
Butyrate Protects against Diet-Induced NASH and Liver Fibrosis and Suppresses Specific Non-Canonical TGF-β Signaling Pathways in Human Hepatic Stellate Cells

 , , , , , and
, , , , , and
Abstract
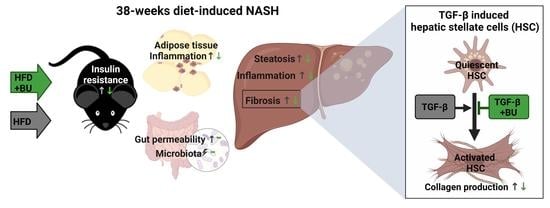
:
1. Introduction
2. Materials and Methods
2.1. Animal Experiment
2.2. Blood Chemistry
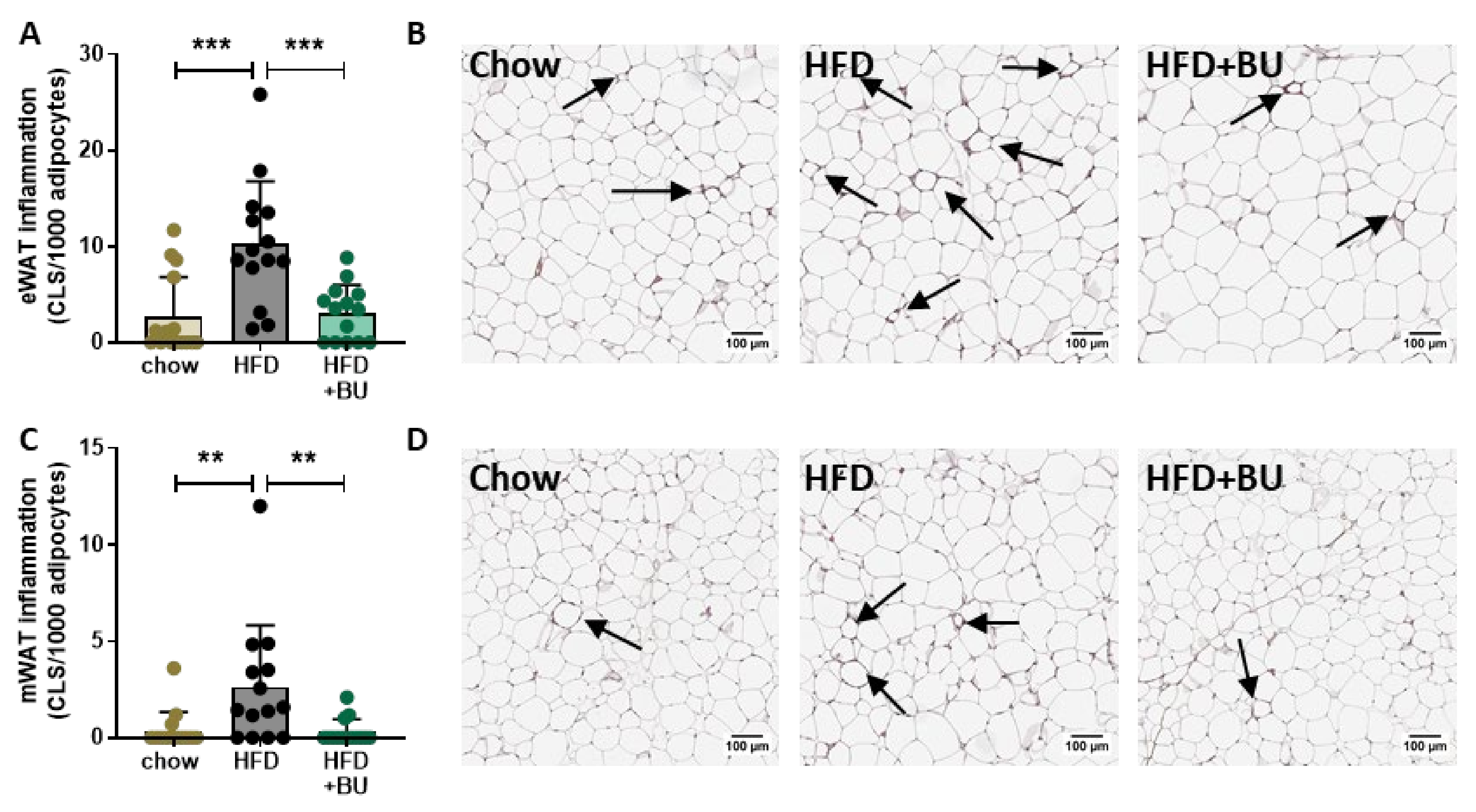
2.3. Adipose Tissue Histopathology Analysis
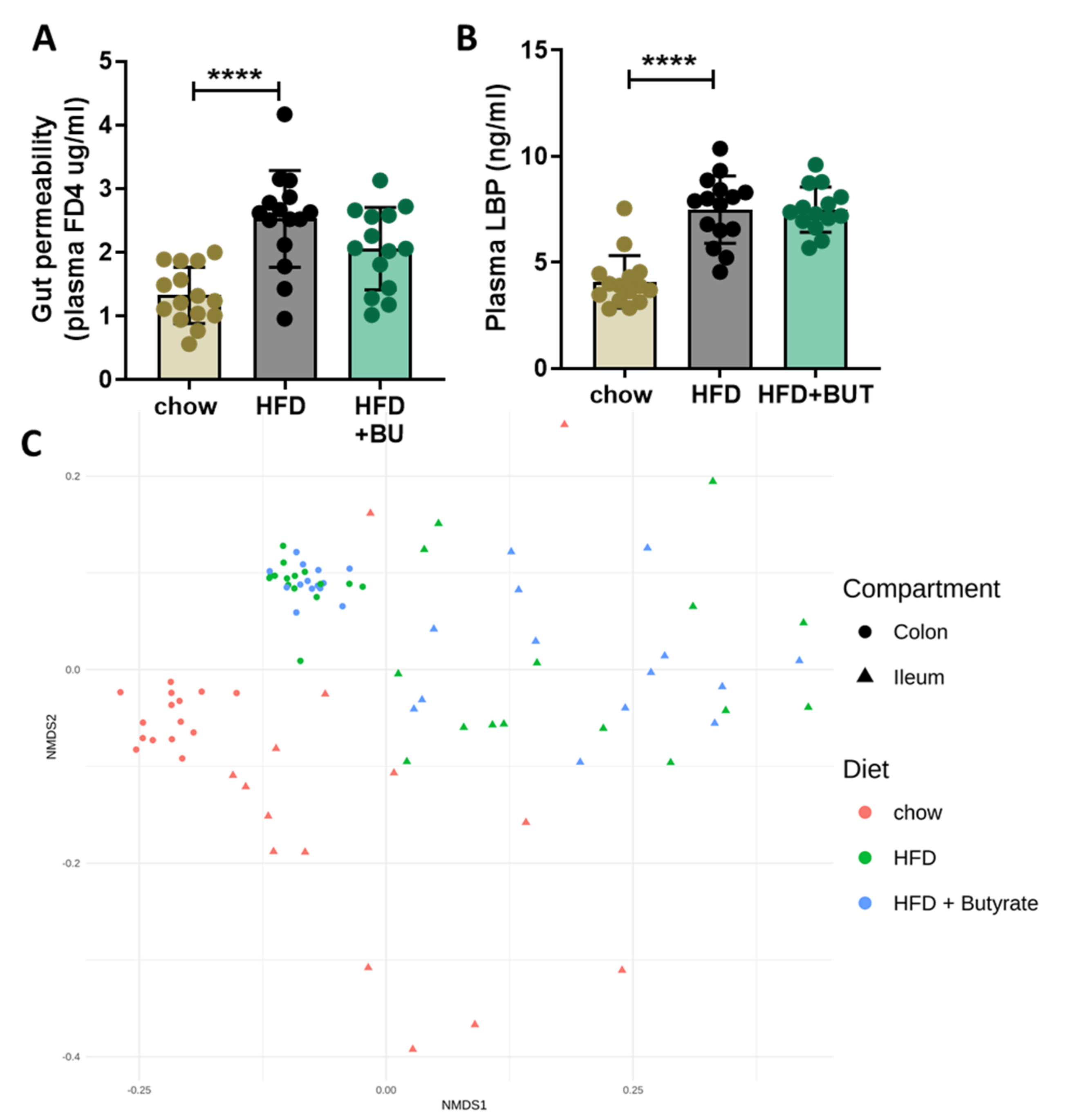
2.4. Gut Permeability and Microbiota Composition Analysis
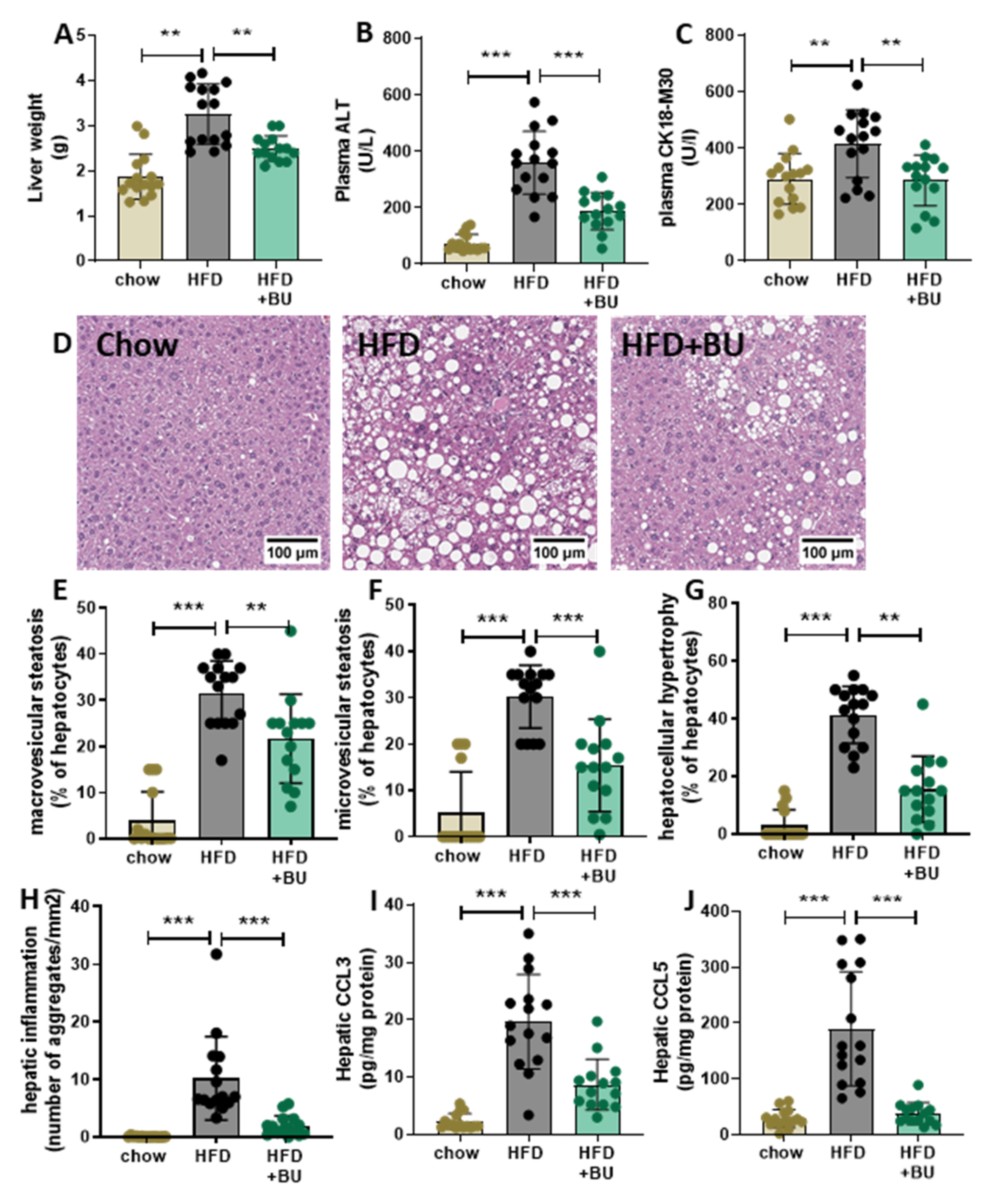
2.5. Liver Histopathology and Biochemistry
2.6. Primary Human Hepatic Stellate Cell Experiments, Gene Expression Analysis, and Protein Expression Using Western Blot Analysis
2.7. Statistics
3. Results
3.1. Butyrate Attenuates HFD-Induced Obesity, Improves Metabolic Risk Factors, and Reduces Adipose Tissue Inflammation
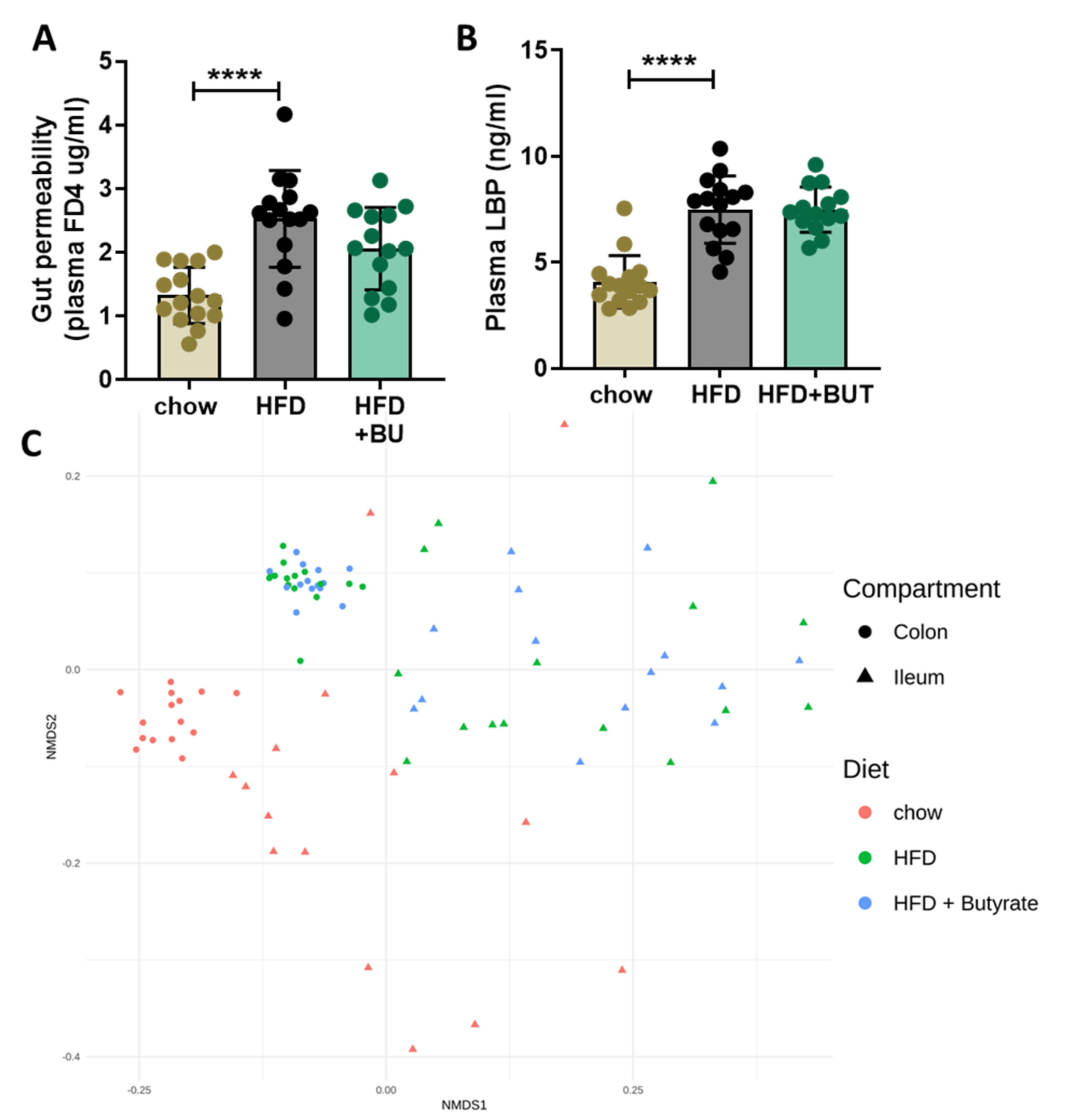
3.2. Butyrate Did Not Affect the HFD-Induced Increase in Gut Permeability or Changes in Microbiota Composition
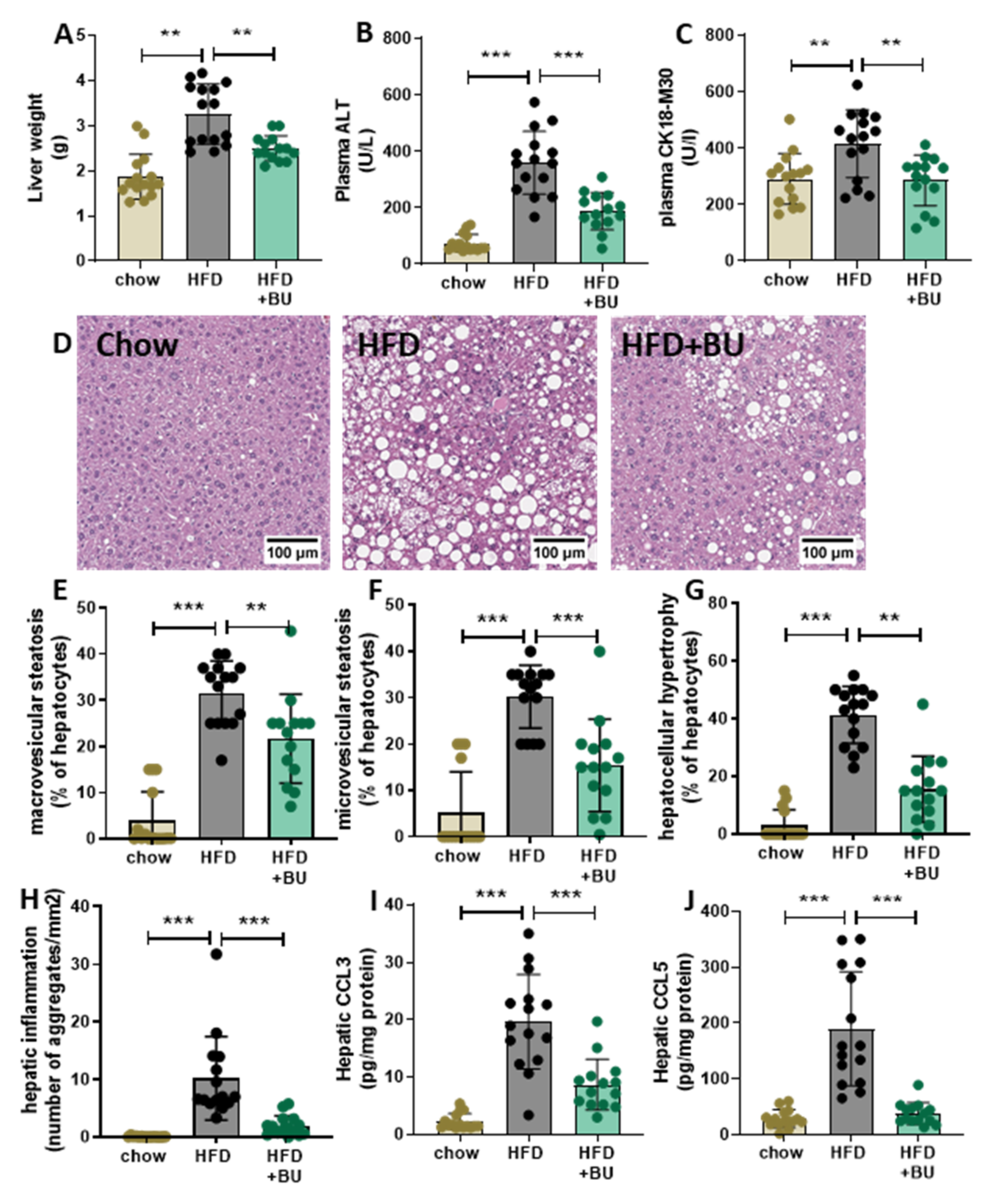
3.3. Butyrate Protected Mice against NASH Development
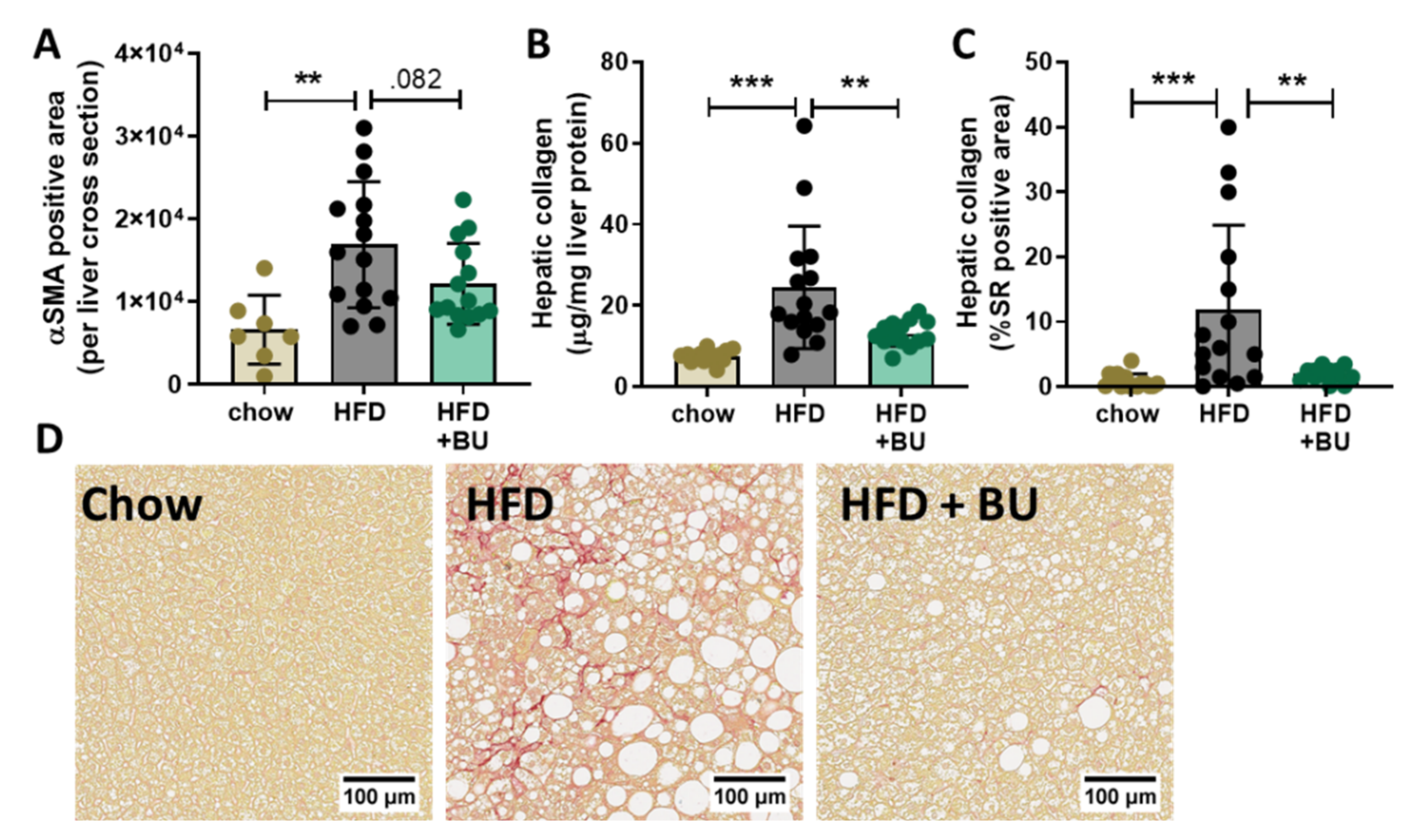
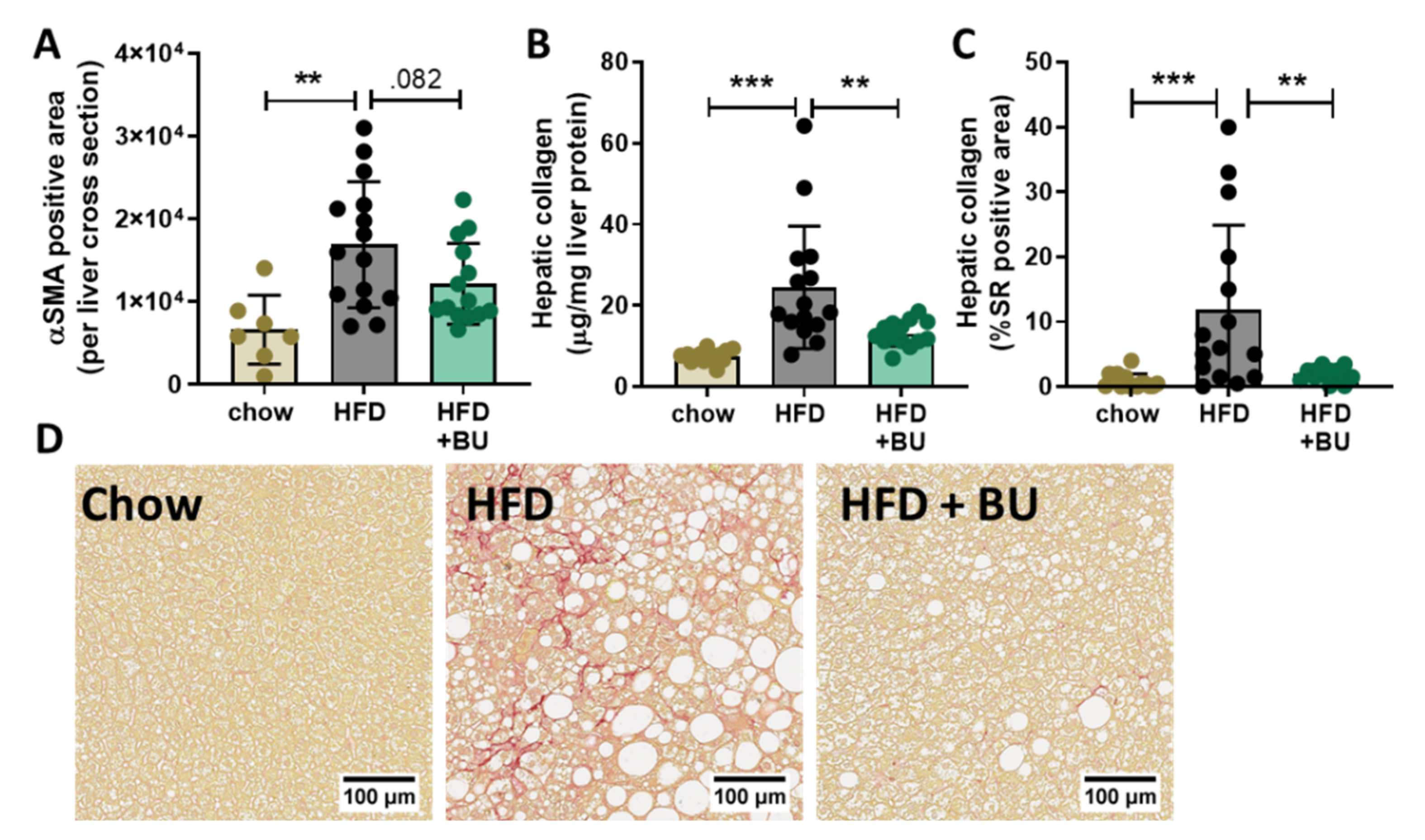
3.4. Butyrate Reduced Liver Fibrosis
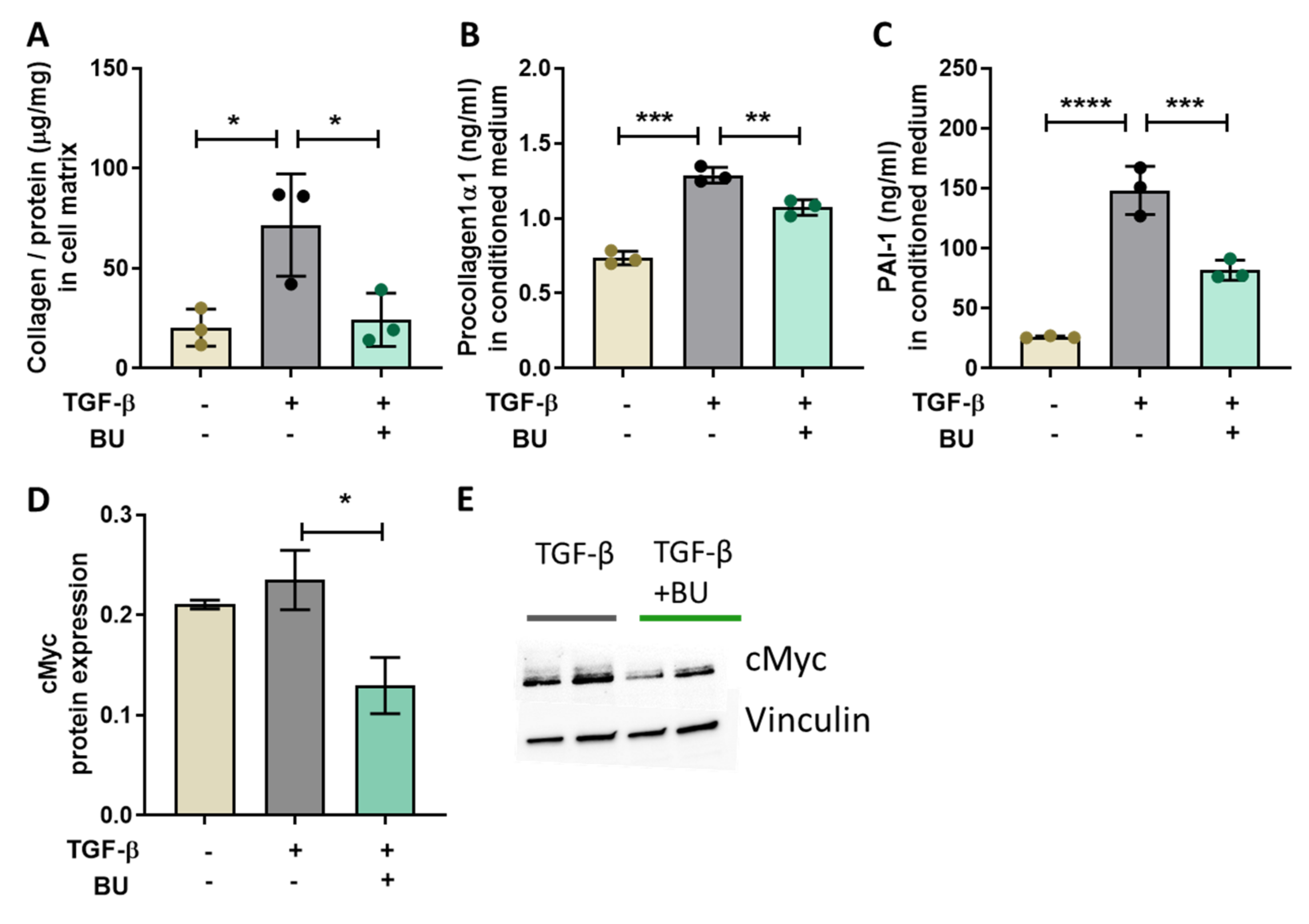
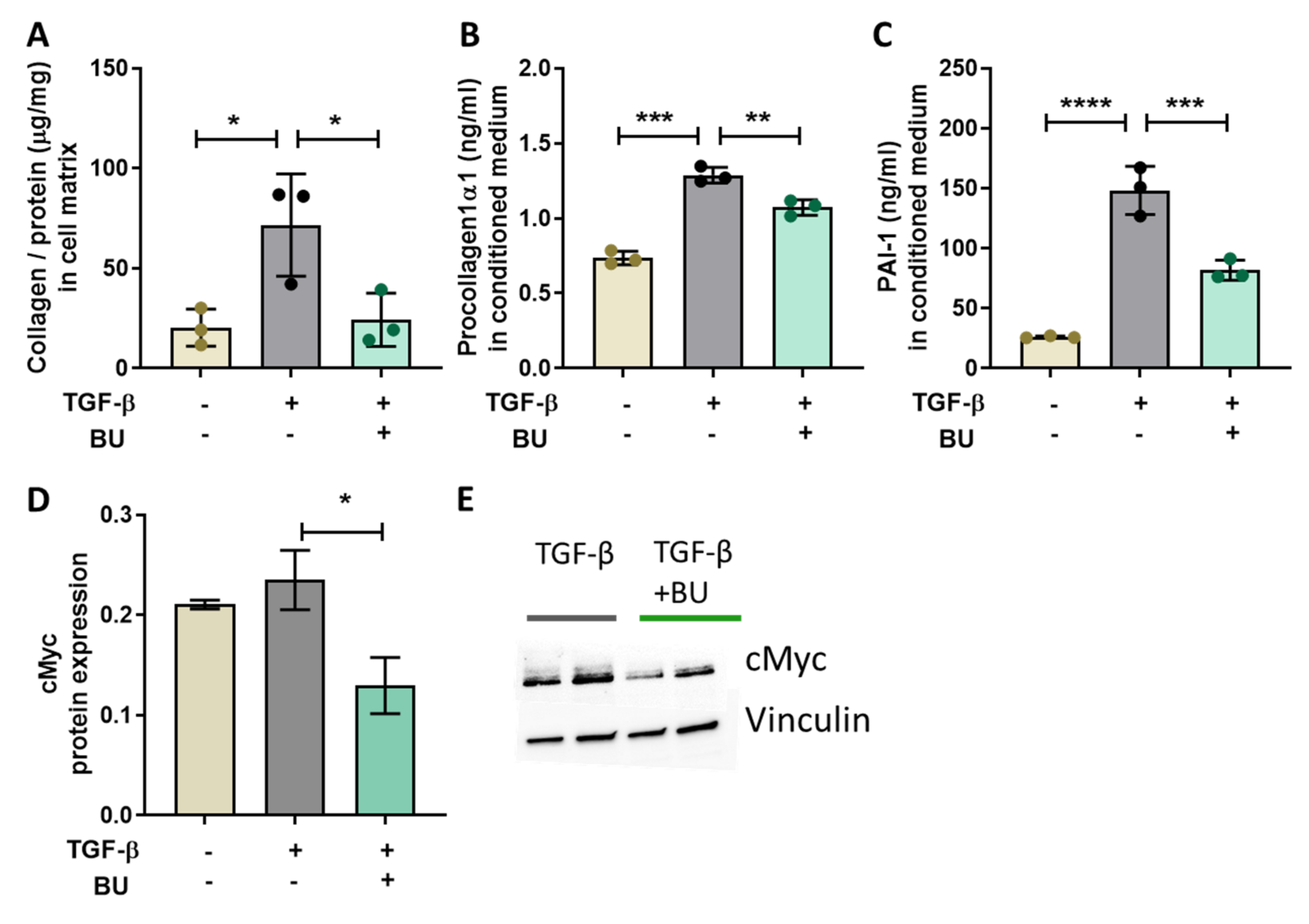
3.5. Butyrate Directly Reduced TGF-β-Induced Collagen Production in Primary Human Hepatic Stellate Cells
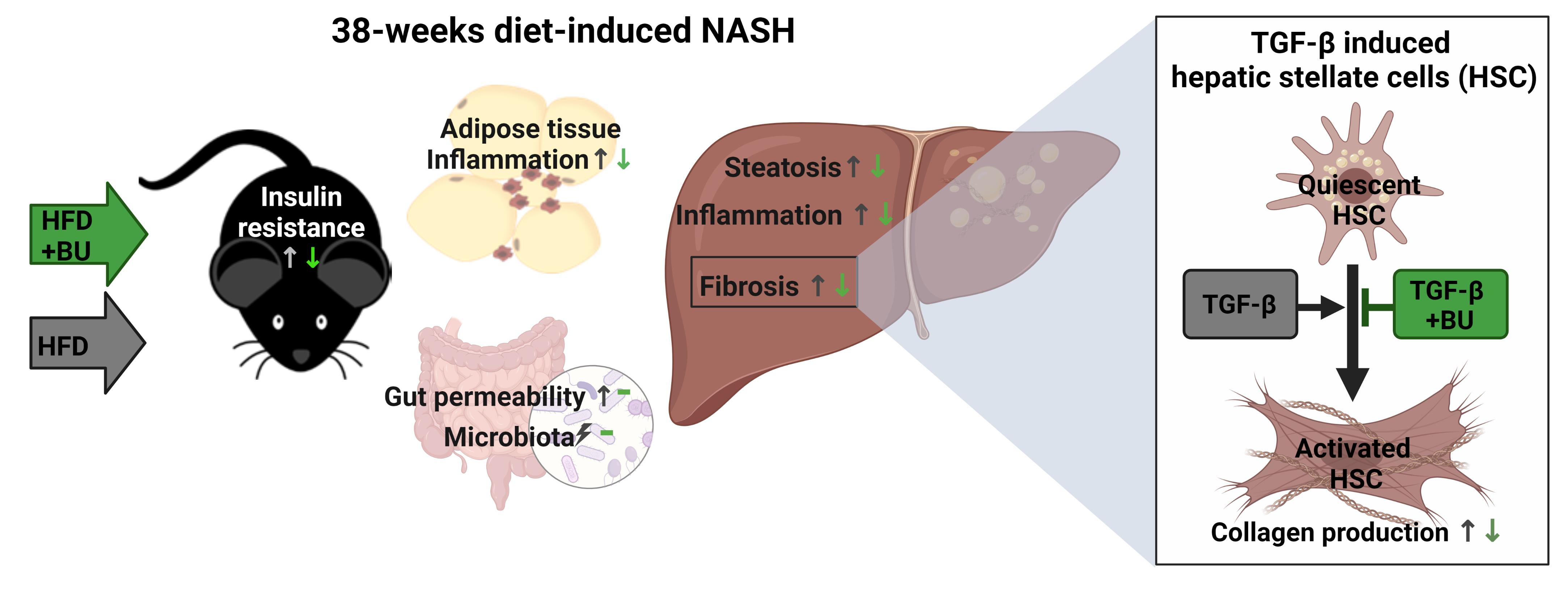
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pelusi, S.; Cespiati, A.; Rametta, R.; Pennisi, G.; Mannisto, V.; Rosso, C.; Baselli, G.; Dongiovanni, P.; Fracanzani, A.L.; Badiali, S.; et al. Prevalence and Risk Factors of Significant Fibrosis in Patients with Nonalcoholic Fatty Liver without Steatohepatitis. Clin. Gastroenterol. Hepatol. 2019, 17, 2310–2319.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilar-Gomez, E.; Calzadilla-Bertot, L.; Wai-Sun Wong, V.; Castellanos, M.; Aller-de la Fuente, R.; Metwally, M.; Eslam, M.; Gonzalez-Fabian, L.; Alvarez-Quiñones Sanz, M.; Conde-Martin, A.F.; et al. Fibrosis Severity as a Determinant of Cause-Specific Mortality in Patients with Advanced Nonalcoholic Fatty Liver Disease: A Multi-National Cohort Study. Gastroenterology 2018, 155, 443–457. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.T.; Kleiner, D.E. Histopathology of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Metabolism 2016, 65, P1080–P1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gart, E.; Lima, E.S.; Schuren, F.; De Ruiter, C.G.F.; Attema, J.; Verschuren, L.; Keijer, J.; Salic, K.; Morrison, M.C. Diet-Independent Correlations between Bacteria and Dysfunction of Gut, Adipose Tissue, and Liver: A Comprehensive Microbiota Analysis in Feces and Mucosa of the Ileum and Colon in Obese Mice with NAFLD. Int. J. Mol. Sci. 2019, 20, 1. [Google Scholar] [CrossRef] [Green Version]
