
Role of Phytoconstituents as PPAR Agonists: Implications for Neurodegenerative Disorders



Abstract
:1. Introduction
2. PPARs Isoforms and Their Distribution across the Nervous System
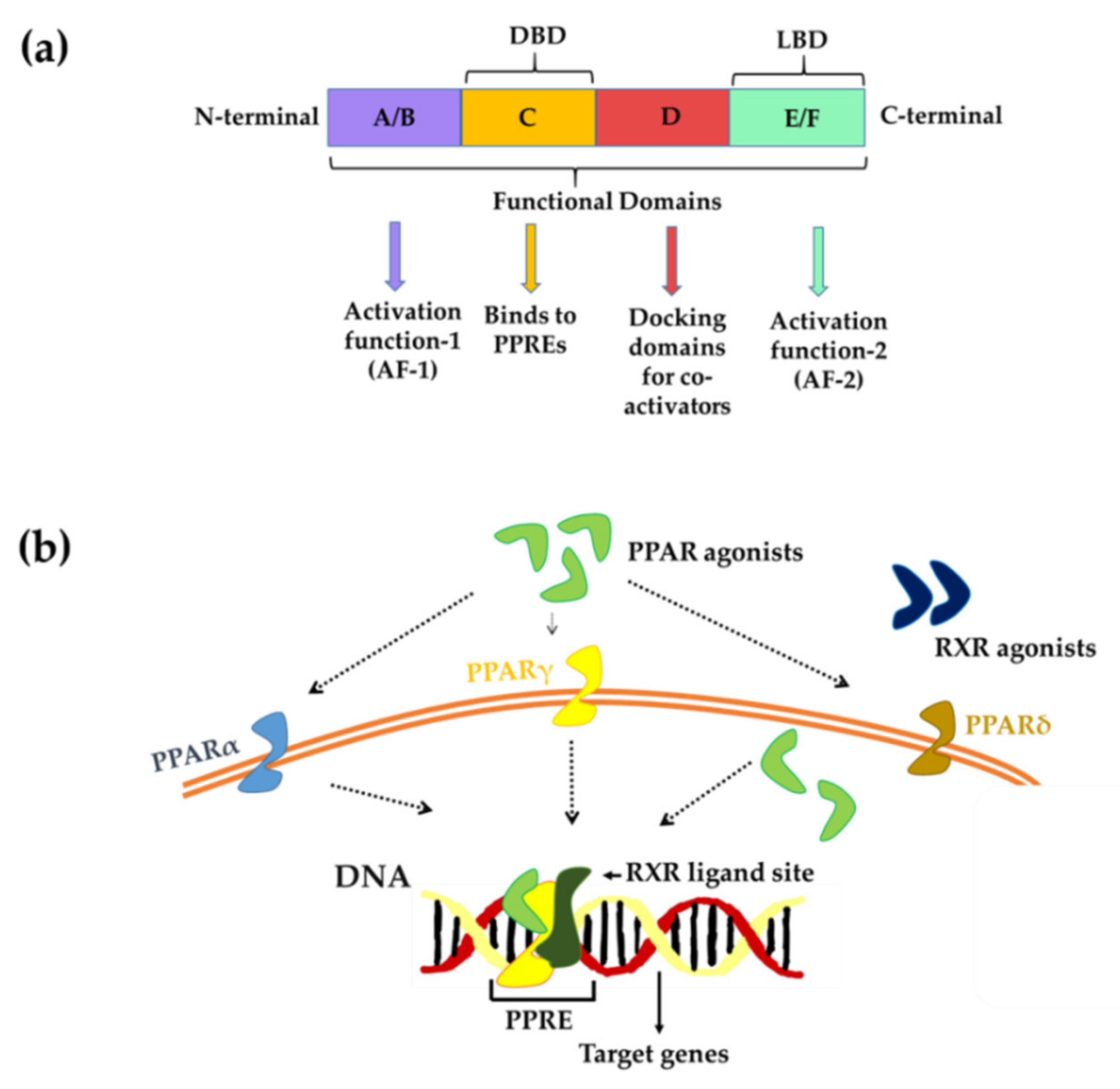
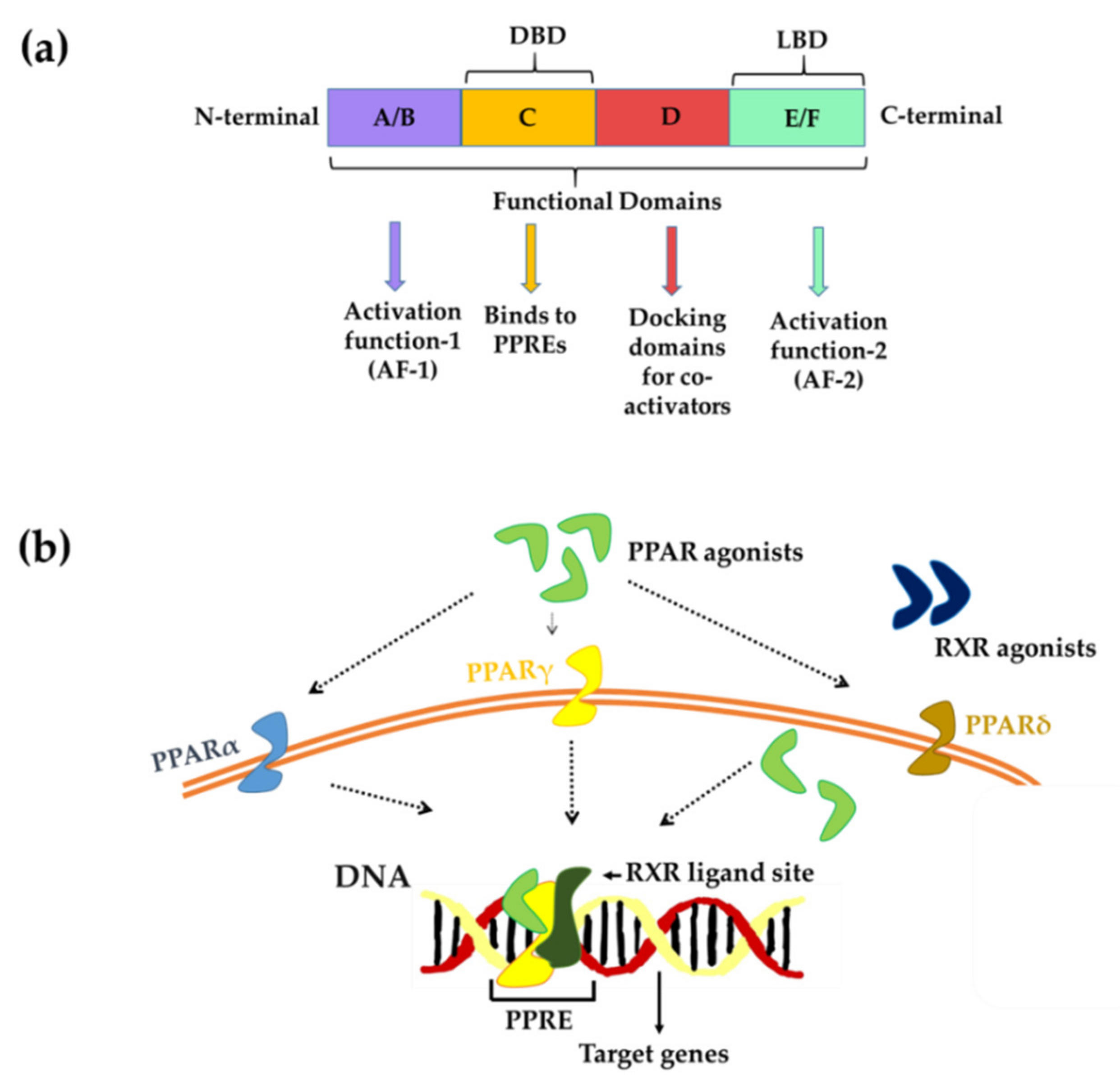
3. PPARs-Structure and Mechanism of Action
4. Neurodegenerative Disorders (NDDs) and Role of PPARs
4.1. Cerebral Ischemia
4.2. Alzheimer’s Disease
4.3. Parkinson’s Disease
4.4. Huntington’s Disease
4.5. Multiple Sclerosis
4.6. Autoimmune Encephalitis
5. Literature Search Strategy
6. Natural Compounds as PPAR Agonists
6.1. Flavonoids
6.1.1. Biochanin A
6.1.2. Icariin
6.1.3. Luteoloside
6.1.4. Naringenin
6.1.5. Chrysin
6.1.6. Cyanidin 3-O-β-Glucopyranoside (Cy-3-G)
6.1.7. Galangin
6.2. Fatty Acids
6.2.1. Poly/Monounsaturated Fatty Acids (PUFAs/MUFAs)
6.2.2. n-3 Fatty Acid-Rich Linseed Oil
6.3. Cannabinoids (CBs)
6.4. Other Compounds Extracted from Plants
6.4.1. Curcumin
6.4.2. Capsaicin
6.4.3. Piperine
6.4.4. Estradiol
7. Phytoconstituents’ Other Modes of Action in NDDs
8. In Vitro Studies from Patient’s Sample and Clinical Data
9. Drosophila in Translational Medicine
10. Conclusions and Future Implications
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kininis, M.; Kraus, W.L. A global view of transcriptional regulation by nuclear receptors: Gene expression, factor localization, and DNA sequence analysis. Nucl. Recept. Signal. 2008, 6, e005. [Google Scholar] [CrossRef] [Green Version]
- Gustafsson, J.-A. Historical overview of nuclear receptors. J. Steroid Biochem. Mol. Biol. 2016, 157, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Grygiel-Górniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications-a review. Nutrition 2014, 13, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Lambert, M.H.; Xu, H.E. Activation of nuclear receptors: A perspective from structural genomics. Structure 2003, 11, 741–746. [Google Scholar] [CrossRef] [Green Version]
- McKenna, N.J.; Lanz, R.B.; O’Malley, B.W. Nuclear receptor coregulators: Cellular and molecular biology. Endocr. Rev. 1999, 20, 321–344. [Google Scholar]
- Nagy, L.; Schwabe, J.W. Mechanism of the nuclear receptor molecular switch. Trends Biochem. Sci. 2004, 29, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Lonard, D.M.; O’Malley, B.W. Nuclear receptor coregulators: Judges, juries, and executioners of cellular regulation. Mol. Cell 2007, 27, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Tenbaum, S.; Baniahmad, A. Nuclear receptors: Structure, function and involvement in disease. Int. J. Biochem. Cell Biol. 1997, 29, 1325–1341. [Google Scholar] [CrossRef]
