
Neutrophils and Platelets: Immune Soldiers Fighting Together in Stroke Pathophysiology



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
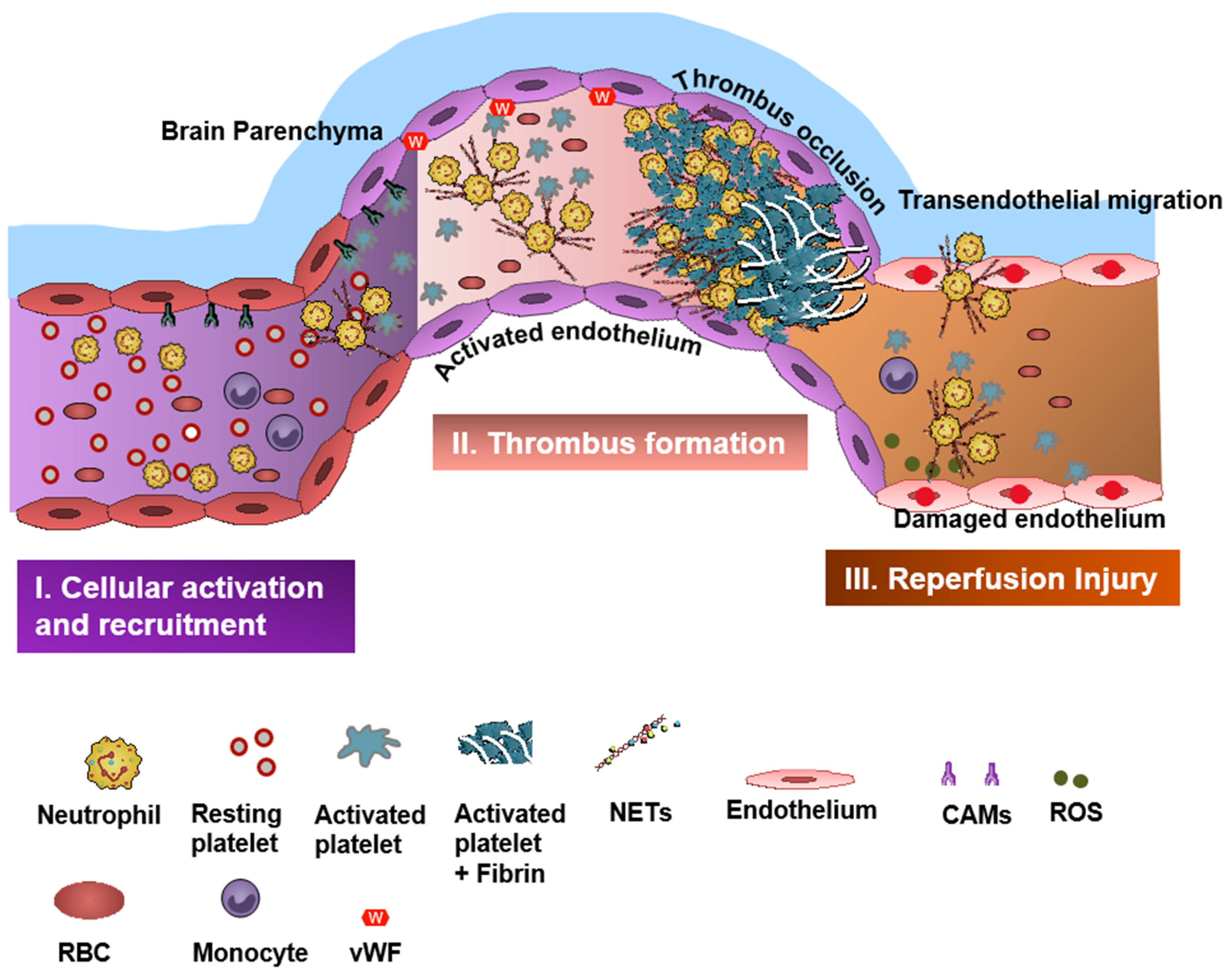
2. Neutrophils in Stroke
3. Neutrophil Serine Proteases and Thromboinflammation
4. Neutrophil Extracellular Traps (NETs) and Stroke
5. Neutrophil-Dependent Oxidative Stress and IS
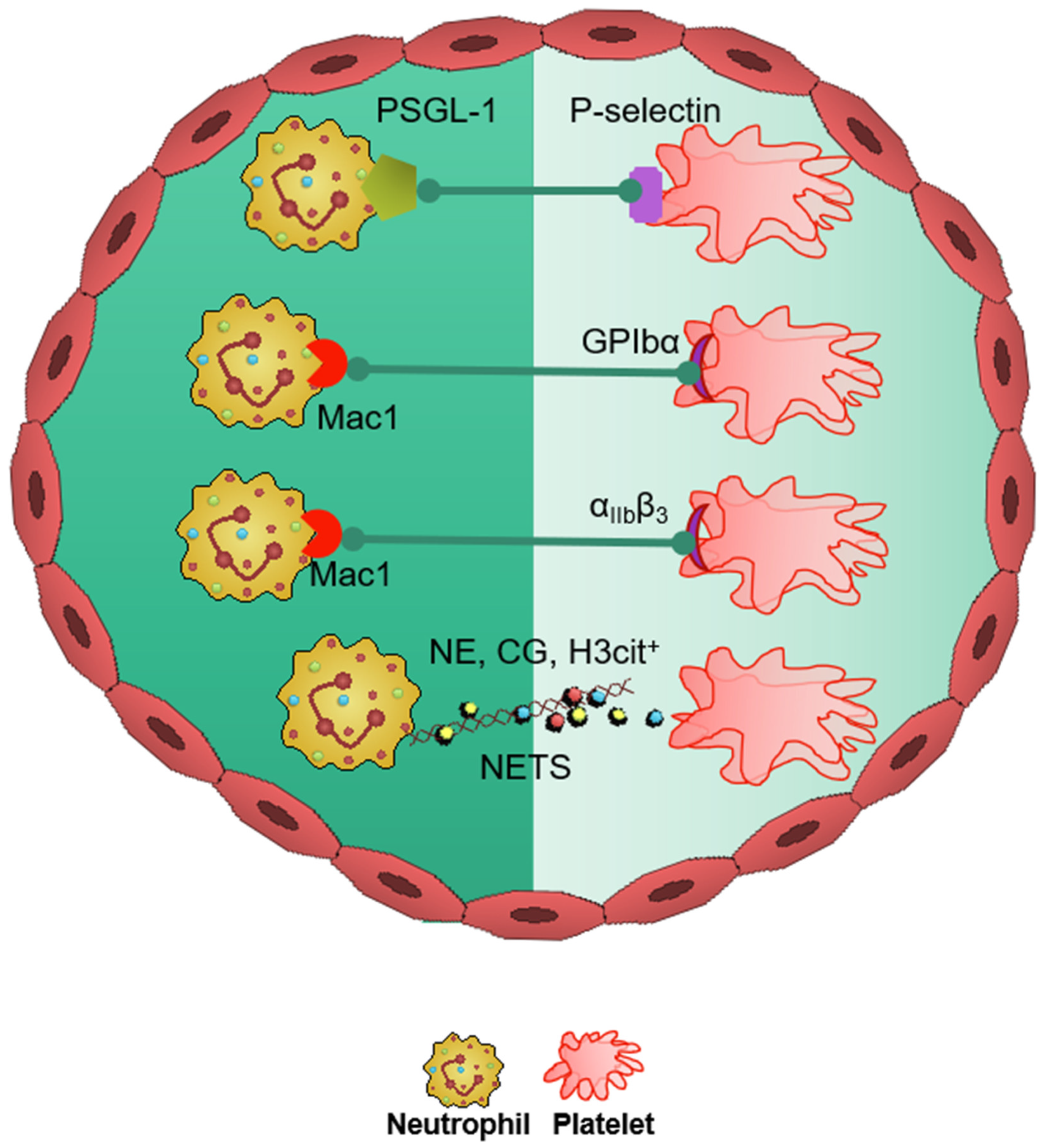
6. Platelets in Stroke
7. Neutrophil- and Platelet-Dependent AnxA1-FPR2/ALX Resolution Axis in Stroke
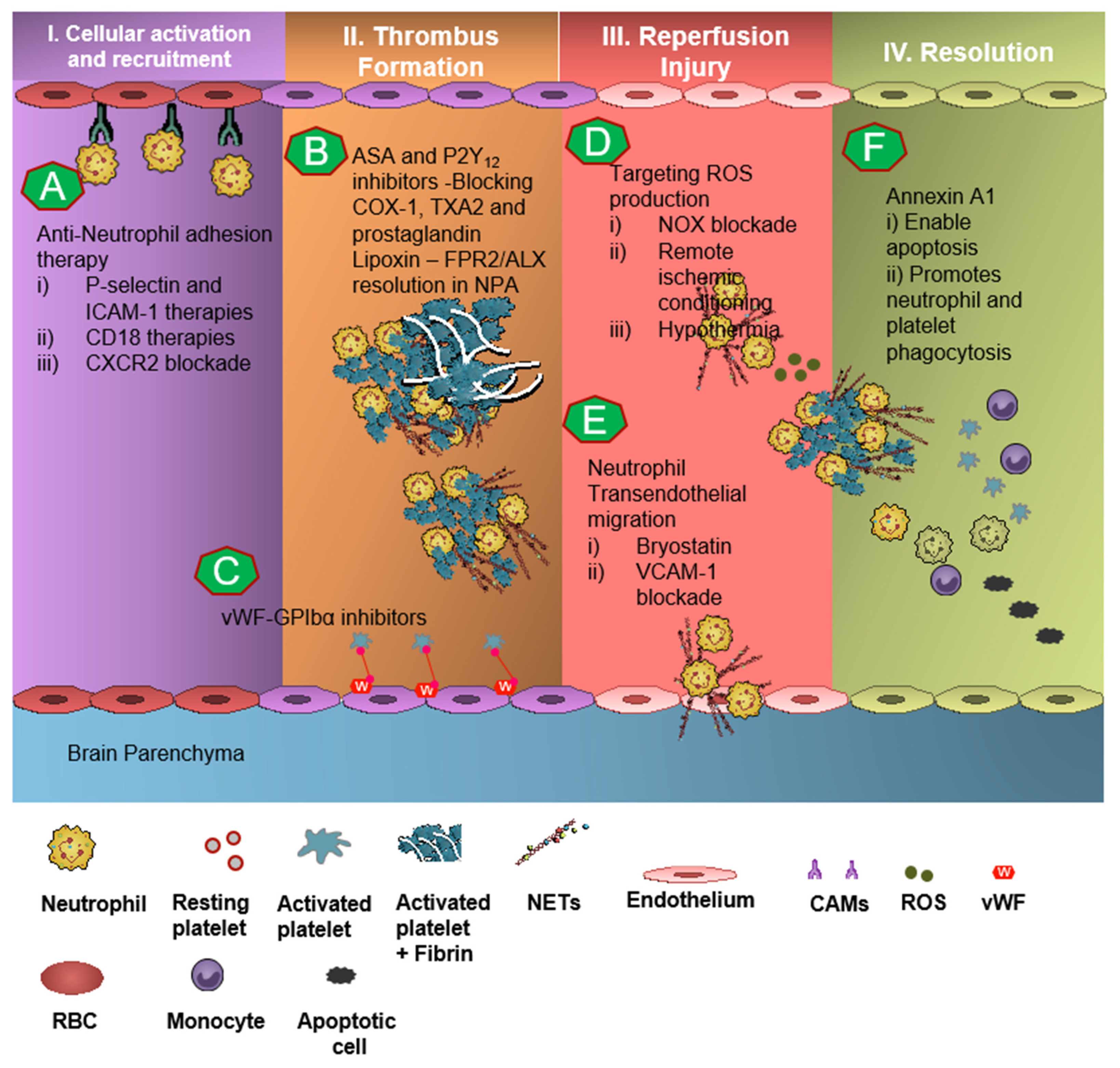
8. Therapeutics in Thromboinflammation
9. Targeting Neutrophil-Dependent Thromboinflammation
10. Targeting Platelet-Dependent Thromboinflammation
11. Concluding Remarks and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Broderick, J.P.; Hill, M.D. Advances in Acute Stroke Treatment 2020. Stroke 2021, 52, 729–734. [Google Scholar] [CrossRef]
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2021 Update: A Report From the American Heart Association. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef]
- Silva, G.S.; Nogueira, R.G. Endovascular Treatment of Acute Ischemic Stroke. Continuum (Minneap Minn) 2020, 26, 310–331. [Google Scholar] [CrossRef] [Green Version]
- Ansari, J.; Senchenkova, E.Y.; Vital, S.A.; Al Yafeai, Z.; Kaur, G.; Sparkenbaugh, E.M.; Orr, A.; Pawlinski, R.; Hebbel, R.P.; Granger, D.N.; et al. Targeting AnxA1/Fpr2/ALX Pathway Regulates Neutrophil Function Promoting Thrombo-Inflammation Resolution in Sickle Cell Disease. Blood 2021, 137, 1538–1549. [Google Scholar] [CrossRef]
- Ansari, J.; Gavins, F.N.E. The impact of thrombo-inflammation on the cerebral microcirculation. Microcirculation 2021, 28, e12689. [Google Scholar] [CrossRef] [PubMed]
- Senchenkova, E.Y.; Ansari, J.; Becker, F.; Vital, S.A.; Al-Yafeai, Z.; Sparkenbaugh, E.M.; Pawlinski, R.; Stokes, K.Y.; Carroll, J.L.; Dragoi, A.M.; et al. A Novel Role for the AnxA1-Fpr2/ALX Signaling Axis as a Key Regulator of Platelet Function to Promote Resolution of Inflammation. Circulation 2019, 140, 319–335. [Google Scholar] [CrossRef]
- Vital, S.A.; Becker, F.; Holloway, P.M.; Russell, J.; Perretti, M.; Granger, D.N.; Gavins, F.N. Formyl-Peptide Receptor 2/3/Lipoxin A4 Receptor Regulates Neutrophil-Platelet Aggregation and Attenuates Cerebral Inflammation: Impact for Therapy in Cardiovascular Disease. Circulation 2016, 133, 2169–2179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perretti, M.; Leroy, X.; Bland, E.J.; Montero-Melendez, T. Resolution Pharmacology: Opportunities for Therapeutic Innovation in Inflammation. Trends Pharmacol. Sci. 2015, 36, 737–755. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Stoll, G.; Kleinschnitz, C.; Nieswandt, B. Combating innate inflammation: A new paradigm for acute treatment of stroke? Ann. N. Y. Acad. Sci. 2010, 1207, 149–154. [Google Scholar] [CrossRef]
- Massberg, S.; Grahl, L.; von Bruehl, M.L.; Manukyan, D.; Pfeiler, S.; Goosmann, C.; Brinkmann, V.; Lorenz, M.; Bidzhekov, K.; Khandagale, A.B.; et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 2010, 16, 887–896. [Google Scholar] [CrossRef]