- Loomba, R.; Friedman, S.L.; Shulman, G.I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 2021, 184, 2537–2564. [Google Scholar] [CrossRef]
- Baiocchini, A.; Montaldo, C.; Conigliaro, A.; Grimaldi, A.; Correani, V.; Mura, F.; Ciccosanti, F.; Rotiroti, N.; Brenna, A.; Montalbano, M.; et al. Extracellular matrix molecular remodeling in human liver fibrosis evolution. PLoS ONE 2016, 11, e0151736. [Google Scholar] [CrossRef] [Green Version]
- Dewidar, B.; Soukupova, J.; Fabregat, I.; Dooley, S. TGF-β in Hepatic Stellate Cell Activation and Liver Fibrogenesis: Updated. Curr. Pathobiol. Rep. 2015, 3, 291–305. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.E. Non-Smad pathways in TGF-β signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, D.; Pan, Q.; Xin, F.Z.; Zhang, R.N.; He, C.X.; Chen, G.Y.; Liu, C.; Chen, Y.W.; Fan, J.G. Sodium butyrate attenuates high-fat diet-induced steatohepatitis in mice by improving gut microbiota and gastrointestinal barrier. World J. Gastroenterol. 2017, 23, 60–75. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Jia, Y.; Hong, J.; Sun, Q.; Gao, S.; Hu, Y.; Zhao, N.; Zhao, R. Sodium butyrate ameliorates high-fat diet-induced NAFLD through PPARα-mediated activation of β oxidation and suppression of inflammation. J. Agric. Food Chem. 2018, 66, 7633–7642. [Google Scholar] [CrossRef]
- Canfora, E.E.; Jocken, J.W.; Blaak, E.E. Short-chain fatty acids in control of body weight and insulin sensitivity. Nat. Rev. Endocrinol. 2015, 11, 577–591. [Google Scholar] [CrossRef] [PubMed]
- Mattace Raso, G.; Simeoli, R.; Russo, R.; Iacono, A.; Santoro, A.; Paciello, O.; Ferrante, M.C.; Canani, R.B.; Calignano, A.; Meli, R. Effects of Sodium Butyrate and Its Synthetic Amide Derivative on Liver Inflammation and Glucose Tolerance in an Animal Model of Steatosis Induced by High Fat Diet. PLoS ONE 2013, 8, e68626. [Google Scholar]
- Arnoldussen, I.A.C.; Wiesmann, M.; Pelgrim, C.E.; Wielemaker, E.M.; Van Duyvenvoorde, W.; Amaral-Santos, P.L.; Verschuren, L.; Keijser, B.J.F.; Heerschap, A.; Kleemann, R.; et al. Butyrate restores HFD-induced adaptations in brain function and metabolism in mid-adult obese mice. Int. J. Obes. 2017, 41, 935–944. [Google Scholar] [CrossRef]
