
Relevance of Autophagy and Mitophagy Dynamics and Markers in Neurodegenerative Diseases





 and
and 
Abstract
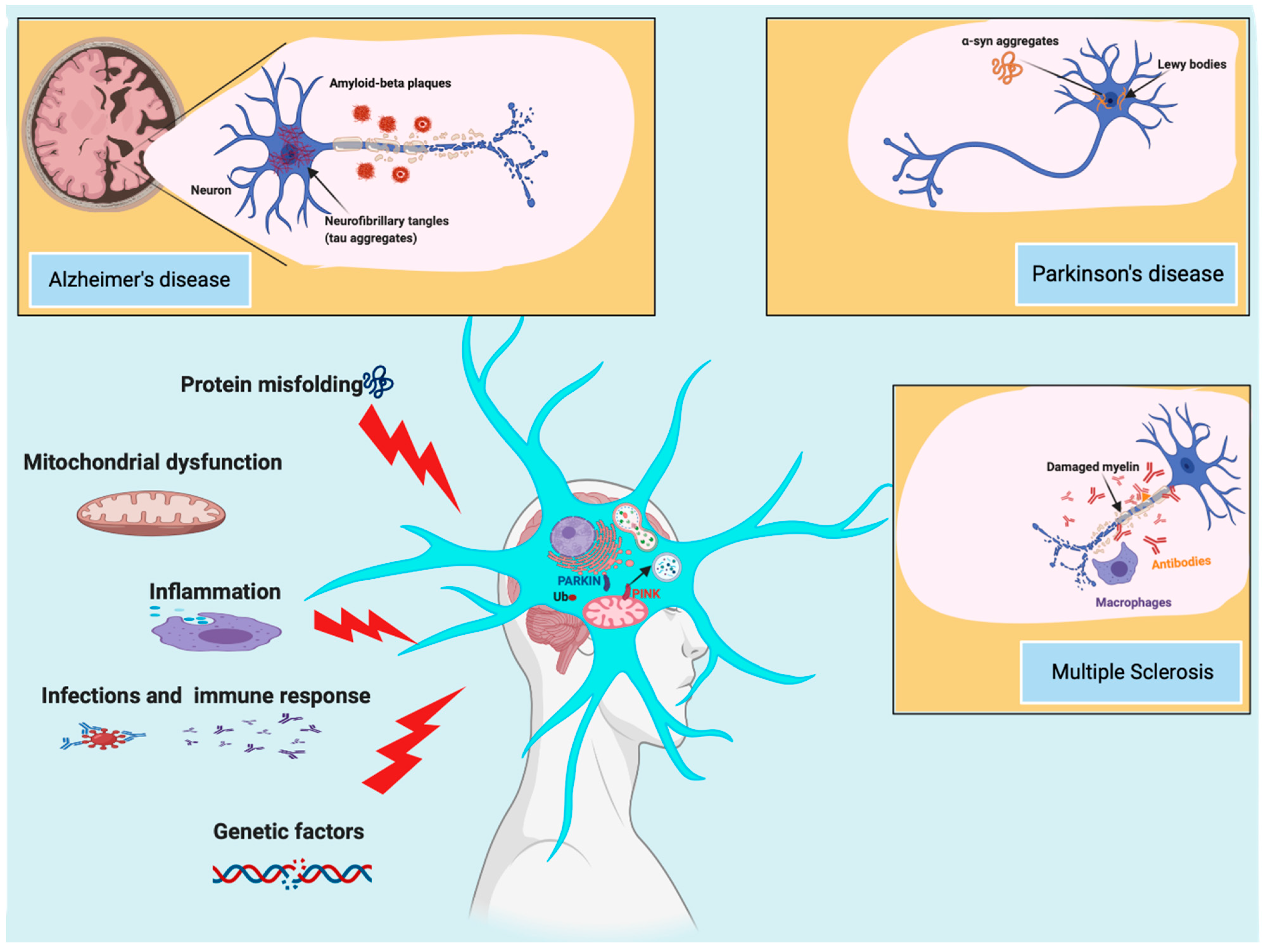
1. Introduction
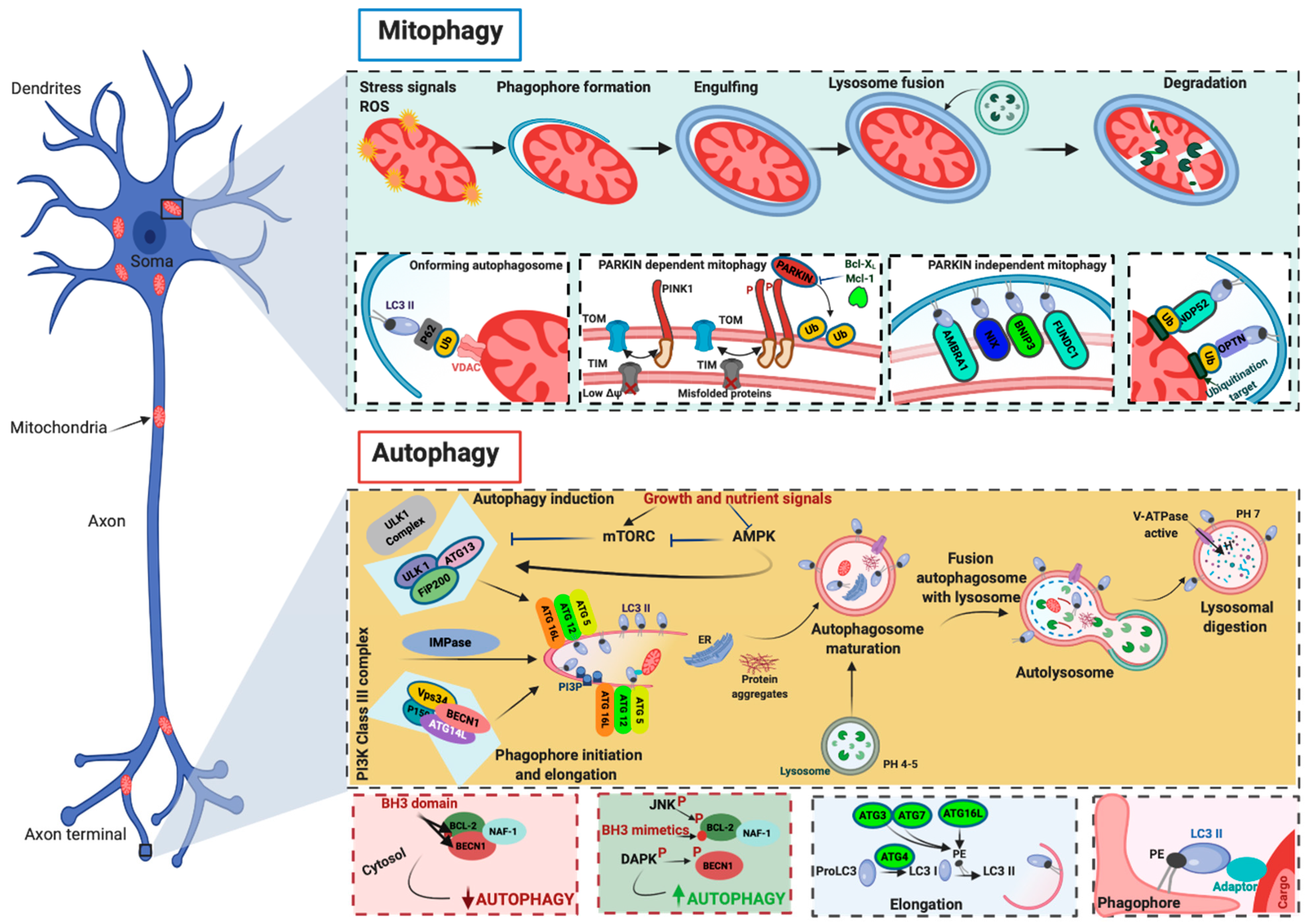
2. A General Overview of Autophagy
3. Mitophagy: The Master Regulator of the Mitochondrial Population
4. Relationship between Autophagy and Mitophagy in MS
5. Involvement of Autophagy Mechanisms in AD Progression
6. Current Knowledge of the Relationship between PD and Autophagy Dynamics
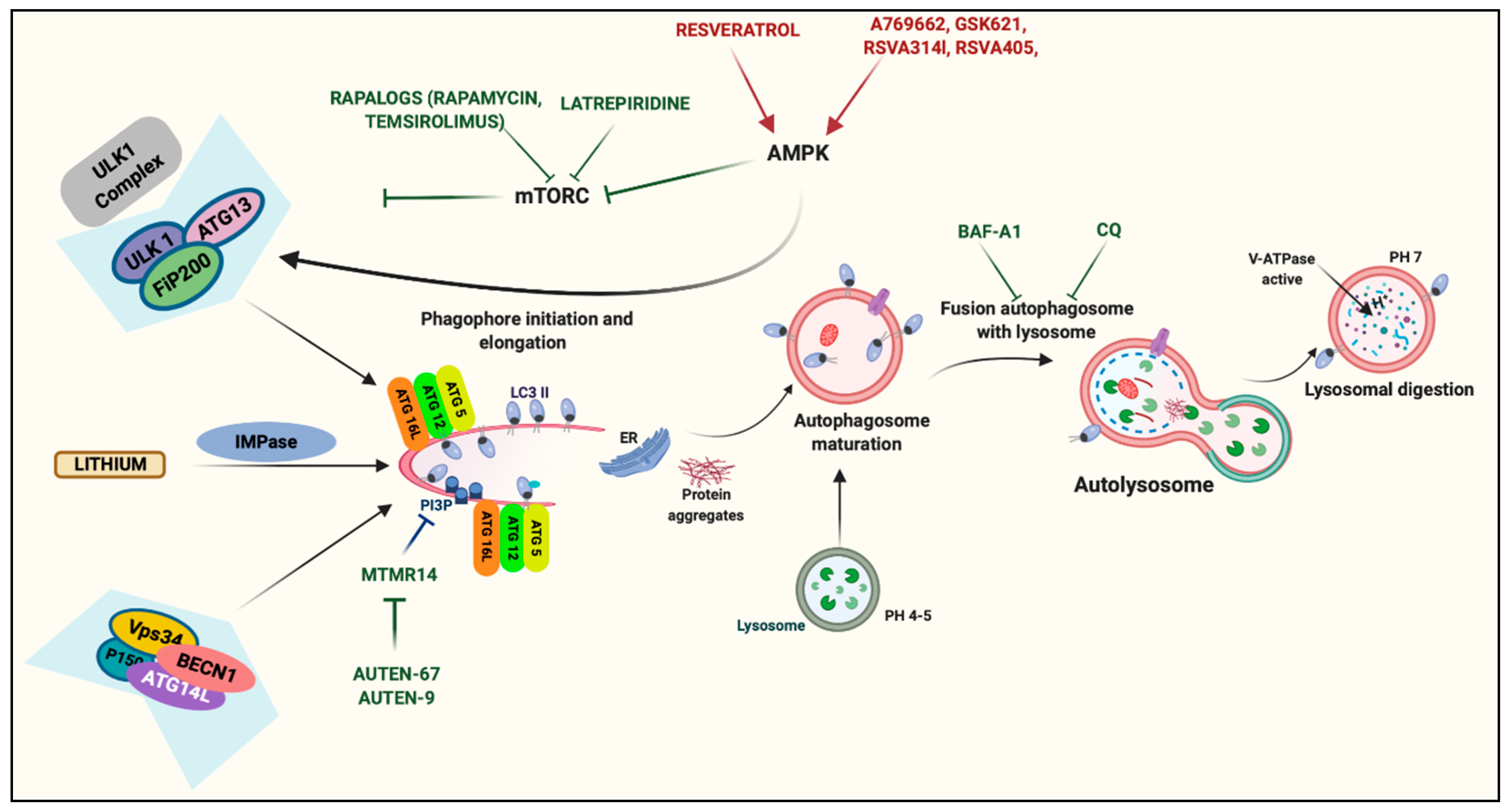
7. Principles and Current Strategies for Targeting Autophagy in Neurodegeneration
8. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harbor Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef]
- Davis, A.A.; Leyns, C.E.G.; Holtzman, D.M. Intercellular Spread of Protein Aggregates in Neurodegenerative Disease. Annu. Rev. Cell Dev. Biol. 2018, 34, 545–568. [Google Scholar] [CrossRef]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Okun, M.S. Diagnosis and Treatment of Parkinson Disease: A Review. JAMA 2020, 323, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Beyer, K.; Ariza, A. Alpha-Synuclein posttranslational modification and alternative splicing as a trigger for neurodegeneration. Mol. Neurobiol. 2013, 47, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Patergnani, S.; Fossati, V.; Bonora, M.; Giorgi, C.; Marchi, S.; Missiroli, S.; Rusielewicz, T.; Wieckowski, M.R.; Pinton, P. Mitochondria in Multiple Sclerosis: Molecular Mechanisms of Pathogenesis. Int. Rev. Cell Mol. Biol. 2017, 328, 49–103. [Google Scholar] [CrossRef]
- Lassmann, H. Pathogenic Mechanisms Associated With Different Clinical Courses of Multiple Sclerosis. Front. Immunol. 2018, 9, 3116. [Google Scholar] [CrossRef]
- Burte, F.; Carelli, V.; Chinnery, P.F.; Yu-Wai-Man, P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat. Rev. Neurol. 2015, 11, 11–24. [Google Scholar] [CrossRef]
- Missiroli, S.; Genovese, I.; Perrone, M.; Vezzani, B.; Vitto, V.A.M.; Giorgi, C. The Role of Mitochondria in Inflammation: From Cancer to Neurodegenerative Disorders. J. Clin. Med. 2020, 9, 740. [Google Scholar] [CrossRef]
- Chitnis, T.; Weiner, H.L. CNS inflammation and neurodegeneration. J. Clin. Investig. 2017, 127, 3577–3587. [Google Scholar] [CrossRef]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Fullgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef]
- Patergnani, S.; Pinton, P. Mitophagy and mitochondrial balance. Methods Mol. Biol. 2015, 1241, 181–194. [Google Scholar] [CrossRef]
- Patergnani, S.; Guzzo, S.; Mangolini, A.; dell’Atti, L.; Pinton, P.; Aguiari, G. The induction of AMPK-dependent autophagy leads to P53 degradation and affects cell growth and migration in kidney cancer cells. Exp. Cell Res. 2020, 395, 112190. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Patergnani, S.; Giorgi, C.; Suarez, J.; Goto, K.; Bononi, A.; Tanji, M.; Novelli, F.; Pastorino, S.; Xu, R.; et al. Asbestos induces mesothelial cell transformation via HMGB1-driven autophagy. Proc. Natl. Acad. Sci. USA 2020, 117, 25543–25552. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.T. Mechanisms of selective autophagy and mitophagy: Implications for neurodegenerative diseases. Neurobiol. Dis. 2019, 122, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Patergnani, S.; Castellazzi, M.; Bonora, M.; Marchi, S.; Casetta, I.; Pugliatti, M.; Giorgi, C.; Granieri, E.; Pinton, P. Autophagy and mitophagy elements are increased in body fluids of multiple sclerosis-affected individuals. J. Neurol. Neurosurg. Psychiatry 2018, 89, 439–441. [Google Scholar] [CrossRef]
- Castellazzi, M.; Patergnani, S.; Donadio, M.; Giorgi, C.; Bonora, M.; Bosi, C.; Brombo, G.; Pugliatti, M.; Seripa, D.; Zuliani, G.; et al. Autophagy and mitophagy biomarkers are reduced in sera of patients with Alzheimer’s disease and mild cognitive impairment. Sci. Rep. 2019, 9, 20009. [Google Scholar] [CrossRef]
- De Duve, C.; Wattiaux, R. Functions of lysosomes. Annu. Rev. Physiol. 1966, 28, 435–492. [Google Scholar] [CrossRef]
- Mijaljica, D.; Prescott, M.; Devenish, R.J. Microautophagy in mammalian cells: Revisiting a 40-year-old conundrum. Autophagy 2011, 7, 673–682. [Google Scholar] [CrossRef]
- Sahu, R.; Kaushik, S.; Clement, C.C.; Cannizzo, E.S.; Scharf, B.; Follenzi, A.; Potolicchio, I.; Nieves, E.; Cuervo, A.M.; Santambrogio, L. Microautophagy of cytosolic proteins by late endosomes. Dev. Cell 2011, 20, 131–139. [Google Scholar] [CrossRef]
- Morshed, S.; Tasnin, M.N.; Ushimaru, T. ESCRT machinery plays a role in microautophagy in yeast. BMC Mol. Cell Biol. 2020, 21, 70. [Google Scholar] [CrossRef]
- Sato, M.; Seki, T.; Konno, A.; Hirai, H.; Kurauchi, Y.; Hisatsune, A.; Katsuki, H. Fluorescent-based evaluation of chaperone-mediated autophagy and microautophagy activities in cultured cells. Genes Cells Devoted Mol. Cell. Mech. 2016, 21, 861–873. [Google Scholar] [CrossRef] [PubMed]
