IRE1α-XBP1s pathway promotes prostate cancer by activating c-MYC signaling. Nat. Commun. 2019, 10, 323. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, N.; Dolgikh, N.; Logue, S.; Patterson, J.B.; Zeng, Q.; Gorman, A.M.; Samali, A.; Fulda, S. The IRE1 and PERK arms of the unfolded protein response promote survival of rhabdomyosarcoma cells. Cancer Lett. 2020, 490, 76–88. [Google Scholar] [CrossRef]
- Mimura, N.; Fulciniti, M.; Gorgun, G.; Tai, Y.T.; Cirstea, D.; Santo, L.; Hu, Y.; Fabre, C.; Minami, J.; Ohguchi, H.; et al. Blockade of XBP1 splicing by inhibition of IRE1α is a promising therapeutic option in multiple myeloma. Blood 2012, 119, 5772–5781. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Tam, A.B.; Alagappan, M.; Hay, M.P.; Gupta, A.; Kozak, M.M.; Solow-Cordero, D.E.; Lum, P.Y.; Denko, N.C.; Giaccia, A.J.; et al. Acridine derivatives as inhibitors of the IRE1α-XBP1 pathway are cytotoxic to human multiple myeloma. Mol. Cancer Ther. 2016, 15, 2055–2065. [Google Scholar] [CrossRef]
- Sanches, M.; Duffy, N.M.; Talukdar, M.; Thevakumaran, N.; Chiovitti, D.; Canny, M.D.; Lee, K.; Kurinov, I.; Uehling, D.; Al-Awar, R.; et al. Structure and mechanism of action of the hydroxy-aryl-aldehyde class of IRE1 endoribonuclease inhibitors. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.H.A.; Ranatunga, S.; Kriss, C.L.; Cubitt, C.L.; Tao, J.; Pinilla-Ibarz, J.A.; Del Valle, J.R.; Hu, C.C.A. Inhibition of ER stress-associated IRE-1/XBP-1 pathway reduces leukemic cell survival. J. Clin. Investig. 2014, 124, 2585–2598. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Tang, C.H.A.; Song, J.H.; Mancuso, A.; Del Valle, J.R.; Cao, J.; Xiang, Y.; Dang, C.V.; Lan, R.; Sanchez, D.J.; et al. IRE1α RNase-dependent lipid homeostasis promotes survival in Myc-transformed cancers. J. Clin. Investig. 2018, 128, 1300–1316. [Google Scholar] [CrossRef]
- Zhang, T.; Li, N.; Sun, C.; Jin, Y.; Sheng, X. MYC and the unfolded protein response in cancer: Synthetic lethal partners in crime? EMBO Mol. Med. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, R.L.; Zhang, Y.; Lee, K.P.K.; Harding, H.P.; Haynes, C.M.; Price, J.; Sicheri, F.; Ron, D. Flavonol Activation Defines an Unanticipated Ligand-Binding Site in the Kinase-RNase Domain of IRE1. Mol. Cell 2010, 38, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Papa, F.R.; Zhang, C.; Shokat, K.; Waiter, P. Bypassing a Kinase Activity with an ATP-Competitive Drug. Science 2003, 302, 1533–1537. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Lerner, A.G.; Vande Walle, L.; Upton, J.P.; Xu, W.; Hagen, A.; Backes, B.J.; Oakes, S.A.; Papa, F.R. IRE1α Kinase Activation Modes Control Alternate Endoribonuclease Outputs to Determine Divergent Cell Fates. Cell 2009, 138, 562–575. [Google Scholar] [CrossRef] [PubMed]
- Ferri, E.; Le Thomas, A.; Wallweber, H.A.; Day, E.S.; Walters, B.T.; Kaufman, S.E.; Braun, M.G.; Clark, K.R.; Beresini, M.H.; Mortara, K.; et al. Activation of the IRE1 RNase through remodeling of the kinase front pocket by ATP-competitive ligands. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Grandjean, J.M.D.; Madhavan, A.; Cech, L.; Seguinot, B.O.; Paxman, R.J.; Smith, E.; Scampavia, L.; Powers, E.T.; Cooley, C.B.; Plate, L.; et al. Pharmacologic IRE1/XBP1s activation confers targeted ER proteostasis reprogramming. Nat. Chem. Biol. 2020, 16, 1052–1061. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siwecka, N.; Rozpędek-Kamińska, W.; Wawrzynkiewicz, A.; Pytel, D.; Diehl, J.A.; Majsterek, I. The Structure, Activation and Signaling of IRE1 and Its Role in Determining Cell Fate. Biomedicines 2021, 9, 156. https://doi.org/10.3390/biomedicines9020156
Siwecka N, Rozpędek-Kamińska W, Wawrzynkiewicz A, Pytel D, Diehl JA, Majsterek I. The Structure, Activation and Signaling of IRE1 and Its Role in Determining Cell Fate. Biomedicines. 2021; 9(2):156. https://doi.org/10.3390/biomedicines9020156
Chicago/Turabian StyleSiwecka, Natalia, Wioletta Rozpędek-Kamińska, Adam Wawrzynkiewicz, Dariusz Pytel, J. Alan Diehl, and Ireneusz Majsterek. 2021. "The Structure, Activation and Signaling of IRE1 and Its Role in Determining Cell Fate" Biomedicines 9, no. 2: 156. https://doi.org/10.3390/biomedicines9020156
APA StyleSiwecka, N., Rozpędek-Kamińska, W., Wawrzynkiewicz, A., Pytel, D., Diehl, J. A., & Majsterek, I. (2021). The Structure, Activation and Signaling of IRE1 and Its Role in Determining Cell Fate. Biomedicines, 9(2), 156. https://doi.org/10.3390/biomedicines9020156