PPARα-Selective Antagonist GW6471 Inhibits Cell Growth in Breast Cancer Stem Cells Inducing Energy Imbalance and Metabolic Stress

 ,
, 
 ,
, 

 ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Flow Cytometer Analysis
2.3. Cell Viability MTS Assay
2.4. Cell Cycle and Apoptosis Analysis by FACS
2.5. 3D Spheroid Assay
2.6. IncuCyte Cytotox Green Assay
2.7. IncuCyte Caspases 3/7 Assay
2.8. Western Blotting
2.9. Subcellular Protein Fractionation
2.10. Immunofluorescence
2.11. YAP/TAZ Immunofluorescence Quantification
2.12. Lipids Extraction
2.13. Thin Layer Chromatography
2.14. Quantitative Real Time-PCR
2.15. Glucose Uptake
2.16. L-Lactate Assay
2.17. IncuCyte Single Spheroid Invasion Assay
2.18. Statistical Analysis
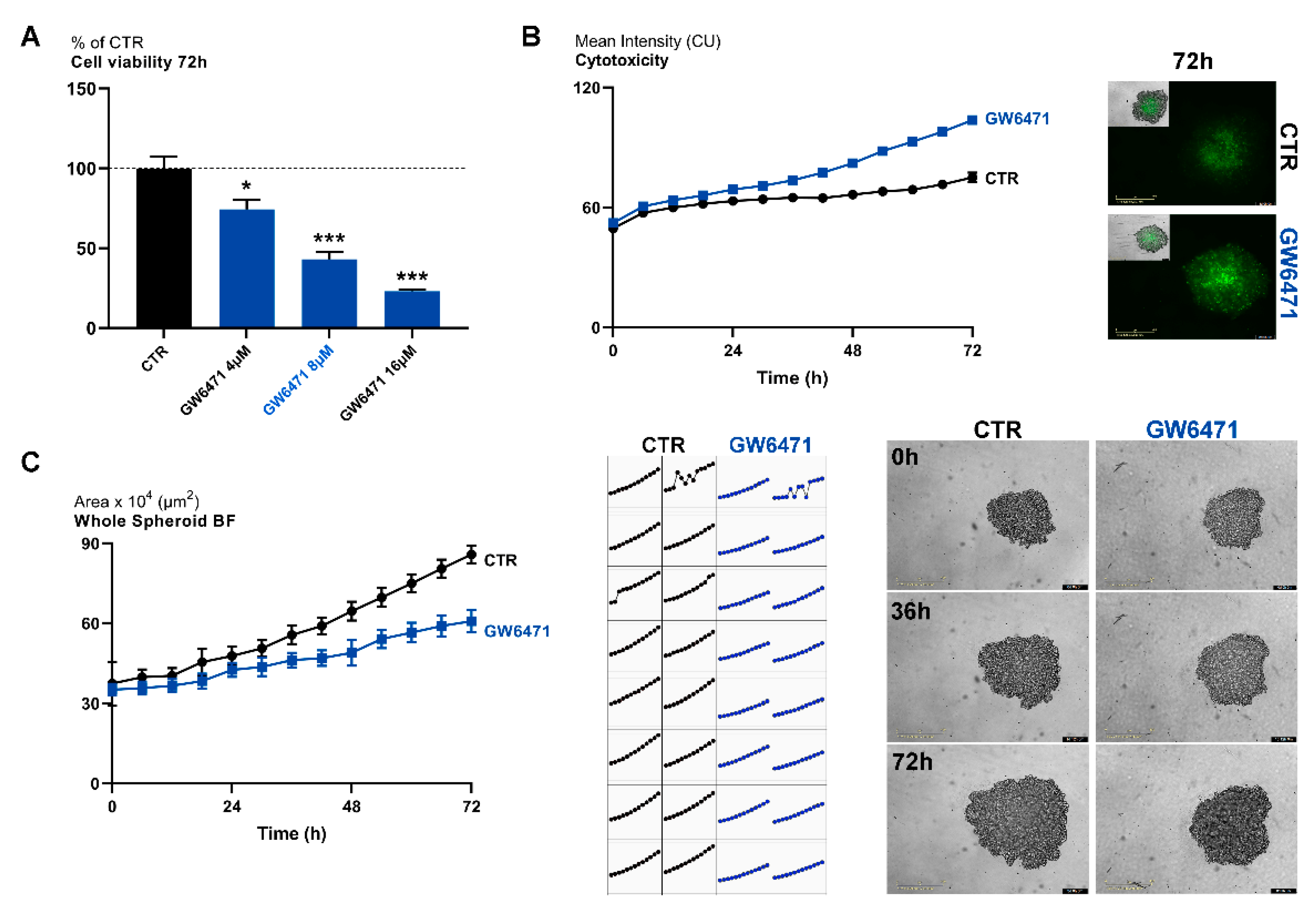
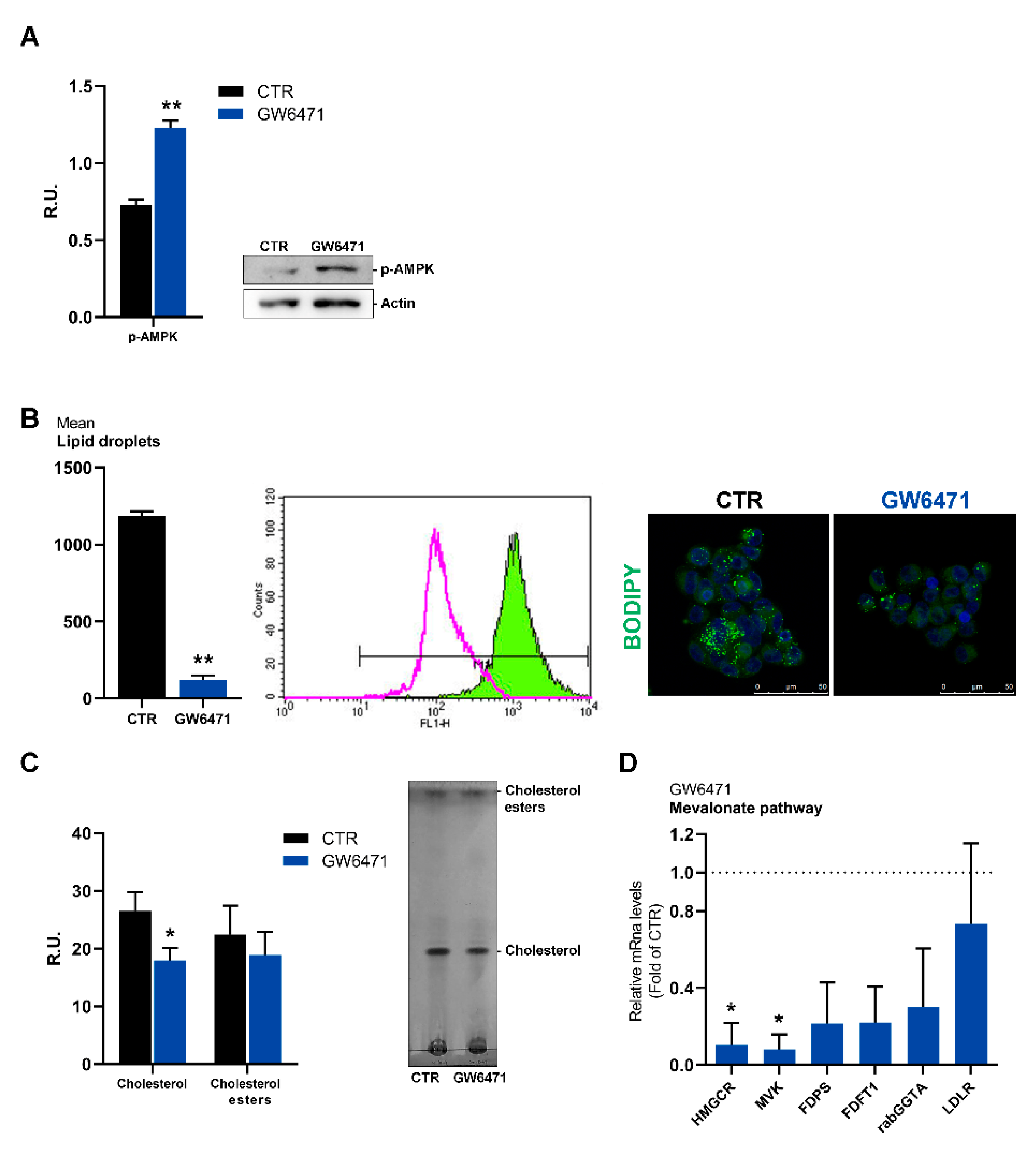
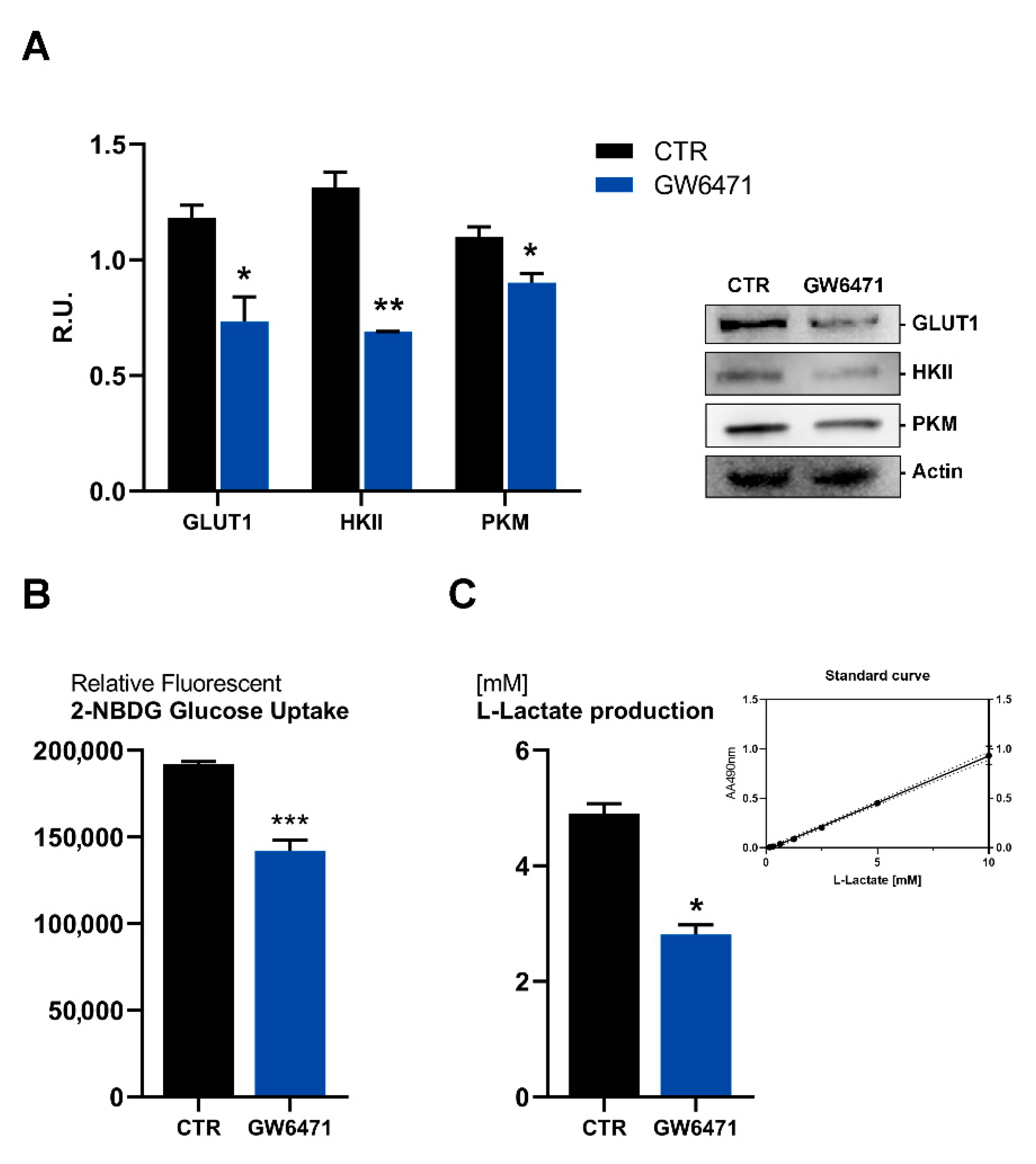
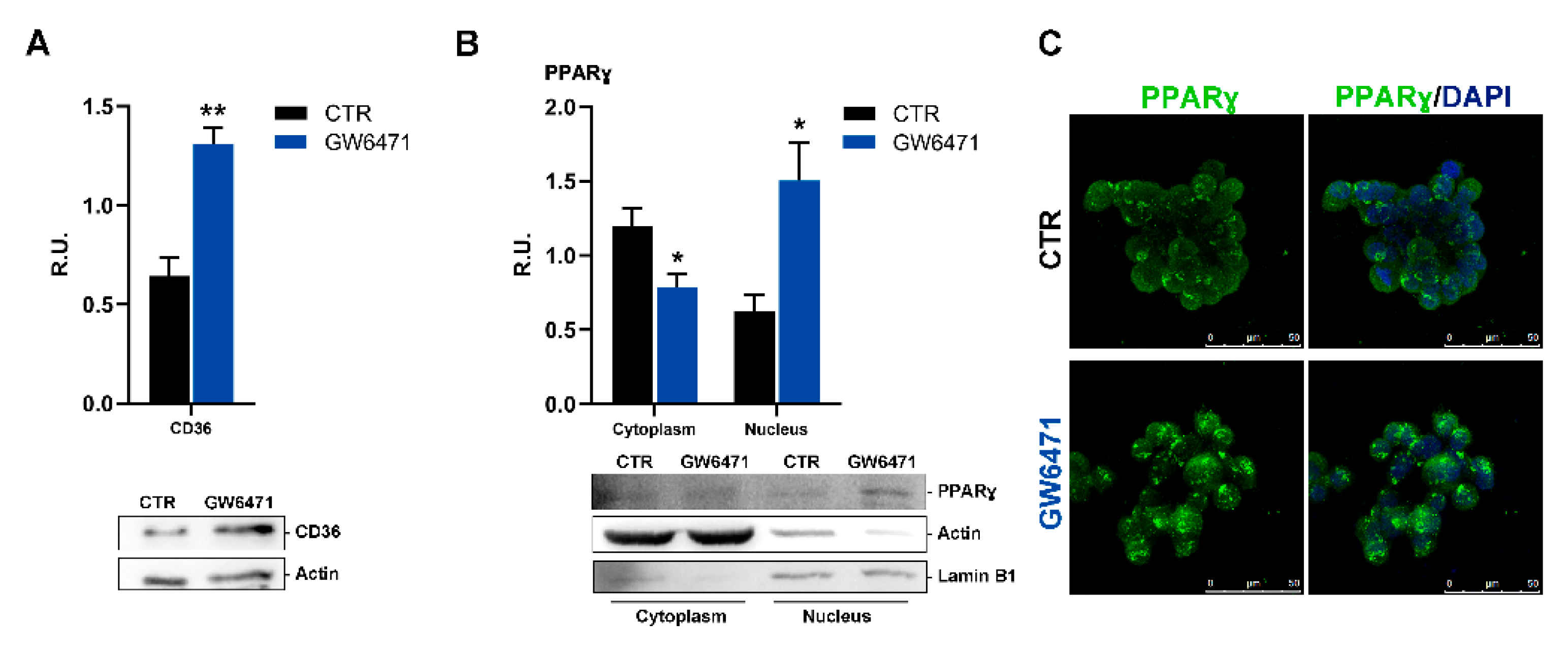
3. Results
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hurvitz, S.; Mead, M. Triple-Negative Breast Cancer: Advancements in Characterization and Treatment Approach. Curr. Opin. Obstet. Gynecol. 2016, 28, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Marra, A.; Trapani, D.; Viale, G.; Criscitiello, C.; Curigliano, G. Practical Classification of Triple-Negative Breast Cancer: Intratumoral Heterogeneity, Mechanisms of Drug Resistance, and Novel Therapies. NPJ Breast Cancer 2020, 6, 54. [Google Scholar] [CrossRef] [PubMed]
- O’Conor, C.J.; Chen, T.; González, I.; Cao, D.; Peng, Y. Cancer Stem Cells in Triple-Negative Breast Cancer: A Potential Target and Prognostic Marker. Biomark. Med. 2018, 12, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.K.; Desai, N.S. Cancer Stem Cells: Acquisition, Characteristics, Therapeutic Implications, Targeting Strategies and Future Prospects. Stem Cell Rev. Rep. 2019, 15, 331–355. [Google Scholar] [CrossRef] [PubMed]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.-G.; Lee, S.-H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer Stem Cells (CSCs) in Drug Resistance and Their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018, 2018, 5416923. [Google Scholar] [CrossRef]
- Akbar Samadani, A.; Keymoradzdeh, A.; Shams, S.; Soleymanpour, A.; Elham Norollahi, S.; Vahidi, S.; Rashidy-Pour, A.; Ashraf, A.; Mirzajani, E.; Khanaki, K.; et al. Mechanisms of Cancer Stem Cell Therapy. Clin. Chim. Acta 2020, 510, 581–592. [Google Scholar] [CrossRef]