- Olsvik, H.L.; Svenning, S.; Abudu, Y.P.; Brech, A.; Stenmark, H.; Johansen, T.; Mejlvang, J. Endosomal microautophagy is an integrated part of the autophagic response to amino acid starvation. Autophagy 2019, 15, 182–183. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Seki, T.; Konno, A.; Hirai, H.; Kurauchi, Y.; Hisatsune, A.; Katsuki, H. Rapamycin activates mammalian microautophagy. J. Pharmacol. Sci. 2019, 140, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.A.; Terasawa, M.; Mostofa, M.G.; Ushimaru, T. The TORC1-Nem1/Spo7-Pah1/lipin axis regulates microautophagy induction in budding yeast. Biochem. Biophys. Res. Commun. 2018, 504, 505–512. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef]
- Chang, K.; Kang, P.; Liu, Y.; Huang, K.; Miao, T.; Sagona, A.P.; Nezis, I.P.; Bodmer, R.; Ocorr, K.; Bai, H. TGFB-INHB/activin signaling regulates age-dependent autophagy and cardiac health through inhibition of MTORC2. Autophagy 2020, 16, 1807–1822. [Google Scholar] [CrossRef]
- Aspernig, H.; Heimbucher, T.; Qi, W.; Gangurde, D.; Curic, S.; Yan, Y.; Donner von Gromoff, E.; Baumeister, R.; Thien, A. Mitochondrial Perturbations Couple mTORC2 to Autophagy in C. elegans. Cell Rep. 2019, 29, 1399–1409.e5. [Google Scholar] [CrossRef]
- Jhanwar-Uniyal, M.; Wainwright, J.V.; Mohan, A.L.; Tobias, M.E.; Murali, R.; Gandhi, C.D.; Schmidt, M.H. Diverse signaling mechanisms of mTOR complexes: mTORC1 and mTORC2 in forming a formidable relationship. Adv. Biol. Regul. 2019, 72, 51–62. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Shang, L.; Chen, S.; Du, F.; Li, S.; Zhao, L.; Wang, X. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc. Natl. Acad. Sci. USA 2011, 108, 4788–4793. [Google Scholar] [CrossRef] [PubMed]
- Bach, M.; Larance, M.; James, D.E.; Ramm, G. The serine/threonine kinase ULK1 is a target of multiple phosphorylation events. Biochem. J. 2011, 440, 283–291. [Google Scholar] [CrossRef]
- Egan, D.F.; Chun, M.G.; Vamos, M.; Zou, H.; Rong, J.; Miller, C.J.; Lou, H.J.; Raveendra-Panickar, D.; Yang, C.C.; Sheffler, D.J.; et al. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol. Cell 2015, 59, 285–297. [Google Scholar] [CrossRef]
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef]
- Wong, P.M.; Feng, Y.; Wang, J.; Shi, R.; Jiang, X. Regulation of autophagy by coordinated action of mTORC1 and protein phosphatase 2A. Nat. Commun. 2015, 6, 8048. [Google Scholar] [CrossRef]
- Torii, S.; Yoshida, T.; Arakawa, S.; Honda, S.; Nakanishi, A.; Shimizu, S. Identification of PPM1D as an essential Ulk1 phosphatase for genotoxic stress-induced autophagy. EMBO Rep. 2016, 17, 1552–1564. [Google Scholar] [CrossRef]
- Park, J.M.; Seo, M.; Jung, C.H.; Grunwald, D.; Stone, M.; Otto, N.M.; Toso, E.; Ahn, Y.; Kyba, M.; Griffin, T.J.; et al. ULK1 phosphorylates Ser30 of BECN1 in association with ATG14 to stimulate autophagy induction. Autophagy 2018, 14, 584–597. [Google Scholar] [CrossRef]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef]
- Tooze, S.A.; Yoshimori, T. The origin of the autophagosomal membrane. Nat. Cell Biol. 2010, 12, 831–835. [Google Scholar] [CrossRef]
- Xu, H.D.; Qin, Z.H. Beclin 1, Bcl-2 and Autophagy. Adv. Exp. Med. Biol. 2019, 1206, 109–126. [Google Scholar] [CrossRef] [PubMed]
- Kuhlbrandt, W. Structure and function of mitochondrial membrane protein complexes. BMC Biol. 2015, 13, 89. [Google Scholar] [CrossRef] [PubMed]
- Gammage, P.A.; Frezza, C. Mitochondrial DNA: The overlooked oncogenome? BMC Biol. 2019, 17, 53. [Google Scholar] [CrossRef] [PubMed]
- Chaban, Y.; Boekema, E.J.; Dudkina, N.V. Structures of mitochondrial oxidative phosphorylation supercomplexes and mechanisms for their stabilisation. Biochim. Biophys. Acta 2014, 1837, 418–426. [Google Scholar] [CrossRef]
- Rimessi, A.; Previati, M.; Nigro, F.; Wieckowski, M.R.; Pinton, P. Mitochondrial reactive oxygen species and inflammation: Molecular mechanisms, diseases and promising therapies. Int. J. Biochem. Cell Biol. 2016, 81, 281–293. [Google Scholar] [CrossRef]
- Patergnani, S.; Danese, A.; Bouhamida, E.; Aguiari, G.; Previati, M.; Pinton, P.; Giorgi, C. Various Aspects of Calcium Signaling in the Regulation of Apoptosis, Autophagy, Cell Proliferation, and Cancer. Int. J. Mol. Sci. 2020, 21, 8323. [Google Scholar] [CrossRef]
- Giorgi, C.; Danese, A.; Missiroli, S.; Patergnani, S.; Pinton, P. Calcium Dynamics as a Machine for Decoding Signals. Trends Cell Biol. 2018, 28, 258–273. [Google Scholar] [CrossRef]
- Perrone, M.; Caroccia, N.; Genovese, I.; Missiroli, S.; Modesti, L.; Pedriali, G.; Vezzani, B.; Vitto, V.A.M.; Antenori, M.; Lebiedzinska-Arciszewska, M.; et al. The role of mitochondria-associated membranes in cellular homeostasis and diseases. Int. Rev. Cell Mol. Biol. 2020, 350, 119–196. [Google Scholar] [CrossRef]
- Patergnani, S.; Missiroli, S.; Marchi, S.; Giorgi, C. Mitochondria-Associated Endoplasmic Reticulum Membranes Microenvironment: Targeting Autophagic and Apoptotic Pathways in Cancer Therapy. Front. Oncol. 2015, 5, 173. [Google Scholar] [CrossRef]