- De Meyer, S.F.; Denorme, F.; Langhauser, F.; Geuss, E.; Fluri, F.; Kleinschnitz, C. Thromboinflammation in Stroke Brain Damage. Stroke 2016, 47, 1165–1172. [Google Scholar] [CrossRef] [Green Version]
- Jickling, G.C.; Liu, D.; Ander, B.P.; Stamova, B.; Zhan, X.; Sharp, F.R. Targeting neutrophils in ischemic stroke: Translational insights from experimental studies. J. Cereb. Blood Flow Metab. 2015, 35, 888–901. [Google Scholar] [CrossRef] [Green Version]
- Cerletti, C.; Tamburrelli, C.; Izzi, B.; Gianfagna, F.; de Gaetano, G. Platelet-leukocyte interactions in thrombosis. Thromb. Res. 2012, 129, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, T.V.; Salter, J.W.; Chidlow, J.H.; Ballantyne, C.M.; Kevil, C.G.; Granger, D.N. Contributions of LFA-1 and Mac-1 to brain injury and microvascular dysfunction induced by transient middle cerebral artery occlusion. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2555–H2560. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Mao, D.; Lü, S.; Tong, C.; Zhang, Y.; Long, M. Distinct binding affinities of Mac-1 and LFA-1 in neutrophil activation. J. Immunol. 2013, 190, 4371–4381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldsmith, H.L.; Spain, S. Margination of leukocytes in blood flow through small tubes. Microvasc. Res. 1984, 27, 204–222. [Google Scholar] [CrossRef]
- Enzmann, G.; Kargaran, S.; Engelhardt, B. Ischemia-reperfusion injury in stroke: Impact of the brain barriers and brain immune privilege on neutrophil function. Ther. Adv. Neurol. Disord. 2018, 11, 1756286418794184. [Google Scholar] [CrossRef] [Green Version]
- Ansari, J.; Gavins, F.N.E. Ischemia-Reperfusion Injury in Sickle Cell Disease: From Basics to Therapeutics. Am. J. Pathol. 2019, 189, 706–718. [Google Scholar] [CrossRef] [Green Version]
- Ritter, L.S.; Stempel, K.M.; Coull, B.M.; McDonagh, P.F. Leukocyte-platelet aggregates in rat peripheral blood after ischemic stroke and reperfusion. Biol. Res. Nurs. 2005, 6, 281–288. [Google Scholar] [CrossRef]
- Noubouossie, D.F.; Reeves, B.N.; Strahl, B.D.; Key, N.S. Neutrophils: Back in the thrombosis spotlight. Blood 2019, 133, 2186–2197. [Google Scholar] [CrossRef]
- Harris, M.G.; Hulseberg, P.; Ling, C.; Karman, J.; Clarkson, B.D.; Harding, J.S.; Zhang, M.; Sandor, A.; Christensen, K.; Nagy, A.; et al. Immune privilege of the CNS is not the consequence of limited antigen sampling. Sci. Rep. 2014, 4, 4422. [Google Scholar] [CrossRef] [Green Version]
- Ousman, S.S.; Kubes, P. Immune surveillance in the central nervous system. Nat. Neurosci. 2012, 15, 1096–1101. [Google Scholar] [CrossRef]
- Manda-Handzlik, A.; Demkow, U. The Brain Entangled: The Contribution of Neutrophil Extracellular Traps to the Diseases of the Central Nervous System. Cells 2019, 8, 1477. [Google Scholar] [CrossRef] [Green Version]
- Allen, C.; Thornton, P.; Denes, A.; McColl, B.W.; Pierozynski, A.; Monestier, M.; Pinteaux, E.; Rothwell, N.J.; Allan, S.M. Neutrophil cerebrovascular transmigration triggers rapid neurotoxicity through release of proteases associated with decondensed DNA. J. Immunol. 2012, 189, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Buck, B.H.; Liebeskind, D.S.; Saver, J.L.; Bang, O.Y.; Yun, S.W.; Starkman, S.; Ali, L.K.; Kim, D.; Villablanca, J.P.; Salamon, N.; et al. Early neutrophilia is associated with volume of ischemic tissue in acute stroke. Stroke 2008, 39, 355–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, K.; Wang, A.; Zhang, X.; Meng, X.; Chen, P.; Li, H.; Wang, Y. Neutrophil to lymphocyte ratio and adverse clinical outcomes in patients with ischemic stroke. Ann. Transl. Med. 2021, 9, 1047. [Google Scholar] [CrossRef] [PubMed]