- Fidoamore, A.; Cristiano, L.; Laezza, C.; Galzio, R.; Benedetti, E.; Cinque, B.; Antonosante, A.; d’Angelo, M.; Castelli, V.; Cifone, M.G.; et al. Energy Metabolism in Glioblastoma Stem Cells: PPARα a Metabolic Adaptor to Intratumoral Microenvironment. Oncotarget 2017, 8, 108430–108450. [Google Scholar] [CrossRef]
- Luo, Y.; Chen, L.; Wang, G.; Qian, G.; Liu, X.; Xiao, Y.; Wang, X.; Qian, K. PPARα Gene Is a Diagnostic and Prognostic Biomarker in Clear Cell Renal Cell Carcinoma by Integrated Bioinformatics Analysis. J. Cancer 2019, 10, 2319–2331. [Google Scholar] [CrossRef]
- Suchanek, K.M.; May, F.J.; Robinson, J.A.; Lee, W.J.; Holman, N.A.; Monteith, G.R.; Roberts-Thomson, S.J. Peroxisome Proliferator-Activated Receptor Alpha in the Human Breast Cancer Cell Lines MCF-7 and MDA-MB-231. Mol. Carcinog. 2002, 34, 165–171. [Google Scholar] [CrossRef]
- Ma, Y.; Temkin, S.M.; Hawkridge, A.M.; Guo, C.; Wang, W.; Wang, X.-Y.; Fang, X. Fatty Acid Oxidation: An Emerging Facet of Metabolic Transformation in Cancer. Cancer Lett. 2018, 435, 92–100. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, M.P.; Liesa, M. The Role of Mitochondrial Fat Oxidation in Cancer Cell Proliferation and Survival. Cells 2020, 9, 2600. [Google Scholar] [CrossRef] [PubMed]
- Antonosante, A.; d’Angelo, M.; Castelli, V.; Catanesi, M.; Iannotta, D.; Giordano, A.; Ippoliti, R.; Benedetti, E.; Cimini, A. The Involvement of PPARs in the Peculiar Energetic Metabolism of Tumor Cells. Int. J. Mol. Sci. 2018, 19, 1907. [Google Scholar] [CrossRef] [PubMed]
- Kamphorst, J.J.; Cross, J.R.; Fan, J.; de Stanchina, E.; Mathew, R.; White, E.P.; Thompson, C.B.; Rabinowitz, J.D. Hypoxic and Ras-Transformed Cells Support Growth by Scavenging Unsaturated Fatty Acids from Lysophospholipids. Proc. Natl. Acad. Sci. USA 2013, 110, 8882–8887. [Google Scholar] [CrossRef] [PubMed]
- Gruenbacher, G.; Thurnher, M. Mevalonate Metabolism in Immuno-Oncology. Front. Immunol. 2017, 8, 1714. [Google Scholar] [CrossRef]
- Gruenbacher, G.; Thurnher, M. Mevalonate Metabolism in Cancer Stemness and Trained Immunity. Front. Oncol. 2018, 8, 394. [Google Scholar] [CrossRef]