- Bouter, K.E.C.; Bakker, G.J.; Levin, E.; Hartstra, A.V.; Kootte, R.S.; Udayappan, S.D.; Katiraei, S.; Bahler, L.; Gilijamse, P.W.; Tremaroli, V.; et al. Differential metabolic effects of oral butyrate treatment in lean versus metabolic syndrome subjects article. Clin. Transl. Gastroenterol. 2018, 9, 155. [Google Scholar] [CrossRef]
- Cerdó, T.; García-Santos, J.A.; Bermúdez, M.G.; Campoy, C. The role of probiotics and prebiotics in the prevention and treatment of obesity. Nutrients 2019, 11, 635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Yi, C.X.; Katiraei, S.; Kooijman, S.; Zhou, E.; Chung, C.K.; Gao, Y.; Van Den Heuvel, J.K.; Meijer, O.C.; Berbée, J.F.P.; et al. Butyrate reduces appetite and activates brown adipose tissue via the gut-brain neural circuit. Gut 2018, 67, 1269–1279. [Google Scholar] [CrossRef] [Green Version]
- Mueller, A.M.; Kleemann, R.; Gart, E.; van Duyvenvoorde, W.; Verschuren, L.; Caspers, M.; Menke, A.; Krömmelbein, N.; Salic, K.; Burmeister, Y.; et al. Cholesterol Accumulation as a Driver of Hepatic Inflammation Under Translational Dietary Conditions Can Be Attenuated by a Multicomponent Medicine. Front. Endocrinol. 2021, 12, 156. [Google Scholar] [CrossRef] [PubMed]
- Morrison, M.C.; Verschuren, L.; Salic, K.; Verheij, J.; Menke, A.; Wielinga, P.Y.; Iruarrizaga-Lejarreta, M.; Gole, L.; Yu, W.; Turner, S.; et al. Obeticholic Acid Modulates Serum Metabolites and Gene Signatures Characteristic of Human NASH and Attenuates Inflammation and Fibrosis Progression in Ldlr-/-.Leiden Mice. Hepatol. Commun. 2018, 2, 1513–1532. [Google Scholar] [CrossRef] [Green Version]
- Van Koppen, A.; Verschuren, L.; van den Hoek, A.M.; Verheij, J.; Morrison, M.C.; Li, K.; Nagabukuro, H.; Costessi, A.; Caspers, M.P.M.; van den Broek, T.J.; et al. Uncovering a Predictive Molecular Signature for the Onset of NASH-Related Fibrosis in a Translational NASH Mouse Model. Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 83–98.e10. [Google Scholar] [CrossRef] [Green Version]
- Van den Hoek, A.M.; de Jong, J.C.B.C.; Worms, N.; van Nieuwkoop, A.; Voskuilen, M.; Menke, A.L.; Lek, S.; Caspers, M.P.M.; Verschuren, L.; Kleemann, R. Diet and exercise reduce pre-existing NASH and fibrosis and have additional beneficial effects on the vasculature, adipose tissue and skeletal muscle via organ-crosstalk. Metabolism 2021, 124, 154873. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.J.; Nedergaard, A.F.; Sun, S.; Veidal, S.S.; Larsen, L.; Zheng, Q.; Suetta, C.; Henriksen, K.; Christiansen, C.; Karsdal, M.A.; et al. The neo-epitope specific PRO-C3 ELISA measures true formation of type III collagen associated with liver and muscle parameters. Am. J. Transl. Res. 2013, 5, 303–315. [Google Scholar] [PubMed]