- Ying, Y.; Yu, F.; Luo, Y.; Feng, X.; Liao, D.; Wei, M.; Li, X.; Huang, Q.; Liu, Z.; Zhang, L.; et al. Neutrophil-to-Lymphocyte Ratio as a Predictive Biomarker for Stroke Severity and Short-Term Prognosis in Acute Ischemic Stroke With Intracranial Atherosclerotic Stenosis. Front. Neurol. 2021, 12, 705949. [Google Scholar] [CrossRef]
- Jiang, X.; Andjelkovic, A.V.; Zhu, L.; Yang, T.; Bennett, M.V.L.; Chen, J.; Keep, R.F.; Shi, Y. Blood-brain barrier dysfunction and recovery after ischemic stroke. Prog. Neurobiol. 2018, 163–164, 144–171. [Google Scholar] [CrossRef] [PubMed]
- Rosell, A.; Cuadrado, E.; Ortega-Aznar, A.; Hernandez-Guillamon, M.; Lo, E.H.; Montaner, J. MMP-9-positive neutrophil infiltration is associated to blood-brain barrier breakdown and basal lamina type IV collagen degradation during hemorrhagic transformation after human ischemic stroke. Stroke 2008, 39, 1121–1126. [Google Scholar] [CrossRef] [Green Version]
- Faraday, N.; Schunke, K.; Saleem, S.; Fu, J.; Wang, B.; Zhang, J.; Morrell, C.; Dore, S. Cathepsin G-dependent modulation of platelet thrombus formation in vivo by blood neutrophils. PLoS ONE 2013, 8, e71447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woloszynek, J.C.; Hu, Y.; Pham, C.T. Cathepsin G-regulated release of formyl peptide receptor agonists modulate neutrophil effector functions. J. Biol. Chem. 2012, 287, 34101–34109. [Google Scholar] [CrossRef] [Green Version]
- Papayannopoulos, V.; Staab, D.; Zychlinsky, A. Neutrophil elastase enhances sputum solubilization in cystic fibrosis patients receiving Dnase therapy. PloS ONE 2011, 6, e28526. [Google Scholar] [CrossRef] [PubMed]
- Pham, C.T. Neutrophil serine proteases: Specific regulators of inflammation. Nat. Rev. Immunol. 2006, 6, 541–550. [Google Scholar] [CrossRef]
- Burgener, S.S.; Leborgne, N.G.F.; Snipas, S.J.; Salvesen, G.S.; Bird, P.I.; Benarafa, C. Cathepsin G Inhibition by Serpinb1 and Serpinb6 Prevents Programmed Necrosis in Neutrophils and Monocytes and Reduces GSDMD-Driven Inflammation. Cell Rep. 2019, 27, 3646–3656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, R.; Iribarren, P.; Zhang, N.; Zhou, Y.; Gong, W.; Cho, E.H.; Lockett, S.; Chertov, O.; Bednar, F.; Rogers, T.J.; et al. Identification of neutrophil granule protein cathepsin G as a novel chemotactic agonist for the G protein-coupled formyl peptide receptor. J. Immunol. 2004, 173, 428–436. [Google Scholar] [CrossRef] [Green Version]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Martinod, K.; Wagner, D.D. Thrombosis: Tangled up in NETs. Blood 2014, 123, 2768–2776. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef] [Green Version]