- Mancini, R.; Noto, A.; Pisanu, M.E.; De Vitis, C.; Maugeri-Saccà, M.; Ciliberto, G. Metabolic Features of Cancer Stem Cells: The Emerging Role of Lipid Metabolism. Oncogene 2018, 37, 2367–2378. [Google Scholar] [CrossRef]
- Friesen, J.A.; Rodwell, V.W. The 3-Hydroxy-3-Methylglutaryl Coenzyme-A (HMG-CoA) Reductases. Genome Biol. 2004, 5, 248. [Google Scholar] [CrossRef]
- Fatehi Hassanabad, A. Current Perspectives on Statins as Potential Anti-Cancer Therapeutics: Clinical Outcomes and Underlying Molecular Mechanisms. Transl. Lung Cancer Res. 2019, 8, 692–699. [Google Scholar] [CrossRef]
- Zhang, Q.; Dong, J.; Yu, Z. Pleiotropic Use of Statins as Non-Lipid-Lowering Drugs. Int. J. Biol Sci. 2020, 16, 2704–2711. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, J.; Lai, X.-N.; Jiao, X.-Q.; Xiong, J.-P.; Xiong, L.-X. Focus on Cdc42 in Breast Cancer: New Insights, Target Therapy Development and Non-Coding RNAs. Cells 2019, 8, 146. [Google Scholar] [CrossRef]
- Kim, D.; Rhee, S. Matrix Metalloproteinase-2 Regulates MDA-MB-231 Breast Cancer Cell Invasion Induced by Active Mammalian Diaphanous-Related Formin 1. Mol. Med. Rep. 2016, 14, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Fritz, G.; Just, I.; Kaina, B. Rho GTPases Are Over-Expressed in Human Tumors. Int. J. Cancer 1999, 81, 682–687. [Google Scholar] [CrossRef]
- Clendening, J.W.; Pandyra, A.; Boutros, P.C.; El Ghamrasni, S.; Khosravi, F.; Trentin, G.A.; Martirosyan, A.; Hakem, A.; Hakem, R.; Jurisica, I.; et al. Dysregulation of the Mevalonate Pathway Promotes Transformation. Proc. Natl. Acad. Sci. USA 2010, 107, 15051–15056. [Google Scholar] [CrossRef] [PubMed]
- Pampalakis, G.; Obasuyi, O.; Papadodima, O.; Chatziioannou, A.; Zoumpourlis, V.; Sotiropoulou, G. The KLK5 Protease Suppresses Breast Cancer by Repressing the Mevalonate Pathway. Oncotarget 2014, 5, 2390–2403. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. Links between Metabolism and Cancer. Genes Dev. 2012, 26, 877–890. [Google Scholar] [CrossRef] [PubMed]
- Yadav, U.P.; Singh, T.; Kumar, P.; Sharma, P.; Kaur, H.; Sharma, S.; Singh, S.; Kumar, S.; Mehta, K. Metabolic Adaptations in Cancer Stem Cells. Front. Oncol. 2020, 10, 1010. [Google Scholar] [CrossRef]