- Leeming, D.J.; Nielsen, M.J.; Dai, Y.; Veidal, S.S.; Vassiliadis, E.; Zhang, C.; He, Y.; Vainer, B.; Zheng, Q.; Karsdal, M.A. Enzyme-linked immunosorbent serum assay specific for the 7S domain of collagen type IV (P4NP 7S): A marker related to the extracellular matrix remodeling during liver fibrogenesis. Hepatol. Res. 2012, 42, 482–493. [Google Scholar] [CrossRef]
- Sand, J.M.; Larsen, L.; Hogaboam, C.; Martinez, F.; Han, M. Larsen, M.R.; Nawrocki, A.; Zheng, Q.; Karsdal, M.A.; Leeming, D.J. MMP Mediated Degradation of Type IV Collagen Alpha 1 and Alpha 3 Chains Reflects Basement Membrane Remodeling in Experimental and Clinical Fibrosis-Validation of Two Novel Biomarker Assays. PLoS ONE 2013, 8, 84934. [Google Scholar]
- Skovgård Veidal, S.; Karsdal, M.A.; Vassiliadis, E.; Nawrocki, A.; Larsen, M.R.; Nguyen, H.T.; Hägglund, P.; Luo, Y.; Zheng, Q.; Vainer, B.; et al. MMP Mediated Degradation of Type VI Collagen Is Highly Associated with Liver Fibrosis-Identification and Validation of a Novel Biochemical Marker Assay. PLoS ONE 2011, 6, e24753. [Google Scholar]
- Galarraga, M.; Campión, J.; Munõz-Barrutia, A.; Boqué, N.; Moreno, H.; Martínez, J.A.; Milagro, F.; Ortiz-de-Solórzano, C. Adiposoft: Automated software for the analysis of white adipose tissue cellularity in histological sections. J. Lipid Res. 2012, 53, 2791–2796. [Google Scholar] [CrossRef] [Green Version]
- Liang, W.; Menke, A.L.; Driessen, A.; Koek, G.H.; Lindeman, J.H.; Stoop, R.; Havekes, L.M.; Kleemann, R.; Van Den Hoek, A.M. Establishment of a general NAFLD scoring system for rodent models and comparison to human liver pathology. PLoS ONE 2014, 9, e115922. [Google Scholar]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Krämer, A.; Green, J.; Pollard, J.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Gart, E.; Salic, K.; Morrison, M.C.; Caspers, M.; Van Duyvenvoorde, W.; Heijnk, M.; Giera, M.; Bobeldijk-Pastorova, I.; Keijer, J.; Storsve, A.B.; et al. Krill Oil Treatment Increases Distinct PUFAs and Oxylipins in Adipose Tissue and Liver and Attenuates Obesity-Associated Inflammation via Direct and Indirect Mechanisms. Nutrients 2021, 13, 2836. [Google Scholar] [CrossRef] [PubMed]
- Mannaerts, I.; Leite, S.B.; Verhulst, S.; Claerhout, S.; Eysackers, N.; Thoen, L.F.R.; Hoorens, A.; Reynaert, H.; Halder, G.; Van Grunsven, L.A. The Hippo pathway effector YAP controls mouse hepatic stellate cell activation. J. Hepatol. 2015, 63, 679–688. [Google Scholar] [CrossRef]