- Marchi, S.; Giorgi, C.; Galluzzi, L.; Pinton, P. Ca2+ Fluxes and Cancer. Mol. Cell 2020, 78, 1055–1069. [Google Scholar] [CrossRef]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Mankowski, R.T.; Burman, J.L.; Donisi, L.; Kim, J.S.; Marzetti, E.; Leeuwenburgh, C. Mitochondrial quality control mechanisms as molecular targets in cardiac ageing. Nat. Rev. Cardiol. 2018, 15, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Sato, M. Multiple ways to prevent transmission of paternal mitochondrial DNA for maternal inheritance in animals. J. Biochem. 2017, 162, 247–253. [Google Scholar] [CrossRef]
- Porter, G.A., Jr.; Hom, J.; Hoffman, D.; Quintanilla, R.; de Mesy Bentley, K.; Sheu, S.S. Bioenergetics, mitochondria, and cardiac myocyte differentiation. Prog. Pediatric Cardiol. 2011, 31, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Sin, J.; Andres, A.M.; Taylor, D.J.; Weston, T.; Hiraumi, Y.; Stotland, A.; Kim, B.J.; Huang, C.; Doran, K.S.; Gottlieb, R.A. Mitophagy is required for mitochondrial biogenesis and myogenic differentiation of C2C12 myoblasts. Autophagy 2016, 12, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Ishihara-Paul, L.; Hulihan, M.M.; Kachergus, J.; Upmanyu, R.; Warren, L.; Amouri, R.; Elango, R.; Prinjha, R.K.; Soto, A.; Kefi, M.; et al. PINK1 mutations and parkinsonism. Neurology 2008, 71, 896–902. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Djarmati, A.; Hedrich, K.; Schafer, N.; Scaglione, C.; Marchese, R.; Kock, N.; Schule, B.; Hiller, A.; Lohnau, T.; et al. PINK1, Parkin, and DJ-1 mutations in Italian patients with early-onset parkinsonism. Eur. J. Hum. Genet. 2005, 13, 1086–1093. [Google Scholar] [CrossRef]
- Hatano, Y.; Li, Y.; Sato, K.; Asakawa, S.; Yamamura, Y.; Tomiyama, H.; Yoshino, H.; Asahina, M.; Kobayashi, S.; Hassin-Baer, S.; et al. Novel PINK1 mutations in early-onset parkinsonism. Ann. Neurol. 2004, 56, 424–427. [Google Scholar] [CrossRef]
- Ibanez, P.; Lesage, S.; Lohmann, E.; Thobois, S.; De Michele, G.; Borg, M.; Agid, Y.; Durr, A.; Brice, A.; French Parkinson’s Disease Genetics Study Group. Mutational analysis of the PINK1 gene in early-onset parkinsonism in Europe and North Africa. Brain J. Neurol. 2006, 129, 686–694. [Google Scholar] [CrossRef]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef]
- Deas, E.; Plun-Favreau, H.; Gandhi, S.; Desmond, H.; Kjaer, S.; Loh, S.H.; Renton, A.E.; Harvey, R.J.; Whitworth, A.J.; Martins, L.M.; et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum. Mol. Genet. 2011, 20, 867–879. [Google Scholar] [CrossRef]
- Yamano, K.; Youle, R.J. PINK1 is degraded through the N-end rule pathway. Autophagy 2013, 9, 1758–1769. [Google Scholar] [CrossRef]
- Hasson, S.A.; Kane, L.A.; Yamano, K.; Huang, C.H.; Sliter, D.A.; Buehler, E.; Wang, C.; Heman-Ackah, S.M.; Hessa, T.; Guha, R.; et al. High-content genome-wide RNAi screens identify regulators of parkin upstream of mitophagy. Nature 2013, 504, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M.; Jin, S.M.; Kane, L.A.; Youle, R.J. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev. Cell 2012, 22, 320–333. [Google Scholar] [CrossRef]
- Okatsu, K.; Oka, T.; Iguchi, M.; Imamura, K.; Kosako, H.; Tani, N.; Kimura, M.; Go, E.; Koyano, F.; Funayama, M.; et al. PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat. Commun. 2012, 3, 1016. [Google Scholar] [CrossRef] [PubMed]
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deak, M.; et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012, 2, 120080. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Holmstrom, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef]
- Yuan, Y.; Zheng, Y.; Zhang, X.; Chen, Y.; Wu, X.; Wu, J.; Shen, Z.; Jiang, L.; Wang, L.; Yang, W.; et al. BNIP3L/NIX-mediated mitophagy protects against ischemic brain injury independent of PARK2. Autophagy 2017, 13, 1754–1766. [Google Scholar] [CrossRef]
- Shi, R.Y.; Zhu, S.H.; Li, V.; Gibson, S.B.; Xu, X.S.; Kong, J.M. BNIP3 interacting with LC3 triggers excessive mitophagy in delayed neuronal death in stroke. CNS Neurosci. Ther. 2014, 20, 1045–1055. [Google Scholar] [CrossRef]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef]
- Strappazzon, F.; Nazio, F.; Corrado, M.; Cianfanelli, V.; Romagnoli, A.; Fimia, G.M.; Campello, S.; Nardacci, R.; Piacentini, M.; Campanella, M.; et al. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 2015, 22, 419–432. [Google Scholar] [CrossRef]
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sorensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F.; et al. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014, 83, 278–286. [Google Scholar] [CrossRef]
- Lublin, F.D. New multiple sclerosis phenotypic classification. Eur. Neurol. 2014, 72 (Suppl. S1), 1–5. [Google Scholar] [CrossRef] [PubMed]