- Benedetti, E.; d’Angelo, M.; Ammazzalorso, A.; Gravina, G.L.; Laezza, C.; Antonosante, A.; Panella, G.; Cinque, B.; Cristiano, L.; Dhez, A.C.; et al. PPARα Antagonist AA452 Triggers Metabolic Reprogramming and Increases Sensitivity to Radiation Therapy in Human Glioblastoma Primary Cells: RADIO-SENSITIZATION OF GLIOBLASTOMA BY PPARα ANTAGONIST AA452. J. Cell. Physiol. 2017, 232, 1458–1466. [Google Scholar] [CrossRef] [PubMed]
- Abu Aboud, O.; Wettersten, H.I.; Weiss, R.H. Inhibition of PPARα Induces Cell Cycle Arrest and Apoptosis, and Synergizes with Glycolysis Inhibition in Kidney Cancer Cells. PLoS ONE 2013, 8, e71115. [Google Scholar] [CrossRef]
- Abu Aboud, O.; Donohoe, D.; Bultman, S.; Fitch, M.; Riiff, T.; Hellerstein, M.; Weiss, R.H. PPARα Inhibition Modulates Multiple Reprogrammed Metabolic Pathways in Kidney Cancer and Attenuates Tumor Growth. Am. J. Physiol. Cell Physiol. 2015, 308, C890–C898. [Google Scholar] [CrossRef]
- Castelli, V.; Piroli, A.; Marinangeli, F.; d’Angelo, M.; Benedetti, E.; Ippoliti, R.; Zis, P.; Varrassi, G.; Giordano, A.; Paladini, A.; et al. Local Anesthetics Counteract Cell Proliferation and Migration of Human Triple-negative Breast Cancer and Melanoma Cells. J. Cell. Physiol. 2020, 235, 3474–3484. [Google Scholar] [CrossRef] [PubMed]
- Brandolini, L.; Cristiano, L.; Fidoamore, A.; De Pizzol, M.; Di Giacomo, E.; Florio, T.M.; Confalone, G.; Galante, A.; Cinque, B.; Benedetti, E.; et al. Targeting CXCR1 on Breast Cancer Stem Cells: Signaling Pathways and Clinical Application Modelling. Oncotarget 2015, 6, 43375–43394. [Google Scholar] [CrossRef] [PubMed]
- Antonosante, A.; Brandolini, L.; d’Angelo, M.; Benedetti, E.; Castelli, V.; Maestro, M.D.; Luzzi, S.; Giordano, A.; Cimini, A.; Allegretti, M. Autocrine CXCL8-Dependent Invasiveness Triggers Modulation of Actin Cytoskeletal Network and Cell Dynamics. Aging 2020, 12, 1928–1951. [Google Scholar] [CrossRef] [PubMed]
- Mueller, E.; Sarraf, P.; Tontonoz, P.; Evans, R.M.; Martin, K.J.; Zhang, M.; Fletcher, C.; Singer, S.; Spiegelman, B.M. Terminal Differentiation of Human Breast Cancer through PPAR Gamma. Mol. Cell 1998, 1, 465–470. [Google Scholar] [CrossRef]
- Zhu, L.; Yu, X.; Xing, S.; Jin, F.; Yang, W.-J. Involvement of AMP-Activated Protein Kinase (AMPK) in Regulation of Cell Membrane Potential in a Gastric Cancer Cell Line. Sci. Rep. 2018, 8, 6028. [Google Scholar] [CrossRef] [PubMed]