- Resche-Rigon, M.; Gronemeyer, H. Therapeutic potential of selective modulators of nuclear receptor action. Curr. Opin. Chem. Biol. 1998, 2, 501–507. [Google Scholar] [CrossRef]
- Aranda, A.; Pascual, A. Nuclear hormone receptors and gene expression. Physiol. Rev. 2001, 81, 1269–1304. [Google Scholar] [CrossRef]
- Decara, J.; Rivera, P.; López-Gambero, A.J.; Serrano, A.; Pavón, F.J.; Baixeras, E.; Rodríguez de Fonseca, F.; Suárez, J. Peroxisome proliferator-activated receptors: Experimental targeting for the treatment of inflammatory bowel diseases. Front. Pharmacol. 2020, 11, 730. [Google Scholar] [CrossRef] [PubMed]
- Braissant, O.; Foufelle, F.; Scotto, C.; Dauça, M.; Wahli, W. Differential expression of peroxisome proliferator-activated receptors (PPARs): Tissue distribution of PPAR-alpha,-beta, and-gamma in the adult rat. Endocrinology 1996, 137, 354–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, S.; Farioli-Vecchioli, S.; Ceru, M. Immunolocalization of peroxisome proliferator-activated receptors and retinoid X receptors in the adult rat CNS. Neuroscience 2004, 123, 131–145. [Google Scholar] [CrossRef]
- Galan-Rodriguez, B.; Suarez, J.; Gonzalez-Aparicio, R.; Bermudez-Silva, F.; Maldonado, R.; Robledo, P.; de Fonseca, F.R.; Fernandez-Espejo, E. Oleoylethanolamide exerts partial and dose-dependent neuroprotection of substantia nigra dopamine neurons. Neuropharmacology 2009, 56, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Cullingford, T.E.; Bhakoo, K.; Peuchen, S.; Dolphin, C.T.; Patel, R.; Clark, J.B. Distribution of mRNAs Encoding the Peroxisome Proliferator-Activated Receptor α, β, and γ and the Retinoid X Receptor α, β, and γ in Rat Central Nervous System. J. Neurochem. 1998, 70, 1366–1375. [Google Scholar] [CrossRef] [PubMed]
- Kainu, T.; Wikström, A.; Gustafsson, J.-A.; Pelto-Huikko, M. Localization of the peroxisome proliferator-activated receptor in the brain. Neuroreport 1994, 5, 2481–2485. [Google Scholar] [CrossRef]
- Xu, J.; Storer, P.D.; Chavis, J.A.; Racke, M.K.; Drew, P.D. Agonists for the peroxisome proliferator-activated receptor-α and the retinoid X receptor inhibit inflammatory responses of microglia. J. Neurosci. Res. 2005, 81, 403–411. [Google Scholar] [CrossRef]