- Alirezaei, M.; Fox, H.S.; Flynn, C.T.; Moore, C.S.; Hebb, A.L.; Frausto, R.F.; Bhan, V.; Kiosses, W.B.; Whitton, J.L.; Robertson, G.S.; et al. Elevated ATG5 expression in autoimmune demyelination and multiple sclerosis. Autophagy 2009, 5, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Paunovic, V.; Petrovic, I.V.; Milenkovic, M.; Janjetovic, K.; Pravica, V.; Dujmovic, I.; Milosevic, E.; Martinovic, V.; Mesaros, S.; Drulovic, J.; et al. Autophagy-independent increase of ATG5 expression in T cells of multiple sclerosis patients. J. Neuroimmunol. 2018, 319, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Becher, J.; Simula, L.; Volpe, E.; Procaccini, C.; La Rocca, C.; D’Acunzo, P.; Cianfanelli, V.; Strappazzon, F.; Caruana, I.; Nazio, F.; et al. AMBRA1 Controls Regulatory T-Cell Differentiation and Homeostasis Upstream of the FOXO3-FOXP3 Axis. Dev. Cell 2018, 47, 592–607.e6. [Google Scholar] [CrossRef] [PubMed]
- Akatsuka, H.; Kuga, S.; Masuhara, K.; Davaadorj, O.; Okada, C.; Iida, Y.; Okada, Y.; Fukunishi, N.; Suzuki, T.; Hosomichi, K.; et al. AMBRA1 is involved in T cell receptor-mediated metabolic reprogramming through an ATG7-independent pathway. Biochem. Biophys. Res. Commun. 2017, 491, 1098–1104. [Google Scholar] [CrossRef]
- Astier, A.L. T-cell regulation by CD46 and its relevance in multiple sclerosis. Immunology 2008, 124, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Joubert, P.E.; Meiffren, G.; Gregoire, I.P.; Pontini, G.; Richetta, C.; Flacher, M.; Azocar, O.; Vidalain, P.O.; Vidal, M.; Lotteau, V.; et al. Autophagy induction by the pathogen receptor CD46. Cell Host Microbe 2009, 6, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Soldan, S.S.; Fogdell-Hahn, A.; Brennan, M.B.; Mittleman, B.B.; Ballerini, C.; Massacesi, L.; Seya, T.; McFarland, H.F.; Jacobson, S. Elevated serum and cerebrospinal fluid levels of soluble human herpesvirus type 6 cellular receptor, membrane cofactor protein, in patients with multiple sclerosis. Ann. Neurol. 2001, 50, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Wu, Z.Y.; Fang, F.; Guo, L.; Chen, D.; Chen, J.X.; Stern, D.; Taylor, G.A.; Jiang, H.; Yan, S.S. Genetic deficiency of Irgm1 (LRG-47) suppresses induction of experimental autoimmune encephalomyelitis by promoting apoptosis of activated CD4+ T cells. FASEB J. 2010, 24, 1583–1592. [Google Scholar] [CrossRef]
- Singh, S.B.; Ornatowski, W.; Vergne, I.; Naylor, J.; Delgado, M.; Roberts, E.; Ponpuak, M.; Master, S.; Pilli, M.; White, E.; et al. Human IRGM regulates autophagy and cell-autonomous immunity functions through mitochondria. Nat. Cell Biol. 2010, 12, 1154–1165. [Google Scholar] [CrossRef]
- Guo, X.; Zhang, W.; Wang, C.; Zhang, B.; Li, R.; Zhang, L.; Zhao, K.; Li, Y.; Tian, L.; Li, B.; et al. IRGM promotes the PINK1-mediated mitophagy through the degradation of Mitofilin in SH-SY5Y cells. FASEB J. 2020, 34, 14768–14779. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Parillon, X.; Zeng, S.; Han, S.; Eissa, N.T. Deficiency of autophagy in dendritic cells protects against experimental autoimmune encephalomyelitis. J. Biol. Chem. 2014, 289, 26525–26532. [Google Scholar] [CrossRef]
- Bonora, M.; De Marchi, E.; Patergnani, S.; Suski, J.M.; Celsi, F.; Bononi, A.; Giorgi, C.; Marchi, S.; Rimessi, A.; Duszynski, J.; et al. Tumor necrosis factor-alpha impairs oligodendroglial differentiation through a mitochondria-dependent process. Cell Death Differ. 2014, 21, 1198–1208. [Google Scholar] [CrossRef]
- Holman, S.P.; Lobo, A.S.; Novorolsky, R.J.; Nichols, M.; Fiander, M.D.J.; Konda, P.; Kennedy, B.E.; Gujar, S.; Robertson, G.S. Neuronal mitochondrial calcium uniporter deficiency exacerbates axonal injury and suppresses remyelination in mice subjected to experimental autoimmune encephalomyelitis. Exp. Neurol. 2020, 333, 113430. [Google Scholar] [CrossRef]
- Castellazzi, M.; Patergnani, S.; Donadio, M.; Giorgi, C.; Bonora, M.; Fainardi, E.; Casetta, I.; Granieri, E.; Pugliatti, M.; Pinton, P. Correlation between auto/mitophagic processes and magnetic resonance imaging activity in multiple sclerosis patients. J. Neuroinflamm. 2019, 16, 131. [Google Scholar] [CrossRef]
- Albanese, M.; Zagaglia, S.; Landi, D.; Boffa, L.; Nicoletti, C.G.; Marciani, M.G.; Mandolesi, G.; Marfia, G.A.; Buttari, F.; Mori, F.; et al. Cerebrospinal fluid lactate is associated with multiple sclerosis disease progression. J. Neuroinflamm. 2016, 13, 36. [Google Scholar] [CrossRef]
- Joodi Khanghah, O.; Nourazarian, A.; Khaki-Khatibi, F.; Nikanfar, M.; Laghousi, D.; Vatankhah, A.M.; Moharami, S. Evaluation of the Diagnostic and Predictive Value of Serum Levels of ANT1, ATG5, and Parkin in Multiple Sclerosis. Clin. Neurol. Neurosurg. 2020, 197, 106197. [Google Scholar] [CrossRef]
- Hassanpour, M.; Cheraghi, O.; Laghusi, D.; Nouri, M.; Panahi, Y. The relationship between ANT1 and NFL with autophagy and mitophagy markers in patients with multiple sclerosis. J. Clin. Neurosci. 2020, 78, 307–312. [Google Scholar] [CrossRef]