- Sanli, T.; Steinberg, G.R.; Singh, G.; Tsakiridis, T. AMP-Activated Protein Kinase (AMPK) beyond Metabolism: A Novel Genomic Stress Sensor Participating in the DNA Damage Response Pathway. Cancer Biol. Ther. 2014, 15, 156–169. [Google Scholar] [CrossRef] [PubMed]
- Olzmann, J.A.; Carvalho, P. Dynamics and Functions of Lipid Droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef] [PubMed]
- Petan, T. Lipid Droplets in Cancer. In Reviews of Physiology, Biochemistry and Pharmacology; Springer: Berlin/Heidelberg, Germany, 2020. [Google Scholar]
- Cruz, A.L.S.; Barreto, E.D.A.; Fazolini, N.P.B.; Viola, J.P.B.; Bozza, P.T. Lipid Droplets: Platforms with Multiple Functions in Cancer Hallmarks. Cell Death Dis. 2020, 11, 105. [Google Scholar] [CrossRef]
- Bujold, K.; Rhainds, D.; Jossart, C.; Febbraio, M.; Marleau, S.; Ong, H. CD36-Mediated Cholesterol Efflux Is Associated with PPARγ Activation via a MAPK-Dependent COX-2 Pathway in Macrophages. Cardiovasc. Res. 2009, 83, 457–464. [Google Scholar] [CrossRef]
- Kim, K.Y.; Kim, S.S.; Cheon, H.G. Differential Anti-Proliferative Actions of Peroxisome Proliferator-Activated Receptor-Gamma Agonists in MCF-7 Breast Cancer Cells. Biochem. Pharmacol. 2006, 72, 530–540. [Google Scholar] [CrossRef]
- Youssef, J.; Badr, M. Peroxisome Proliferator-Activated Receptors and Cancer: Challenges and Opportunities. Br. J. Pharmacol. 2011, 164, 68–82. [Google Scholar] [CrossRef] [PubMed]
- Jarrar, M.H.; Baranova, A. PPARgamma Activation by Thiazolidinediones (TZDs) May Modulate Breast Carcinoma Outcome: The Importance of Interplay with TGFbeta Signalling. J. Cell. Mol. Med. 2007, 11, 71–87. [Google Scholar] [CrossRef] [PubMed]
- Basu-Roy, U.; Han, E.; Rattanakorn, K.; Gadi, A.; Verma, N.; Maurizi, G.; Gunaratne, P.H.; Coarfa, C.; Kennedy, O.D.; Garabedian, M.J.; et al. PPARγ Agonists Promote Differentiation of Cancer Stem Cells by Restraining YAP Transcriptional Activity. Oncotarget 2016, 7, 60954–60970. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, H.; Li, Y.; Xia, D.; Yang, L.; Ma, Y.; Li, H. The Role of YAP/TAZ Activity in Cancer Metabolic Reprogramming. Mol. Cancer 2018, 17, 134. [Google Scholar] [CrossRef] [PubMed]