- Laridan, E.; Denorme, F.; Desender, L.; Francois, O.; Andersson, T.; Deckmyn, H.; Vanhoorelbeke, K.; De Meyer, S.F. Neutrophil extracellular traps in ischemic stroke thrombi. Ann. Neurol. 2017, 82, 223–232. [Google Scholar] [CrossRef]
- Zuo, Y.; Zuo, M.; Yalavarthi, S.; Gockman, K.; Madison, J.A.; Shi, H.; Woodard, W.; Lezak, S.P.; Lugogo, N.L.; Knight, J.S.; et al. Neutrophil extracellular traps and thrombosis in COVID-19. J. Thromb. Thrombolysis 2021, 51, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Perdomo, J.; Leung, H.H.L.; Ahmadi, Z.; Yan, F.; Chong, J.J.H.; Passam, F.H.; Chong, B.H. Neutrophil activation and NETosis are the major drivers of thrombosis in heparin-induced thrombocytopenia. Nat. Commun. 2019, 10, 1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauracher, L.M.; Posch, F.; Martinod, K.; Grilz, E.; Daullary, T.; Hell, L.; Brostjan, C.; Zielinski, C.; Ay, C.; Wagner, D.D.; et al. Citrullinated histone H3, a biomarker of neutrophil extracellular trap formation, predicts the risk of venous thromboembolism in cancer patients. J. Thromb. Haemost. 2018, 16, 508–518. [Google Scholar] [CrossRef]
- Zhou, P.; Li, T.; Jin, J.; Liu, Y.; Li, B.; Sun, Q.; Tian, J.; Zhao, H.; Liu, Z.; Ma, S.; et al. Interactions between neutrophil extracellular traps and activated platelets enhance procoagulant activity in acute stroke patients with ICA occlusion. EbioMedicine 2020, 53, 102671. [Google Scholar] [CrossRef]
- Kang, L.; Yu, H.; Yang, X.; Zhu, Y.; Bai, X.; Wang, R.; Cao, Y.; Xu, H.; Luo, H.; Lu, L.; et al. Neutrophil extracellular traps released by neutrophils impair revascularization and vascular remodeling after stroke. Nat. Commun. 2020, 11, 2488. [Google Scholar] [CrossRef]
- Chan, P.H. Oxygen radicals in focal cerebral ischemia. Brain Pathol. 1994, 4, 59–65. [Google Scholar] [CrossRef]
- Lorenzano, S.; Rost, N.S.; Khan, M.; Li, H.; Batista, L.M.; Chutinet, A.; Green, R.E.; Thankachan, T.K.; Thornell, B.; Muzikansky, A.; et al. Early molecular oxidative stress biomarkers of ischemic penumbra in acute stroke. Neurology 2019, 93, e1288–e1298. [Google Scholar] [CrossRef] [PubMed]
- Cook-Mills, J.M.; Marchese, M.E.; Abdala-Valencia, H. Vascular cell adhesion molecule-1 expression and signaling during disease: Regulation by reactive oxygen species and antioxidants. Antioxid. Redox Signal. 2011, 15, 1607–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigo, R.; Fernandez-Gajardo, R.; Gutierrez, R.; Matamala, J.M.; Carrasco, R.; Miranda-Merchak, A.; Feuerhake, W. Oxidative stress and pathophysiology of ischemic stroke: Novel therapeutic opportunities. CNS Neurol. Disord. Drug Targets 2013, 12, 698–714. [Google Scholar] [CrossRef] [PubMed]
- Hoda, M.N.; Siddiqui, S.; Herberg, S.; Periyasamy-Thandavan, S.; Bhatia, K.; Hafez, S.S.; Johnson, M.H.; Hill, W.D.; Ergul, A.; Fagan, S.C.; et al. Remote ischemic perconditioning is effective alone and in combination with intravenous tissue-type plasminogen activator in murine model of embolic stroke. Stroke 2012, 43, 2794–2799. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Liu, X.; Luo, Y.; Ji, X. Therapeutic hypothermia for stroke: Where to go? Exp. Neurol. 2015, 272, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Ohkura, N.; Hiraishi, S.; Itabe, H.; Hamuro, T.; Kamikubo, Y.; Takano, T.; Matsuda, J.; Horie, S. Oxidized phospholipids in oxidized low-density lipoprotein reduce the activity of tissue factor pathway inhibitor through association with its carboxy-terminal region. Antioxid. Redox Signal. 2004, 6, 705–712. [Google Scholar] [CrossRef]