- Kisseleva, T.; Cong, M.; Paik, Y.H.; Scholten, D.; Jiang, C.; Benner, C.; Iwaisako, K.; Moore-Morris, T.; Scott, B.; Tsukamoto, H.; et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 9448–9453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, A.K.; Vaughan, D.E. PAI-1 in Tissue Fibrosis. J. Cell. Physiol. 2012, 227, 493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noguchi, R.; Kaji, K.; Namisaki, T.; Moriya, K.; Kawaratani, H.; Kitade, M.; Takaya, H.; Aihara, Y.; Douhara, A.; Asada, K.; et al. Novel oral plasminogen activator inhibitor.1 inhibitor TM5275 attenuates hepatic fibrosis under metabolic syndrome via suppression of activated hepatic stellate cells in rats. Mol. Med. Rep. 2020, 22, 2948–2956. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, S.; Saito, A.; Nagase, T. YAP/TAZ Signaling as a Molecular Link between Fibrosis and Cancer. Int. J. Mol. Sci. 2018, 19, 3674. [Google Scholar] [CrossRef] [Green Version]
- Higgins, C.E.; Tang, J.; Higgins, S.P.; Gifford, C.C.; Mian, B.M.; Jones, D.M.; Zhang, W.; Costello, A.; Conti, D.J.; Samarakoon, R.; et al. The Genomic Response to TGF-β1 Dictates Failed Repair and Progression of Fibrotic Disease in the Obstructed Kidney. Front. Cell Dev. Biol. 2021, 9, 1716. [Google Scholar] [CrossRef]
- Shen, Y.; Miao, N.; Wang, B.; Xu, J.; Gan, X.; Xu, D.; Zhou, L.; Xue, H.; Zhang, W.; Yang, L.; et al. c-Myc promotes renal fibrosis by inducing integrin αv-mediated transforming growth factor-β signaling. Kidney Int. 2017, 92, 888–899. [Google Scholar] [CrossRef] [PubMed]
- Nevzorova, Y.A.; Hu, W.; Cubero, F.J.; Haas, U.; Freimuth, J.; Tacke, F.; Trautwein, C.; Liedtke, C. Overexpression of c-myc in hepatocytes promotes activation of hepatic stellate cells and facilitates the onset of liver fibrosis. Biochim. Biophys. Acta - Mol. Basis Dis. 2013, 1832, 1765–1775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumann, A.; Jin, C.J.; Brandt, A.; Sellmann, C.; Nier, A.; Burkard, M.; Venturelli, S.; Bergheim, I. Oral supplementation of sodium butyrate attenuates the progression of non-alcoholic steatohepatitis. Nutrients 2020, 12, 951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, C.J.; Sellmann, C.; Engstler, A.J.; Ziegenhardt, D.; Bergheim, I. Supplementation of sodium butyrate protects mice from the development of non-alcoholic steatohepatitis (NASH). Br. J. Nutr. 2015, 114, 1745–1755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J.; Lv, L.; Wu, W.; Li, Y.; Shi, D.; Fang, D.; Guo, F.; Jiang, H.; Yan, R.; Ye, W.; et al. Butyrate Protects Mice Against Methionine–Choline-Deficient Diet-Induced Non-alcoholic Steatohepatitis by Improving Gut Barrier Function, Attenuating Inflammation and Reducing Endotoxin Levels. Front. Microbiol. 