- Velasco-Velázquez, M.A.; Popov, V.M.; Lisanti, M.P.; Pestell, R.G. The Role of Breast Cancer Stem Cells in Metastasis and Therapeutic Implications. Am. J. Pathol. 2011, 179, 2–11. [Google Scholar] [CrossRef]
- Latruffe, N.; Cherkaoui Malki, M.; Nicolas-Frances, V.; Clemencet, M.C.; Jannin, B.; Berlot, J.P. Regulation of the Peroxisomal Beta-Oxidation-Dependent Pathway by Peroxisome Proliferator-Activated Receptor Alpha and Kinases. Biochem. Pharmacol. 2000, 60, 1027–1032. [Google Scholar] [CrossRef]
- Miziorko, H.M. Enzymes of the Mevalonate Pathway of Isoprenoid Biosynthesis. Arch. Biochem. Biophys. 2011, 505, 131–143. [Google Scholar] [CrossRef]
- Laezza, C.; D’Alessandro, A.; Di Croce, L.; Picardi, P.; Ciaglia, E.; Pisanti, S.; Malfitano, A.M.; Comegna, M.; Faraonio, R.; Gazzerro, P.; et al. P53 Regulates the Mevalonate Pathway in Human Glioblastoma Multiforme. Cell Death Dis. 2015, 6, e1909. [Google Scholar] [CrossRef] [PubMed]
- Hershey, B.J.; Vazzana, R.; Joppi, D.L.; Havas, K.M. Lipid Droplets Define a Sub-Population of Breast Cancer Stem Cells. J. Clin. Med. 2019, 9, 87. [Google Scholar] [CrossRef]
- Velasco-Velázquez, M.A.; Homsi, N.; De La Fuente, M.; Pestell, R.G. Breast Cancer Stem Cells. Int. J. Biochem. Cell Biol. 2012, 44, 573–577. [Google Scholar] [CrossRef]
- Liu, S.; Dontu, G.; Wicha, M.S. Mammary Stem Cells, Self-Renewal Pathways, and Carcinogenesis. Breast Cancer Res. 2005, 7, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt Signaling in Cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.; Ablett, M.P.; Spence, K.; Landberg, G.; Sims, A.H.; Clarke, R.B. Wnt Pathway Activity in Breast Cancer Sub-Types and Stem-like Cells. PLoS ONE 2013, 8, e67811. [Google Scholar] [CrossRef] [PubMed]
- Khramtsov, A.I.; Khramtsova, G.F.; Tretiakova, M.; Huo, D.; Olopade, O.I.; Goss, K.H. Wnt/Beta-Catenin Pathway Activation Is Enriched in Basal-like Breast Cancers and Predicts Poor Outcome. Am. J. Pathol. 2010, 176, 2911–2920. [Google Scholar] [CrossRef] [PubMed]
- Shang, S.; Hua, F.; Hu, Z.-W. The Regulation of β-Catenin Activity and Function in Cancer: Therapeutic Opportunities. Oncotarget 2017, 8, 33972–33989. [Google Scholar] [CrossRef] [PubMed]