- Harberts, E.; Datta, D.; Chen, S.; Wohler, J.E.; Oh, U.; Jacobson, S. Translocator protein 18 kDa (TSPO) expression in multiple sclerosis patients. J. Neuroimmune Pharmacol. 2013, 8, 51–57. [Google Scholar] [CrossRef][Green Version]
- Nohara, S.; Ishii, A.; Yamamoto, F.; Yanagiha, K.; Moriyama, T.; Tozaka, N.; Miyake, Z.; Yatsuga, S.; Koga, Y.; Hosaka, T.; et al. GDF-15, a mitochondrial disease biomarker, is associated with the severity of multiple sclerosis. J. Neurol. Sci. 2019, 405, 116429. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, B.; Carinci, M.; Patergnani, S.; Pasquin, M.P.; Guarino, A.; Aziz, N.; Pinton, P.; Simonato, M.; Giorgi, C. The Dichotomous Role of Inflammation in the CNS: A Mitochondrial Point of View. Biomolecules 2020, 10, 1437. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.J.; Zhang, X.; Chen, W.W. Role of oxidative stress in Alzheimer’s disease. Biomed. Rep. 2016, 4, 519–522. [Google Scholar] [CrossRef] [PubMed]
- Mark, R.J.; Pang, Z.; Geddes, J.W.; Uchida, K.; Mattson, M.P. Amyloid beta-peptide impairs glucose transport in hippocampal and cortical neurons: Involvement of membrane lipid peroxidation. J. Neurosci. 1997, 17, 1046–1054. [Google Scholar] [CrossRef]
- De Wilde, M.C.; Vellas, B.; Girault, E.; Yavuz, A.C.; Sijben, J.W. Lower brain and blood nutrient status in Alzheimer’s disease: Results from meta-analyses. Alzheimer’s Dement. 2017, 3, 416–431. [Google Scholar] [CrossRef]
- Luth, H.J.; Munch, G.; Arendt, T. Aberrant expression of NOS isoforms in Alzheimer’s disease is structurally related to nitrotyrosine formation. Brain Res. 2002, 953, 135–143. [Google Scholar] [CrossRef]
- Kerr, J.S.; Adriaanse, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci. 2017, 40, 151–166. [Google Scholar] [CrossRef]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive involvement of autophagy in Alzheimer disease: An immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef]
- Nilsson, P.; Loganathan, K.; Sekiguchi, M.; Matsuba, Y.; Hui, K.; Tsubuki, S.; Tanaka, M.; Iwata, N.; Saito, T.; Saido, T.C. Abeta secretion and plaque formation depend on autophagy. Cell Rep. 2013, 5, 61–69. [Google Scholar] [CrossRef]
- Piras, A.; Collin, L.; Gruninger, F.; Graff, C.; Ronnback, A. Autophagic and lysosomal defects in human tauopathies: Analysis of post-mortem brain from patients with familial Alzheimer disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol. Commun. 2016, 4, 22. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Rispoli, J.; Kaphzan, H.; Klann, E.; Chen, E.I.; Kim, J.; Komatsu, M.; Abeliovich, A. Macroautophagy deficiency mediates age-dependent neurodegeneration through a phospho-tau pathway. Mol. Neurodegener. 2012, 7, 48. [Google Scholar] [CrossRef]
- Kesharwani, R.; Sarmah, D.; Kaur, H.; Mounika, L.; Verma, G.; Pabbala, V.; Kotian, V.; Kalia, K.; Borah, A.; Dave, K.R.; et al. Interplay between Mitophagy and Inflammasomes in Neurological Disorders. ACS Chem. Neurosci. 2019, 10, 2195–2208. [Google Scholar] [CrossRef] [PubMed]
- Martin-Maestro, P.; Gargini, R.; Perry, G.; Avila, J.; Garcia-Escudero, V. PARK2 enhancement is able to compensate mitophagy alterations found in sporadic Alzheimer’s disease. Hum. Mol. Genet. 2016, 25, 792–806. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Schwagerl, A.L.; Mohan, P.S.; Cataldo, A.M.; Vonsattel, J.P.; Kowall, N.W.; Nixon, R.A. Elevated levels of the endosomal-lysosomal proteinase cathepsin D in cerebrospinal fluid in Alzheimer disease. J. Neurochem. 1995, 64, 443–446. [Google Scholar] [CrossRef]
- Sundelof, J.; Sundstrom, J.; Hansson, O.; Eriksdotter-Jonhagen, M.; Giedraitis, V.; Larsson, A.; Degerman-Gunnarsson, M.; Ingelsson, M.; Minthon, L.; Blennow, K.; et al. Higher cathepsin B levels in plasma in Alzheimer’s disease compared to healthy controls. J. Alzheimer’s Dis. 2010, 22, 1223–1230. [Google Scholar] [CrossRef]
- Armstrong, A.; Mattsson, N.; Appelqvist, H.; Janefjord, C.; Sandin, L.; Agholme, L.; Olsson, B.; Svensson, S.; Blennow, K.; Zetterberg, H.; et al. Lysosomal network proteins as potential novel CSF biomarkers for Alzheimer’s disease. Neuromol. Med. 2014, 16, 150–160. [Google Scholar] [CrossRef]
- Sjodin, S.; Brinkmalm, G.; Ohrfelt, A.; Parnetti, L.; Paciotti, S.; Hansson, O.; Hardy, J.; Blennow, K.; Zetterberg, H.; Brinkmalm, A. Endo-lysosomal proteins and ubiquitin CSF concentrations in Alzheimer’s and Parkinson’s disease. Alzheimer’s Res. Ther. 2019, 11, 82. [Google Scholar] [CrossRef]
- Francois, A.; Julian, A.; Ragot, S.; Dugast, E.; Blanchard, L.; Brishoual, S.; Terro, F.; Chassaing, D.; Page, G.; Paccalin, M. Inflammatory Stress on Autophagy in Peripheral Blood Mononuclear Cells from Patients with Alzheimer’s Disease during 24 Months of Follow-Up. PLoS ONE 2015, 10, e0138326. [Google Scholar] [CrossRef]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef]
- Di Maio, R.; Barrett, P.J.; Hoffman, E.K.; Barrett, C.W.; Zharikov, A.; Borah, A.; Hu, X.; McCoy, J.; Chu, C.T.; Burton, E.A.; et al. alpha-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 2016, 8, 342ra378. [Google Scholar] [CrossRef]