- Bernardo, A.; Bianchi, D.; Magnaghi, V.; Minghetti, L. Peroxisome proliferator-activated receptor-γ agonists promote differentiation and antioxidant defenses of oligodendrocyte progenitor cells. J. Neuropathol. Exp. Neurol. 2009, 68, 797–808. [Google Scholar] [CrossRef]
- Guida, F.; Luongo, L.; Boccella, S.; Giordano, M.; Romano, R.; Bellini, G.; Manzo, I.; Furiano, A.; Rizzo, A.; Imperatore, R. Palmitoylethanolamide induces microglia changes associated with increased migration and phagocytic activity: Involvement of the CB2 receptor. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Esmaeili, M.A.; Yadav, S.; Gupta, R.K.; Waggoner, G.R.; Deloach, A.; Calingasan, N.Y.; Beal, M.F.; Kiaei, M. Preferential PPAR-α activation reduces neuroinflammation, and blocks neurodegeneration in vivo. Hum. Mol. Genet. 2016, 25, 317–327. [Google Scholar] [CrossRef] [Green Version]
- Bordet, R.; Gelé, P.; Duriez, P.; Fruchart, J. PPARs: A New Target for Neuroprotection; Sigma-Aldrich: Burlington, MA, USA, 2006. [Google Scholar]
- Daynes, R.A.; Jones, D.C. Emerging roles of PPARs in inflammation and immunity. Nat. Rev. Immunol. 2002, 2, 748–759. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Quiles, M.; Broekema, M.F.; Kalkhoven, E. PPARgamma in metabolism, immunity, and cancer: Unified and diverse mechanisms of action. Front. Endocrinol. 2021, 12, 624112. [Google Scholar] [CrossRef] [PubMed]
- Warden, A.; Truitt, J.; Merriman, M.; Ponomareva, O.; Jameson, K.; Ferguson, L.B.; Mayfield, R.D.; Harris, R.A. Localization of PPAR isotypes in the adult mouse and human brain. Sci. Rep. 2016, 6, 1–15. [Google Scholar]
- Strosznajder, A.K.; Wójtowicz, S.; Jeżyna, M.J.; Sun, G.Y.; Strosznajder, J.B. Recent Insights on the Role of PPAR-β/δ in Neuroinflammation and Neurodegeneration, and Its Potential Target for Therapy. NeuroMol. Med. 2020, 1–13. [Google Scholar] [CrossRef]
- Hall, M.; Quignodon, L.; Desvergne, B. Peroxisome proliferator-activated receptor β/δ in the brain: Facts and hypothesis. PPAR Res. 2008, 2008, 780452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’sullivan, S. Cannabinoids go nuclear: Evidence for activation of peroxisome proliferator-activated receptors. Br. J. Pharmacol. 2007, 152, 576–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroker, A.J.; Bruning, J.B. Review of the structural and dynamic mechanisms of PPARγ partial agonism. PPAR Res. 2015, 2015, 816856. [Google Scholar] [CrossRef] [Green Version]