- Demirin, H.; Ozhan, H.; Ucgun, T.; Celer, A.; Bulur, S.; Cil, H.; Gunes, C.; Yildirim, H.A. Normal range of mean platelet volume in healthy subjects: Insight from a large epidemiologic study. Thromb. Res. 2011, 128, 358–360. [Google Scholar] [CrossRef] [PubMed]
- Eason, C.T.; Pattison, A.; Howells, D.D.; Mitcheson, J.; Bonner, F.W. Platelet population profiles: Significance of species variation and drug-induced changes. J. Appl. Toxicol. 1986, 6, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Daly, M.E. Determinants of platelet count in humans. Haematologica 2011, 96, 10–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefrancais, E.; Ortiz-Munoz, G.; Caudrillier, A.; Mallavia, B.; Liu, F.; Sayah, D.M.; Thornton, E.E.; Headley, M.B.; David, T.; Coughlin, S.R.; et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature 2017, 544, 105–109. [Google Scholar] [CrossRef]
- Koupenova, M.; Clancy, L.; Corkrey, H.A.; Freedman, J.E. Circulating Platelets as Mediators of Immunity, Inflammation, and Thrombosis. Circ. Res. 2018, 122, 337–351. [Google Scholar] [CrossRef]
- Margraf, A.; Zarbock, A. Platelets in Inflammation and Resolution. J. Immunol. 2019, 203, 2357–2367. [Google Scholar] [CrossRef]
- Pluthero, F.G.; Kahr, W.H.A. The Birth and Death of Platelets in Health and Disease. Physiology (Bethesda) 2018, 33, 225–234. [Google Scholar] [CrossRef]
- Michelson, A.D. Antiplatelet therapies for the treatment of cardiovascular disease. Nat. Rev. Drug Discov. 2010, 9, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, V.R.; Tomer, A. Megakaryocyte development and platelet production. Br. J. Haematol. 2006, 134, 453–466. [Google Scholar] [CrossRef]
- Rawish, E.; Nording, H.; Münte, T.; Langer, H.F. Platelets as Mediators of Neuroinflammation and Thrombosis. Front. Immunol. 2020, 11, 548631. [Google Scholar] [CrossRef]
- Kehrel, B.E.; Fender, A.C. Resolving Thromboinflammation in the Brain After Ischemic Stroke? Circulation 2016, 133, 2128–2131. [Google Scholar] [CrossRef]
- Lievens, D.; Zernecke, A.; Seijkens, T.; Soehnlein, O.; Beckers, L.; Munnix, I.C.; Wijnands, E.; Goossens, P.; van Kruchten, R.; Thevissen, L.; et al. Platelet CD40L mediates thrombotic and inflammatory processes in atherosclerosis. Blood 2010, 116, 4317–4327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, S.; Bodenstein, R.; Chen, Q.; Feil, S.; Feil, R.; Rheinlaender, J.; Schaffer, T.E.; Bohn, E.; Frick, J.S.; Borst, O.; et al. Platelet-derived HMGB1 is a critical mediator of thrombosis. J. Clin. Investig. 2015, 125, 4638–4654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef]
- Rana, A.; Westein, E.; Niego, B.; Hagemeyer, C.E. Shear-Dependent Platelet Aggregation: Mechanisms and Therapeutic Opportunities. Front. Cardiovasc. Med. 2019, 6, 141. [Google Scholar] [CrossRef]
- Meyer, S.F.D.; Stoll, G.; Wagner, D.D.; Kleinschnitz, C. von Willebrand Factor. Stroke 2012, 43, 599–606. [Google Scholar] [CrossRef]