2018, 9, 1967. [Google Scholar] [CrossRef] [Green Version]
- Saunder, D.R. Absorption of Short-Chain Fatty Acids in Human Stomach and Rectum. Nutr. Res. 1991, 8, 55. [Google Scholar] [CrossRef]
- Schmitt, M.G.; Soergel, K.H.; Wood, C.M.; Steff, J.J. Absorption of Short-Chain Fatty Acids from the Human Ileum. Dig. Dis. Sci. 1977, 22, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Gill, R.K.; Saksena, S.; Alrefai, W.A.; Sarwar, Z.; Goldstein, J.L.; Carroll, R.E.; Ramaswamy, K.; Dudeja, P.K. Expression and membrane localization of MCT isoforms along the length of the human intestine. Am. J. Physiol. -Cell Physiol. 2005, 289, C846–C852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guilloteau, P.; Martin, L.; Eeckhaut, V.; Ducatelle, R.; Zabielski, R.; Van Immerseel, F. From the gut to the peripheral tissues: The multiple effects of butyrate. Nutr. Res. Rev. 2010, 23, 366–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.; Song, H.; Wang, Y.; Sheng, Y.; Chen, J. Sodium butyrate protects the intestinal barrier function in peritonitic mice. Int. J. Clin. Exp. Med. 2015, 8, 4000–4007. [Google Scholar] [PubMed]
- Wang, H.; Song, W.; Wu, Q.; Gao, X.; Li, J.; Tan, C.; Zhou, H.; Zhu, J.; He, Y.; Yin, J. Fecal Transplantation from db/db Mice Treated with Sodium Butyrate Attenuates Ischemic Stroke Injury. Microbiol. Spectr. 2021, 9, e00042-21. [Google Scholar] [CrossRef]
- Bai, H.-B.; Yang, P.; Zhang, H.-B.; Liu, Y.-L.; Fang, S.-X.; Xu, X.-Y. Short-chain fatty acid butyrate acid attenuates atherosclerotic plaque formation in apolipoprotein E-knockout mice and the underlying mechanism. Sheng Li Xue Bao 2021, 73, 42–50. [Google Scholar]
- Pelgrim, C.E.; Franx, B.A.A.; Snabel, J.; Kleemann, R.; Arnoldussen, I.A.C.; Kiliaan, A.J. Butyrate reduces HFD-induced adipocyte hypertrophy and metabolic risk factors in Obese LDLr-/-.Leiden mice. Nutrients 2017, 9, 714. [Google Scholar] [CrossRef] [Green Version]
- Ikejima, K.; Takei, Y.; Honda, H.; Hirose, M.; Yoshikawa, M.; Zhang, Y.-J.; Lang, T.; Fukuda, T.; Yamashina, S.; Kitamura, T.; et al. Leptin receptor–mediated signaling regulates hepatic fibrogenesis and remodeling of extracellular matrix in the rat. Gastroenterology 2002, 122, 1399–1410. [Google Scholar] [CrossRef]