- Walter, J.; Bolognin, S.; Antony, P.M.A.; Nickels, S.L.; Poovathingal, S.K.; Salamanca, L.; Magni, S.; Perfeito, R.; Hoel, F.; Qing, X.; et al. Neural Stem Cells of Parkinson’s Disease Patients Exhibit Aberrant Mitochondrial Morphology and Functionality. Stem Cell Rep. 2019, 12, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Ordureau, A.; Sarraf, S.A.; Duda, D.M.; Heo, J.M.; Jedrychowski, M.P.; Sviderskiy, V.O.; Olszewski, J.L.; Koerber, J.T.; Xie, T.; Beausoleil, S.A.; et al. Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol. Cell 2014, 56, 360–375. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Fiesel, F.C.; Truban, D.; Castanedes Casey, M.; Lin, W.L.; Soto, A.I.; Tacik, P.; Rousseau, L.G.; Diehl, N.N.; Heckman, M.G.; et al. Age- and disease-dependent increase of the mitophagy marker phospho-ubiquitin in normal aging and Lewy body disease. Autophagy 2018, 14, 1404–1418. [Google Scholar] [CrossRef] [PubMed]
- McLelland, G.L.; Goiran, T.; Yi, W.; Dorval, G.; Chen, C.X.; Lauinger, N.D.; Krahn, A.I.; Valimehr, S.; Rakovic, A.; Rouiller, I.; et al. Mfn2 ubiquitination by PINK1/parkin gates the p97-dependent release of ER from mitochondria to drive mitophagy. Elife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Dorn, G.W., 2nd. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef]
- Rakovic, A.; Grunewald, A.; Kottwitz, J.; Bruggemann, N.; Pramstaller, P.P.; Lohmann, K.; Klein, C. Mutations in PINK1 and Parkin impair ubiquitination of Mitofusins in human fibroblasts. PLoS ONE 2011, 6, e16746. [Google Scholar] [CrossRef]
- Rodriguez-Arribas, M.; Yakhine-Diop, S.M.S.; Pedro, J.M.B.; Gomez-Suaga, P.; Gomez-Sanchez, R.; Martinez-Chacon, G.; Fuentes, J.M.; Gonzalez-Polo, R.A.; Niso-Santano, M. Mitochondria-Associated Membranes (MAMs): Overview and Its Role in Parkinson’s Disease. Mol. Neurobiol. 2017, 54, 6287–6303. [Google Scholar] [CrossRef] [PubMed]
- Gautier, C.A.; Erpapazoglou, Z.; Mouton-Liger, F.; Muriel, M.P.; Cormier, F.; Bigou, S.; Duffaure, S.; Girard, M.; Foret, B.; Iannielli, A.; et al. The endoplasmic reticulum-mitochondria interface is perturbed in PARK2 knockout mice and patients with PARK2 mutations. Hum. Mol. Genet. 2016, 25, 2972–2984. [Google Scholar] [CrossRef]
- Missiroli, S.; Bonora, M.; Patergnani, S.; Poletti, F.; Perrone, M.; Gafa, R.; Magri, E.; Raimondi, A.; Lanza, G.; Tacchetti, C.; et al. PML at Mitochondria-Associated Membranes Is Critical for the Repression of Autophagy and Cancer Development. Cell Rep. 2016, 16, 2415–2427. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Ottolini, D.; Cali, T.; Negro, A.; Brini, M. The Parkinson disease-related protein DJ-1 counteracts mitochondrial impairment induced by the tumour suppressor protein p53 by enhancing endoplasmic reticulum-mitochondria tethering. Hum. Mol. Genet. 2013, 22, 2152–2168. [Google Scholar] [CrossRef]
- Kara, E.; Ling, H.; Pittman, A.M.; Shaw, K.; de Silva, R.; Simone, R.; Holton, J.L.; Warren, J.D.; Rohrer, J.D.; Xiromerisiou, G.; et al. The MAPT p.A152T variant is a risk factor associated with tauopathies with atypical clinical and neuropathological features. Neurobiol. Aging 2012, 33, 2231.e7–2231.e14. [Google Scholar] [CrossRef][Green Version]
- Lewis, J.; McGowan, E.; Rockwood, J.; Melrose, H.; Nacharaju, P.; Van Slegtenhorst, M.; Gwinn-Hardy, K.; Paul Murphy, M.; Baker, M.; Yu, X.; et al. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat. Genet. 2000, 25, 402–405. [Google Scholar] [CrossRef]
- Walker, R.H.; Friedman, J.; Wiener, J.; Hobler, R.; Gwinn-Hardy, K.; Adam, A.; DeWolfe, J.; Gibbs, R.; Baker, M.; Farrer, M.; et al. A family with a tau P301L mutation presenting with parkinsonism. Parkinsonism Relat. Disord. 2002, 9, 121–123. [Google Scholar] [CrossRef]
- Caballero, B.; Wang, Y.; Diaz, A.; Tasset, I.; Juste, Y.R.; Stiller, B.; Mandelkow, E.M.; Mandelkow, E.; Cuervo, A.M. Interplay of pathogenic forms of human tau with different autophagic pathways. Aging Cell 2018, 17. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.; Lee, S.B.; Lee, H.S.; Yang, H.O.; Park, J.; Kim, J.S.; Oh, E.; Park, S.; Jang, W. Cerebrospinal Fluid Levels of Autophagy-related Proteins Represent Potentially Novel Biomarkers of Early-Stage Parkinson’s Disease. Sci. Rep. 2018, 8, 16866. [Google Scholar] [CrossRef]
- Zhang, X.; Gao, F.; Wang, D.; Li, C.; Fu, Y.; He, W.; Zhang, J. Tau Pathology in Parkinson’s Disease. Front. Neurol. 2018, 9, 809. [Google Scholar] [CrossRef]
- Boman, A.; Svensson, S.; Boxer, A.; Rojas, J.C.; Seeley, W.W.; Karydas, A.; Miller, B.; Kagedal, K.; Svenningsson, P. Distinct Lysosomal Network Protein Profiles in Parkinsonian Syndrome Cerebrospinal Fluid. J. Parkinson’s Dis. 2016, 6, 307–315. [Google Scholar] [CrossRef]
- Klaver, A.C.; Coffey, M.P.; Aasly, J.O.; Loeffler, D.A. CSF lamp2 concentrations are decreased in female Parkinson’s disease patients with LRRK2 mutations. Brain Res. 2018, 1683, 12–16. [Google Scholar] [CrossRef]