- Shankaran, H.; Alexandridis, P.; Neelamegham, S. Aspects of hydrodynamic shear regulating shear-induced platelet activation and self-association of von Willebrand factor in suspension. Blood 2003, 101, 2637–2645. [Google Scholar] [CrossRef]
- Li, Y.; Choi, H.; Zhou, Z.; Nolasco, L.; Pownall, H.J.; Voorberg, J.; Moake, J.L.; Dong, J.F. Covalent regulation of ULVWF string formation and elongation on endothelial cells under flow conditions. J. Thromb. Haemost. 2008, 6, 1135–1143. [Google Scholar] [CrossRef] [Green Version]
- Sorvillo, N.; Mizurini, D.M.; Coxon, C.; Martinod, K.; Tilvawala, R.; Cherpokova, D.; Salinger, A.J.; Seward, R.J.; Staudinger, C.; Weerapana, E.; et al. Plasma Peptidylarginine Deiminase IV Promotes VWF-Platelet String Formation and Accelerates Thrombosis After Vessel Injury. Circ. Res. 2019, 125, 507–519. [Google Scholar] [CrossRef]
- Fullerton, J.N.; Gilroy, D.W. Resolution of inflammation: A new therapeutic frontier. Nat. Rev. Drug Discov. 2016, 15, 551–567. [Google Scholar] [CrossRef]
- Serhan, C.N.; Chiang, N.; Van Dyke, T.E. Resolving inflammation: Dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 2008, 8, 349–361. [Google Scholar] [CrossRef] [Green Version]
- Ansari, J.; Kaur, G.; Gavins, F.N.E. Therapeutic Potential of Annexin A1 in Ischemia Reperfusion Injury. Int. J. Mol. Sci. 2018, 19, 1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kain, V.; Liu, F.; Kozlovskaya, V.; Ingle, K.A.; Bolisetty, S.; Agarwal, A.; Khedkar, S.; Prabhu, S.D.; Kharlampieva, E.; Halade, G.V. Resolution Agonist 15-epi-Lipoxin A4 Programs Early Activation of Resolving Phase in Post-Myocardial Infarction Healing. Sci. Rep. 2017, 7, 9999. [Google Scholar] [CrossRef] [PubMed]
- Goicoechea, M.; Sanchez-Nino, M.D.; Ortiz, A.; Garcia de Vinuesa, S.; Quiroga, B.; Bernis, C.; Morales, E.; Fernandez-Juarez, G.; de Sequera, P.; Verdalles, U.; et al. Low dose aspirin increases 15-epi-lipoxin A4 levels in diabetic chronic kidney disease patients. Prostaglandins Leukot. Essent. Fatty Acids 2017, 125, 8–13. [Google Scholar] [CrossRef]
- Perucci, L.O.; Sugimoto, M.A.; Gomes, K.B.; Dusse, L.M.; Teixeira, M.M.; Sousa, L.P. Annexin A1 and specialized proresolving lipid mediators: Promoting resolution as a therapeutic strategy in human inflammatory diseases. Expert Opin. Ther. Targets 2017, 21, 879–896. [Google Scholar] [CrossRef]
- Chamani, S.; Bianconi, V.; Tasbandi, A.; Pirro, M.; Barreto, G.E.; Jamialahmadi, T.; Sahebkar, A. Resolution of Inflammation in Neurodegenerative Diseases: The Role of Resolvins. Mediat. Inflamm. 2020, 2020, 3267172. [Google Scholar] [CrossRef]
- Patel, H.B.; Kornerup, K.N.; Sampaio, A.L.; D’Acquisto, F.; Seed, M.P.; Girol, A.P.; Gray, M.; Pitzalis, C.; Oliani, S.M.; Perretti, M. The impact of endogenous annexin A1 on glucocorticoid control of inflammatory arthritis. Ann. Rheum. Dis. 2012, 71, 1872–1880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Langmead, C.J.; Riddy, D.M. New Advances in Targeting the Resolution of Inflammation: Implications for Specialized Pro-Resolving Mediator GPCR Drug Discovery. ACS Pharmacol. Transl. Sci. 2020, 3, 88–106. [Google Scholar] [CrossRef] [PubMed]