- Evans, R.M.; Mangelsdorf, D.J. Nuclear receptors, RXR, and the big bang. Cell 2014, 157, 255–266. [Google Scholar] [CrossRef] [Green Version]
- Feige, J.N.; Gelman, L.; Michalik, L.; Desvergne, B.; Wahli, W. From molecular action to physiological outputs: Peroxisome proliferator-activated receptors are nuclear receptors at the crossroads of key cellular functions. Prog. Lipid Res. 2006, 45, 120–159. [Google Scholar] [CrossRef]
- Rigano, D.; Sirignano, C.; Taglialatela-Scafati, O. The potential of natural products for targeting PPARα. Acta Pharm. Sin. B 2017, 7, 427–438. [Google Scholar] [CrossRef]
- Iannotti, F.A.; Vitale, R.M. The endocannabinoid system and PPARs: Focus on their signalling crosstalk, action and transcriptional regulation. Cells 2021, 10, 586. [Google Scholar] [CrossRef] [PubMed]
- Heemels, M.-T. Neurodegenerative diseases. Nature 2016, 539, 179–180. [Google Scholar] [CrossRef] [Green Version]
- Gitler, A.D.; Dhillon, P.; Shorter, J. Neurodegenerative disease: Models, mechanisms, and a new hope. Lancet 2017, 10, 499–502. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.S.; Joh, T.H. Microglia, major player in the brain inflammation: Their roles in the pathogenesis of Parkinson’s disease. Exp. Mol. Med. 2006, 38, 333–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saijo, K.; Crotti, A.; Glass, C.K. Regulation of microglia activation and deactivation by nuclear receptors. Glia 2013, 61, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.S.; Koh, S.-H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 1–12. [Google Scholar] [CrossRef]
- González-Scarano, F.; Baltuch, G. Microglia as mediators of inflammatory and degenerative diseases. Annu. Rev. Neurosci. 1999, 22, 219–240. [Google Scholar] [CrossRef]
- Liu, B.; Hong, J.-S. Role of microglia in inflammation-mediated neurodegenerative diseases: Mechanisms and strategies for therapeutic intervention. J. Pharmacol. Exp. Ther. 2003, 304, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Breunig, J.; Guillot-Sestier, M.-V.; Town, T. Brain injury, neuroinflammation and Alzheimer’s disease. Front. Aging Neurosci. 2013, 5, 26. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Guo, C.; Wu, J. Therapeutic potential of PPARγ natural agonists in liver diseases. J. Cell. Mol. Med. 2020, 24, 2736–2748. [Google Scholar] [CrossRef]