- Miki, Y.; Shimoyama, S.; Kon, T.; Ueno, T.; Hayakari, R.; Tanji, K.; Matsumiya, T.; Tsushima, E.; Mori, F.; Wakabayashi, K.; et al. Alteration of autophagy-related proteins in peripheral blood mononuclear cells of patients with Parkinson’s disease. Neurobiol. Aging 2018, 63, 33–43. [Google Scholar] [CrossRef]
- Pyle, A.; Anugrha, H.; Kurzawa-Akanbi, M.; Yarnall, A.; Burn, D.; Hudson, G. Reduced mitochondrial DNA copy number is a biomarker of Parkinson’s disease. Neurobiol. Aging 2016, 38, 216.e7–216.e10. [Google Scholar] [CrossRef] [PubMed]
- Gui, Y.X.; Xu, Z.P.; Lv, W.; Zhao, J.J.; Hu, X.Y. Evidence for polymerase gamma, POLG1 variation in reduced mitochondrial DNA copy number in Parkinson’s disease. Parkinsonism Relat. Disord. 2015, 21, 282–286. [Google Scholar] [CrossRef]
- Podlesniy, P.; Vilas, D.; Taylor, P.; Shaw, L.M.; Tolosa, E.; Trullas, R. Mitochondrial DNA in CSF distinguishes LRRK2 from idiopathic Parkinson’s disease. Neurobiol. Dis. 2016, 94, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Iacobazzi, V.; Castegna, A.; Infantino, V.; Andria, G. Mitochondrial DNA methylation as a next-generation biomarker and diagnostic tool. Mol. Genet. Metab. 2013, 110, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Siman, R.; Cocca, R.; Dong, Y. The mTOR Inhibitor Rapamycin Mitigates Perforant Pathway Neurodegeneration and Synapse Loss in a Mouse Model of Early-Stage Alzheimer-Type Tauopathy. PLoS ONE 2015, 10, e0142340. [Google Scholar] [CrossRef] [PubMed]
- Berger, Z.; Ravikumar, B.; Menzies, F.M.; Oroz, L.G.; Underwood, B.R.; Pangalos, M.N.; Schmitt, I.; Wullner, U.; Evert, B.O.; O’Kane, C.J.; et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum. Mol. Genet. 2006, 15, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Yu, J.T.; Zhu, X.C.; Zhang, Q.Q.; Cao, L.; Wang, H.F.; Tan, M.S.; Gao, Q.; Qin, H.; Zhang, Y.D.; et al. Temsirolimus attenuates tauopathy in vitro and in vivo by targeting tau hyperphosphorylation and autophagic clearance. Neuropharmacology 2014, 85, 121–130. [Google Scholar] [CrossRef]
- Steele, J.W.; Lachenmayer, M.L.; Ju, S.; Stock, A.; Liken, J.; Kim, S.H.; Delgado, L.M.; Alfaro, I.E.; Bernales, S.; Verdile, G.; et al. Latrepirdine improves cognition and arrests progression of neuropathology in an Alzheimer’s mouse model. Mol. Psychiatry 2013, 18, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Li, X.; Zhu, J.X.; Xie, W.; Le, W.; Fan, Z.; Jankovic, J.; Pan, T. Resveratrol-activated AMPK/SIRT1/autophagy in cellular models of Parkinson’s disease. Neuro-Signals 2011, 19, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Perera, G.; Bhadbhade, M.; Halliday, G.M.; Dzamko, N. Autophagy activation promotes clearance of alpha-synuclein inclusions in fibril-seeded human neural cells. J. Biol. Chem. 2019, 294, 14241–14256. [Google Scholar] [CrossRef]
- Papp, D.; Kovacs, T.; Billes, V.; Varga, M.; Tarnoci, A.; Hackler, L., Jr.; Puskas, L.G.; Liliom, H.; Tarnok, K.; Schlett, K.; et al. AUTEN-67, an autophagy-enhancing drug candidate with potent antiaging and neuroprotective effects. Autophagy 2016, 12, 273–286. [Google Scholar] [CrossRef]
- Kovacs, T.; Billes, V.; Komlos, M.; Hotzi, B.; Manzeger, A.; Tarnoci, A.; Papp, D.; Szikszai, F.; Szinyakovics, J.; Racz, A.; et al. The small molecule AUTEN-99 (autophagy enhancer-99) prevents the progression of neurodegenerative symptoms. Sci. Rep. 2017, 7, 42014. [Google Scholar] [CrossRef]
- Sarkar, S.; Floto, R.A.; Berger, Z.; Imarisio, S.; Cordenier, A.; Pasco, M.; Cook, L.J.; Rubinsztein, D.C. Lithium induces autophagy by inhibiting inositol monophosphatase. J. Cell Biol. 2005, 170, 1101–1111. [Google Scholar] [CrossRef]
- Forlenza, O.V.; De-Paula, V.J.; Diniz, B.S. Neuroprotective effects of lithium: Implications for the treatment of Alzheimer’s disease and related neurodegenerative disorders. ACS Chem. Neurosci. 2014, 5, 443–450. [Google Scholar] [CrossRef]
- Forlenza, O.V.; Diniz, B.S.; Radanovic, M.; Santos, F.S.; Talib, L.L.; Gattaz, W.F. Disease-modifying properties of long-term lithium treatment for amnestic mild cognitive impairment: Randomised controlled trial. Br. J. Psychiatry 2011, 198, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Klucken, J.; Poehler, A.M.; Ebrahimi-Fakhari, D.; Schneider, J.; Nuber, S.; Rockenstein, E.; Schlotzer-Schrehardt, U.; Hyman, B.T.; McLean, P.J.; Masliah, E.; et al. Alpha-synuclein aggregation involves a bafilomycin A 1-sensitive autophagy pathway. Autophagy 2012, 8, 754–766. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Y.; Peng, C.; Shi, H.; Wang, S.; Wang, Q.; Wang, J.Z. Inhibition of autophagy causes tau proteolysis by activating calpain in rat brain. J. Alzheimer’s Dis. 2009, 16, 39–47. [Google Scholar] [CrossRef]
- Thome, R.; Moraes, A.S.; Bombeiro, A.L.; Farias Ados, S.; Francelin, C.; da Costa, T.A.; Di Gangi, R.; dos Santos, L.M.; de Oliveira, A.L.; Verinaud, L. Chloroquine treatment enhances regulatory T cells and reduces the severity of experimental autoimmune encephalomyelitis. PLoS ONE 2013, 8, e65913. [Google Scholar] [CrossRef]