- Vital, S.A.; Senchenkova, E.Y.; Ansari, J.; Gavins, F.N.E. Targeting AnxA1/Formyl Peptide Receptor 2 Pathway Affords Protection against Pathological Thrombo-Inflammation. Cells 2020, 9, 2473. [Google Scholar] [CrossRef] [PubMed]
- Drieu, A.; Levard, D.; Vivien, D.; Rubio, M. Anti-inflammatory treatments for stroke: From bench to bedside. Ther. Adv. Neurol. Disord. 2018, 11, 1756286418789854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prestigiacomo, C.J.; Kim, S.C.; Connolly, E.S., Jr.; Liao, H.; Yan, S.F.; Pinsky, D.J. CD18-mediated neutrophil recruitment contributes to the pathogenesis of reperfused but not nonreperfused stroke. Stroke 1999, 30, 1110–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, H.; Hayashi, T.; Tojo, S.J.; Kitagawa, H.; Kimura, K.; Mizugaki, M.; Itoyama, Y.; Abe, K. Anti-P-selectin antibody attenuates rat brain ischemic injury. Neurosci. Lett. 1999, 265, 163–166. [Google Scholar] [CrossRef]
- Enlimomab Acute Stroke Trial, I. Use of anti-ICAM-1 therapy in ischemic stroke: Results of the Enlimomab Acute Stroke Trial. Neurology 2001, 57, 1428–1434. [Google Scholar] [CrossRef]
- Ansari, J. Targeting Peptidyl Arginine Deiminase 4 and NADPH Oxidase Pathway Regulates Neutrophil Dependent Thrombo-Inflammation. Br. J. Pharmacol. 2020, 178, 409–410. [Google Scholar]
- Carpenter, A.C.; Alexander, J.S. Endothelial PKC delta activation attenuates neutrophil transendothelial migration. Inflamm. Res. 2008, 57, 216–229. [Google Scholar] [CrossRef]
- Yoo, J.; Nichols, A.; Song, J.C.; Mammen, J.; Calvo, I.; Worrell, R.T.; Cuppoletti, J.; Matlin, K.; Matthews, J.B. Bryostatin-1 attenuates TNF-induced epithelial barrier dysfunction: Role of novel PKC isozymes. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 284, G703–G712. [Google Scholar] [CrossRef] [Green Version]
- Tan, Z.; Turner, R.C.; Leon, R.L.; Li, X.; Hongpaisan, J.; Zheng, W.; Logsdon, A.F.; Naser, Z.J.; Alkon, D.L.; Rosen, C.L.; et al. Bryostatin improves survival and reduces ischemic brain injury in aged rats after acute ischemic stroke. Stroke 2013, 44, 3490–3497. [Google Scholar] [CrossRef] [Green Version]
- Hankey, G.J. The benefits of aspirin in early secondary stroke prevention. Lancet 2016, 388, 312–314. [Google Scholar] [CrossRef] [Green Version]
- Awtry, E.H.; Loscalzo, J. Aspirin. Circulation 2000, 101, 1206–1218. [Google Scholar] [CrossRef]
- Rothwell, P.M.; Giles, M.F.; Chandratheva, A.; Marquardt, L.; Geraghty, O.; Redgrave, J.N.; Lovelock, C.E.; Binney, L.E.; Bull, L.M.; Cuthbertson, F.C.; et al. Effect of urgent treatment of transient ischaemic attack and minor stroke on early recurrent stroke (EXPRESS study): A prospective population-based sequential comparison. Lancet 2007, 370, 1432–1442. [Google Scholar] [CrossRef]
- Meadows, T.A.; Bhatt, D.L. Clinical aspects of platelet inhibitors and thrombus formation. Circ. Res. 2007, 100, 1261–1275. [Google Scholar] [CrossRef] [Green Version]
- Hackam, D.G.; Spence, J.D. Antiplatelet Therapy in Ischemic Stroke and Transient Ischemic Attack. Stroke 2019, 50, 773–778. [Google Scholar] [CrossRef]