- Zolezzi, J.M.; Santos, M.J.; Bastías-Candia, S.; Pinto, C.; Godoy, J.A.; Inestrosa, N.C. PPARs in the central nervous system: Roles in neurodegeneration and neuroinflammation. Biology 2017, 92, 2046–2069. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Jiang, X.-F.; Katayama, T.; Osada, S.; Umesono, K.; Namura, S. Brain protection by resveratrol and fenofibrate against stroke requires peroxisome proliferator-activated receptor α in mice. Neurosci. Lett. 2003, 352, 203–206. [Google Scholar] [CrossRef]
- Besson, V.C.; Chen, X.R.; Plotkine, M.; Marchand-Verrecchia, C. Fenofibrate, a peroxisome proliferator-activated receptor α agonist, exerts neuroprotective effects in traumatic brain injury. Neurosci. Lett. 2005, 388, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Cuzzocrea, S.; Mazzon, E.; Di Paola, R.; Peli, A.; Bonato, A.; Britti, D.; Genovese, T.; Muia, C.; Crisafulli, C.; Caputi, A.P. The role of the peroxisome proliferator-activated receptor-α (PPAR-α) in the regulation of acute inflammation. J. Leukoc. Biol. 2006, 79, 999–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cramer, P.E.; Cirrito, J.R.; Wesson, D.W.; Lee, C.D.; Karlo, J.C.; Zinn, A.E.; Casali, B.T.; Restivo, J.L.; Goebel, W.D.; James, M.J. ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. Science 2012, 335, 1503–1506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gold, P.W.; Licinio, J.; Pavlatou, M. Pathological parainflammation and endoplasmic reticulum stress in depression: Potential translational targets through the CNS insulin, klotho and PPAR-γ systems. Mol. Psychiatry 2013, 18, 154–165. [Google Scholar] [CrossRef]
- Landreth, G.E.; Heneka, M.T. Anti-inflammatory actions of peroxisome proliferator-activated receptor gamma agonists in Alzheimer’s disease. Neurobiol. Aging 2001, 22, 937–944. [Google Scholar] [CrossRef]
- Kanakasabai, S.; Pestereva, E.; Chearwae, W.; Gupta, S.K.; Ansari, S.; Bright, J.J. PPARγ agonists promote oligodendrocyte differentiation of neural stem cells by modulating stemness and differentiation genes. PLoS ONE 2012, 7, e50500. [Google Scholar] [CrossRef]
- Chiang, M.-C.; Cheng, Y.-C.; Chen, H.-M.; Liang, Y.-J.; Yen, C.-H. Rosiglitazone promotes neurite outgrowth and mitochondrial function in N2A cells via PPARgamma pathway. Mitochondrion 2014, 14, 7–17. [Google Scholar] [CrossRef]