- Choi, S.S.; Syn, W.K.; Karaca, G.F.; Omenetti, A.; Moylan, C.A.; Witek, R.P.; Agboola, K.M.; Jung, Y.; Michelotti, G.A.; Diehl, A.M. Leptin promotes the myofibroblastic phenotype in hepatic stellate cells by activating the Hedgehog pathway. J. Biol. Chem. 2010, 285, 36551–36560. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chow | HFD | HFD + BU | |
|---|---|---|---|
| Body weight (g) | 42.4 ± 6.3 *** | 56.5 ± 3.9 | 45.3 ± 4.4 *** |
| Average energy intake (kcal/mouse/day) | 12.7 ± 0.5 | 13.4 ± 0.3 | 11.6 ± 0.5 ** |
| Average food efficiency (mg BW gain/kcal) | 3.85 ± 1.72 *** | 7.54 ± 1.14 | 5.73 ± 0.86 *** |
| Cholesterol (mM) | 9.3 ± 2.2 *** | 34.4 ± 6.4 | 35.7 ± 7.5 |
| Triglycerides (mM) | 1.7 ± 0.6 *** | 5.5 ± 2.1 | 6.3 ± 2.5 |
| Glucose (mM) | 7.1 ± 1.1 | 6.6 ± 0.9 | 6.6 ± 1.2 |
| Insulin (ng/mL) | 9.3 ± 16.4 * | 20.3 ± 11.1 | 8.1 ± 4.2 * |
| Leptin (ng/mL) | 25.3 ± 13.3 *** | 59.2 ± 16.0 | 39.3 ± 9.5 *** |
| Adiponectin (ug/mL) | 7.2 ± 1.2 * | 6.1 ± 1.6 | 6.2 ± 0.8 |
| TGF-β vs. Unstimulated | BU+TGF-β vs. TGF-β | |||
|---|---|---|---|---|
| Gene Name: | 2logR | p-Value | 2logR | p-Value |
| Myofibroblast markers | ||||
| ACTA2 | 4.28 | 0.000 | −1.15 | 0.000 |
| SERPINE1 | 2.03 | 0.000 | −0.37 | 0.000 |
| ELN | 2.13 | 0.000 | −1.40 | 0.000 |
| CDH11 | 1.43 | 0.000 | −1.45 | 0.000 |
| DES | 0.70 | 0.007 | 1.62 | 0.000 |
| VIM | −0.02 | 0.682 | 0.79 | 0.000 |
| PALLD | 2.29 | 0.000 | −0.27 | 0.000 |
| TPM1 | 3.24 | 0.000 | 0.13 | 0.002 |
| CFL1 | 0.62 | 0.000 | −0.26 | 0.000 |
| COL1A1 | 0.63 | 0.000 | −0.05 | 0.402 |
| TGF-β vs. Unstimulated | BU+TGF-β vs. TGF-β | |||
|---|---|---|---|---|
| Upstream Regulator: | Z-Score | p-Value | Z-Score | p-Value |
| TGF-β signaling | ||||
| TGFB1 | 9.7 | 0.000 | 1.5 | 0.000 |
| TGFB2 | 5.1 | 0.000 | −0.4 | 0.002 |
| TGFB3 | 5.6 | 0.000 | −0.7 | 0.000 |
| TGFBR1 | 2.6 | 0.000 | −0.2 | 0.030 |
| TGFBR2 | 1.8 | 0.000 | −0.1 | 0.000 |
| TGFBR3 | −1.0 | 0.026 | N/A | N/A |
| Canonical TGF-β signaling | ||||
| SMAD1 | 0.4 | 0.000 | 0.7 | 0.003 |
| SMAD2 | 4.5 | 0.000 | N/A | 1.000 |
| SMAD3 | 2.8 | 0.000 | 2.0 | 0.014 |
| SMAD4 | 3.5 | 0.000 | 1.3 | 0.002 |
| SMAD7 | −1.5 | 0.000 | −0.9 | 0.002 |
| SMAD2/3/4 | 2.6 | 0.001 | N/A | 1.000 |
| SMAD1/5/9 | N/A | 1.000 | N/A | 0.022 |
| Non-canonical TGF- β signaling | ||||
| via ERK | ||||
| RAS | 2.9 | 0.000 | 0.9 | 0.000 |
| RAF | 0.2 | 0.000 | 0.5 | 0.000 |
| MAP2K1 | 1.6 | 0.000 | 2.1 | 0.000 |
| ERK | 3.0 | 0.000 | 3.1 | 0.000 |
| CAV1 | 1.3 | 0.000 | 1.6 | 0.000 |
| via JNK/p38 | ||||