- Jeselsohn, R.; Brown, N.E.; Arendt, L.; Klebba, I.; Hu, M.G.; Kuperwasser, C.; Hinds, P.W. Cyclin D1 Kinase Activity Is Required for the Self-Renewal of Mammary Stem and Progenitor Cells That Are Targets of MMTV-ErbB2 Tumorigenesis. Cancer Cell 2010, 17, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Lecarpentier, Y.; Schussler, O.; Hébert, J.-L.; Vallée, A. Multiple Targets of the Canonical WNT/β-Catenin Signaling in Cancers. Front. Oncol. 2019, 9, 1248. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Hardie, D.G. Sensing of Energy and Nutrients by AMP-Activated Protein Kinase. Am. J. Clin. Nutr. 2011, 93, 891S–896S. [Google Scholar] [CrossRef]
- Bungard, D.; Fuerth, B.J.; Zeng, P.-Y.; Faubert, B.; Maas, N.L.; Viollet, B.; Carling, D.; Thompson, C.B.; Jones, R.G.; Berger, S.L. Signaling Kinase AMPK Activates Stress-Promoted Transcription via Histone H2B Phosphorylation. Science 2010, 329, 1201–1205. [Google Scholar] [CrossRef]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Arkwright, R.T.; Deshmukh, R.; Adapa, N.; Stevens, R.; Zonder, E.; Zhang, Z.; Farshi, P.; Ahmed, R.S.I.; El-Banna, H.A.; Chan, T.-H.; et al. Lessons from Nature: Sources and Strategies for Developing AMPK Activators for Cancer Chemotherapeutics. Anticancer Agents Med. Chem. 2015, 15, 657–671. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Vincent, E.E.; Poffenberger, M.C.; Jones, R.G. The AMP-Activated Protein Kinase (AMPK) and Cancer: Many Faces of a Metabolic Regulator. Cancer Lett. 2015, 356, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Papi, A.; Guarnieri, T.; Storci, G.; Santini, D.; Ceccarelli, C.; Taffurelli, M.; De Carolis, S.; Avenia, N.; Sanguinetti, A.; Sidoni, A.; et al. Nuclear Receptors Agonists Exert Opposing Effects on the Inflammation Dependent Survival of Breast Cancer Stem Cells. Cell Death Differ. 2012, 19, 1208–1219. [Google Scholar] [CrossRef] [PubMed]
- Veliceasa, D.; Schulze-Hoëpfner, F.T.; Volpert, O.V. PPARgamma and Agonists against Cancer: Rational Design of Complementation Treatments. PPAR Res. 2008, 2008, 945275. [Google Scholar] [CrossRef]
- Vella, V.; Nicolosi, M.L.; Giuliano, S.; Bellomo, M.; Belfiore, A.; Malaguarnera, R. PPAR-γ Agonists As Antineoplastic Agents in Cancers with Dysregulated IGF Axis. Front. Endocrinol. 2017, 8. [Google Scholar] [CrossRef]