- Thome, R.; Issayama, L.K.; DiGangi, R.; Bombeiro, A.L.; da Costa, T.A.; Ferreira, I.T.; de Oliveira, A.L.; Verinaud, L. Dendritic cells treated with chloroquine modulate experimental autoimmune encephalomyelitis. Immunol. Cell Biol. 2014, 92, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Sachs, H.H.; Bercury, K.K.; Popescu, D.C.; Narayanan, S.P.; Macklin, W.B. A new model of cuprizone-mediated demyelination/remyelination. ASN Neuro 2014, 6. [Google Scholar] [CrossRef]
- Yamate-Morgan, H.; Lauderdale, K.; Horeczko, J.; Merchant, U.; Tiwari-Woodruff, S.K. Functional Effects of Cuprizone-Induced Demyelination in the Presence of the mTOR-Inhibitor Rapamycin. Neuroscience 2019, 406, 667–683. [Google Scholar] [CrossRef] [PubMed]
- Lisi, L.; Navarra, P.; Cirocchi, R.; Sharp, A.; Stigliano, E.; Feinstein, D.L.; Dello Russo, C. Rapamycin reduces clinical signs and neuropathic pain in a chronic model of experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2012, 243, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Bagherpour, B.; Salehi, M.; Jafari, R.; Bagheri, A.; Kiani-Esfahani, A.; Edalati, M.; Kardi, M.T.; Shaygannejad, V. Promising effect of rapamycin on multiple sclerosis. Mult. Scler. Relat. Disord. 2018, 26, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Hertz, N.T.; Berthet, A.; Sos, M.L.; Thorn, K.S.; Burlingame, A.L.; Nakamura, K.; Shokat, K.M. A neo-substrate that amplifies catalytic activity of parkinson’s-disease-related kinase PINK1. Cell 2013, 154, 737–747. [Google Scholar] [CrossRef]
- Orr, A.L.; Rutaganira, F.U.; de Roulet, D.; Huang, E.J.; Hertz, N.T.; Shokat, K.M.; Nakamura, K. Long-term oral kinetin does not protect against alpha-synuclein-induced neurodegeneration in rodent models of Parkinson’s disease. Neurochem. Int. 2017, 109, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Shiba-Fukushima, K.; Inoshita, T.; Sano, O.; Iwata, H.; Ishikawa, K.I.; Okano, H.; Akamatsu, W.; Imai, Y.; Hattori, N. A Cell-Based High-Throughput Screening Identified Two Compounds that Enhance PINK1-Parkin Signaling. iScience 2020, 23, 101048. [Google Scholar] [CrossRef]
- Moskal, N.; Riccio, V.; Bashkurov, M.; Taddese, R.; Datti, A.; Lewis, P.N.; Angus McQuibban, G. ROCK inhibitors upregulate the neuroprotective Parkin-mediated mitophagy pathway. Nat. Commun. 2020, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Zetterberg, H.; Burnham, S.C. Blood-based molecular biomarkers for Alzheimer’s disease. Mol. Brain 2019, 12, 26. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Neurodegenerative Condition | Marker | Role | Type of Human Biofluid |
|---|---|---|---|
| MS | Parkin | Mitophagy regulator | Serum, CSF |
| ATG5 | Autophagy regulator | Serum, CSF | |
| Mitochondrial adenine nucleotide translocase 1 (ANT1) | Mitochondrial ADP/ATP translocase | Serum, CSF | |
| Translocator protein 18 kDa (TSPO) | Regulator of mPTP opening | Blood PBMCs | |
| Growth/differentiation factor 15 (GDF-15) | Mitochondrial disease marker | Serum | |
| TNFα | Proinflammatory cytokine | Serum, CSF | |
| Lactate | Mitochondrial dysfunction marker | Serum, CSF | |
| AD | BECN1 | Autophagy regulator | Blood PBMCs, serum |
| p62 | Autophagy regulator | Blood PBMCs | |
| LC3 | Autophagy regulator | Blood PBMCs | |
| ATG5 | Autophagy regulator | Plasma, serum | |
| Parkin | Mitophagy regulator | Serum | |
| EEA1, LAMP1, LAMP2, RAB3, and RAB7 | Lysosomal regulators | CSF | |
| PD | LC3B | Autophagy regulator | CSF |
| BECN1 | Autophagy regulator | CSF, blood PBMCs | |
| ATG5 | Autophagy regulator | CSF | |
| LAMP2 | Lysosomal regulator | CSF | |
| ULK1 | Autophagy regulator | Blood PBMCs | |
| ATG5 | Autophagy regulator | Blood PBMCs | |
| ATG4B | Autophagy regulator | Blood PBMCs | |
| ATG16L1 | Autophagy regulator | Blood PBMCs |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giorgi, C.; Bouhamida, E.; Danese, A.; Previati, M.; Pinton, P.; Patergnani, S. Relevance of Autophagy and Mitophagy Dynamics and Markers in Neurodegenerative Diseases. Biomedicines 2021, 9, 149. https://doi.org/10.3390/biomedicines9020149
Giorgi C, Bouhamida E, Danese A, Previati M, Pinton P, Patergnani S. Relevance of Autophagy and Mitophagy Dynamics and Markers in Neurodegenerative Diseases. Biomedicines. 2021; 9(2):149. https://doi.org/10.3390/biomedicines9020149
Chicago/Turabian StyleGiorgi, Carlotta, Esmaa Bouhamida, Alberto Danese, Maurizio Previati, Paolo Pinton, and Simone Patergnani. 2021. "Relevance of Autophagy and Mitophagy Dynamics and Markers in Neurodegenerative Diseases" Biomedicines 9, no. 2: 149. https://doi.org/10.3390/biomedicines9020149
APA StyleGiorgi, C., Bouhamida, E., Danese, A., Previati, M., Pinton, P., & Patergnani, S. (2021). Relevance of Autophagy and Mitophagy Dynamics and Markers in Neurodegenerative Diseases. Biomedicines, 9(2), 149. https://doi.org/10.3390/biomedicines9020149