- Johnston, S.C.; Easton, J.D.; Farrant, M.; Barsan, W.; Conwit, R.A.; Elm, J.J.; Kim, A.S.; Lindblad, A.S.; Palesch, Y.Y. Clopidogrel and Aspirin in Acute Ischemic Stroke and High-Risk TIA. N. Eng. J. Med. 2018, 379, 215–225. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Zhao, X.; Liu, L.; Wang, D.; Wang, C.; Wang, C.; Li, H.; Meng, X.; Cui, L.; et al. Clopidogrel with Aspirin in Acute Minor Stroke or Transient Ischemic Attack. N. Engl. J. Med. 2013, 369, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, S.C.; Amarenco, P.; Denison, H.; Evans, S.R.; Himmelmann, A.; James, S.; Knutsson, M.; Ladenvall, P.; Molina, C.A.; Wang, Y. Ticagrelor and Aspirin or Aspirin Alone in Acute Ischemic Stroke or TIA. N. Engl. J. Med. 2020, 383, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.C.; Amarenco, P.; Albers, G.W.; Denison, H.; Easton, J.D.; Evans, S.R.; Held, P.; Jonasson, J.; Minematsu, K.; Molina, C.A.; et al. Ticagrelor versus Aspirin in Acute Stroke or Transient Ischemic Attack. N. Engl. J. Med. 2016, 375, 35–43. [Google Scholar] [CrossRef]
- Wang, Y.; Meng, X.; Wang, A.; Xie, X.; Pan, Y.; Johnston, S.C.; Li, H.; Bath, P.M.; Dong, Q.; Xu, A.; et al. Ticagrelor versus Clopidogrel in CYP2C19 Loss-of-Function Carriers with Stroke or TIA. N. Engl. J. Med. 2021. [Google Scholar] [CrossRef] [PubMed]
- Diener, H.C.; Cunha, L.; Forbes, C.; Sivenius, J.; Smets, P.; Lowenthal, A. European Stroke Prevention Study. 2. Dipyridamole and acetylsalicylic acid in the secondary prevention of stroke. J. Neurol. Sci. 1996, 143, 1–13. [Google Scholar] [CrossRef]
- Wieberdink, R.G.; van Schie, M.C.; Koudstaal, P.J.; Hofman, A.; Witteman, J.C.; de Maat, M.P.; Leebeek, F.W.; Breteler, M.M. High von Willebrand factor levels increase the risk of stroke: The Rotterdam study. Stroke 2010, 41, 2151–2156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bongers, T.N.; de Maat, M.P.; van Goor, M.L.; Bhagwanbali, V.; van Vliet, H.H.; Gómez García, E.B.; Dippel, D.W.; Leebeek, F.W. High von Willebrand factor levels increase the risk of first ischemic stroke: Influence of ADAMTS13, inflammation, and genetic variability. Stroke 2006, 37, 2672–2677. [Google Scholar] [CrossRef] [PubMed]
- Buchtele, N.; Schwameis, M.; Gilbert, J.C.; Schörgenhofer, C.; Jilma, B. Targeting von Willebrand Factor in Ischaemic Stroke: Focus on Clinical Evidence. Thromb. Haemost. 2018, 118, 959–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ansari, J.; Gavins, F.N.E. Neutrophils and Platelets: Immune Soldiers Fighting Together in Stroke Pathophysiology. Biomedicines 2021, 9, 1945. https://doi.org/10.3390/biomedicines9121945
Ansari J, Gavins FNE. Neutrophils and Platelets: Immune Soldiers Fighting Together in Stroke Pathophysiology. Biomedicines. 2021; 9(12):1945. https://doi.org/10.3390/biomedicines9121945
Chicago/Turabian StyleAnsari, Junaid, and Felicity N. E. Gavins. 2021. "Neutrophils and Platelets: Immune Soldiers Fighting Together in Stroke Pathophysiology" Biomedicines 9, no. 12: 1945. https://doi.org/10.3390/biomedicines9121945
APA StyleAnsari, J., & Gavins, F. N. E. (2021). Neutrophils and Platelets: Immune Soldiers Fighting Together in Stroke Pathophysiology. Biomedicines, 9(12), 1945. https://doi.org/10.3390/biomedicines9121945