- Zaytseva, Y.Y.; Wang, X.; Southard, R.C.; Wallis, N.K.; Kilgore, M.W. Down-Regulation of PPARgamma1 Suppresses Cell Growth and Induces Apoptosis in MCF-7 Breast Cancer Cells. Mol. Cancer 2008, 7, 90. [Google Scholar] [CrossRef]
- Benedetti, E.; Galzio, R.; Cinque, B.; Biordi, L.; D’Amico, M.A.; D’Angelo, B.; Laurenti, G.; Ricci, A.; Festuccia, C.; Cifone, M.G.; et al. Biomolecular Characterization of Human Glioblastoma Cells in Primary Cultures: Differentiating and Antiangiogenic Effects of Natural and Synthetic PPARγ Agonists. J. Cell. Physiol. 2008, 217, 93–102. [Google Scholar] [CrossRef]
- Kuramoto, K.; Yamamoto, M.; Suzuki, S.; Togashi, K.; Sanomachi, T.; Kitanaka, C.; Okada, M. Inhibition of the Lipid Droplet-Peroxisome Proliferator-Activated Receptor α Axis Suppresses Cancer Stem Cell Properties. Genes 2021, 12, 99. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castelli, V.; Catanesi, M.; Alfonsetti, M.; Laezza, C.; Lombardi, F.; Cinque, B.; Cifone, M.G.; Ippoliti, R.; Benedetti, E.; Cimini, A.; et al. PPARα-Selective Antagonist GW6471 Inhibits Cell Growth in Breast Cancer Stem Cells Inducing Energy Imbalance and Metabolic Stress. Biomedicines 2021, 9, 127. https://doi.org/10.3390/biomedicines9020127
Castelli V, Catanesi M, Alfonsetti M, Laezza C, Lombardi F, Cinque B, Cifone MG, Ippoliti R, Benedetti E, Cimini A, et al. PPARα-Selective Antagonist GW6471 Inhibits Cell Growth in Breast Cancer Stem Cells Inducing Energy Imbalance and Metabolic Stress. Biomedicines. 2021; 9(2):127. https://doi.org/10.3390/biomedicines9020127
Chicago/Turabian StyleCastelli, Vanessa, Mariano Catanesi, Margherita Alfonsetti, Chiara Laezza, Francesca Lombardi, Benedetta Cinque, Maria Grazia Cifone, Rodolfo Ippoliti, Elisabetta Benedetti, Annamaria Cimini, and et al. 2021. "PPARα-Selective Antagonist GW6471 Inhibits Cell Growth in Breast Cancer Stem Cells Inducing Energy Imbalance and Metabolic Stress" Biomedicines 9, no. 2: 127. https://doi.org/10.3390/biomedicines9020127
APA StyleCastelli, V., Catanesi, M., Alfonsetti, M., Laezza, C., Lombardi, F., Cinque, B., Cifone, M. G., Ippoliti, R., Benedetti, E., Cimini, A., & d’Angelo, M. (2021). PPARα-Selective Antagonist GW6471 Inhibits Cell Growth in Breast Cancer Stem Cells Inducing Energy Imbalance and Metabolic Stress. Biomedicines, 9(2), 127. https://doi.org/10.3390/biomedicines9020127