| MAP3K7 | 0.6 | 0.000 | N/A | 1.000 |
| JNK | 3.4 | 0.000 | 0.0 | 0.000 |
| MAPK11 | 0.6 | 0.030 | 2.4 | 0.120 |
| MAPK13 | −0.8 | 0.039 | 1.3 | 0.037 |
| MAPK14 | 1.4 | 0.000 | 2.7 | 0.000 |
| AP1 | 2.2 | 0.000 | 2.7 | 0.026 |
| via Rho-like GTPases | ||||
| RHOA | 1.2 | 0.001 | −1.0 | 0.001 |
| ROCK | 1.5 | 0.000 | −0.9 | 0.000 |
| CDC42 | N/A | 1.000 | N/A | 1.000 |
| RAC1 | 2.7 | 0.000 | N/A | 1.000 |
| PAK1 | 1.4 | 0.006 | −1.0 | 0.019 |
| PAK2 | −0.3 | 0.000 | −1.6 | 0.006 |
| via PI3K/AKT | ||||
| AKT | 3.3 | 0.000 | 0.2 | 0.000 |
| PTEN | −2.0 | 0.000 | 1.2 | 0.000 |
| GSK3B | 0.6 | 0.011 | −1.1 | 0.042 |
| MDM2 | N/A | 1.000 | −0.7 | 0.008 |
| MYC | 2.2 | 0.000 | −6.2 | 0.000 |
| MTORC1 | 0.7 | 0.003 | N/A | 1.000 |
| HIPPO signaling | ||||
| YAP1 | 2.5 | 0.000 | −2.1 | 0.000 |
| WWTR1 | 0.8 | 0.000 | −1.8 | 0.000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gart, E.; van Duyvenvoorde, W.; Toet, K.; Caspers, M.P.M.; Verschuren, L.; Nielsen, M.J.; Leeming, D.J.; Souto Lima, E.; Menke, A.; Hanemaaijer, R.; et al. Butyrate Protects against Diet-Induced NASH and Liver Fibrosis and Suppresses Specific Non-Canonical TGF-β Signaling Pathways in Human Hepatic Stellate Cells. Biomedicines 2021, 9, 1954. https://doi.org/10.3390/biomedicines9121954
Gart E, van Duyvenvoorde W, Toet K, Caspers MPM, Verschuren L, Nielsen MJ, Leeming DJ, Souto Lima E, Menke A, Hanemaaijer R, et al. Butyrate Protects against Diet-Induced NASH and Liver Fibrosis and Suppresses Specific Non-Canonical TGF-β Signaling Pathways in Human Hepatic Stellate Cells. Biomedicines. 2021; 9(12):1954. https://doi.org/10.3390/biomedicines9121954
Chicago/Turabian StyleGart, Eveline, Wim van Duyvenvoorde, Karin Toet, Martien P. M. Caspers, Lars Verschuren, Mette Juul Nielsen, Diana Julie Leeming, Everton Souto Lima, Aswin Menke, Roeland Hanemaaijer, and et al. 2021. "Butyrate Protects against Diet-Induced NASH and Liver Fibrosis and Suppresses Specific Non-Canonical TGF-β Signaling Pathways in Human Hepatic Stellate Cells" Biomedicines 9, no. 12: 1954. https://doi.org/10.3390/biomedicines9121954
APA StyleGart, E., van Duyvenvoorde, W., Toet, K., Caspers, M. P. M., Verschuren, L., Nielsen, M. J., Leeming, D. J., Souto Lima, E., Menke, A., Hanemaaijer, R., Keijer, J., Salic, K., Kleemann, R., & Morrison, M. C. (2021). Butyrate Protects against Diet-Induced NASH and Liver Fibrosis and Suppresses Specific Non-Canonical TGF-β Signaling Pathways in Human Hepatic Stellate Cells. Biomedicines, 9(12), 1954. https://doi.org/10.3390/biomedicines9121954