- Landreth, G. PPARgamma agonists as new therapeutic agents for the treatment of Alzheimer’s disease. Exp. Neurol. 2006, 199, 245–248. [Google Scholar] [CrossRef]
- Chen, L.-W.; Horng, L.-Y.; Wu, C.-L.; Sung, H.-C.; Wu, R.-T. Activating mitochondrial regulator PGC-1α expression by astrocytic NGF is a therapeutic strategy for Huntington’s disease. Neuropharmacology 2012, 63, 719–732. [Google Scholar] [CrossRef]
- Martin, H.L.; Mounsey, R.B.; Mustafa, S.; Sathe, K.; Teismann, P. Pharmacological manipulation of peroxisome proliferator-activated receptor γ (PPARγ) reveals a role for anti-oxidant protection in a model of Parkinson’s disease. Exp. Neurol. 2012, 235, 528–538. [Google Scholar] [CrossRef] [Green Version]
- Schnegg, C.I.; Robbins, M.E. Neuroprotective mechanisms of PPARδ: Modulation of oxidative stress and inflammatory processes. PPAR Res. 2011, 2011, 373560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savage, S.R.; McCollum, G.W.; Yang, R.; Penn, J.S. RNA-seq identifies a role for the PPARβ/δ inverse agonist GSK0660 in the regulation of TNFα-induced cytokine signaling in retinal endothelial cells. Mol. Vis. 2015, 21, 568. [Google Scholar] [PubMed]
- Bai, B.; Yan, Z.; Hao, Y.; Zhang, Z.; Li, G.; Dekker, J.; Qiu, C. A randomised controlled multimodal intervention trial in patients with ischaemic stroke in Shandong, China: Design and rationale. Lancet 2017, 390, S13. [Google Scholar] [CrossRef] [Green Version]
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N. Heart disease and stroke statistics—2021 update: A report from the American Heart Association. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef]
- Ekdahl, C.T.; Claasen, J.-H.; Bonde, S.; Kokaia, Z.; Lindvall, O. Inflammation is detrimental for neurogenesis in adult brain. Proc. Natl. Acad. Sci. USA 2003, 100, 13632–13637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, X.-Y.; Liu, L.; Yang, Q.-W. Functions and mechanisms of microglia/macrophages in neuroinflammation and neurogenesis after stroke. Prog. Neurobiol. 2016, 142, 23–44. [Google Scholar] [CrossRef] [PubMed]
- Culman, J.; Zhao, Y.; Gohlke, P.; Herdegen, T. PPAR-γ: Therapeutic target for ischemic stroke. Trends Pharmacol. Sci. 2007, 28, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Victor, N.A.; Wanderi, E.W.; Gamboa, J.; Zhao, X.; Aronowski, J.; Deininger, K.; Lust, W.D.; Landreth, G.E.; Sundararajan, S. Altered PPARγ expression and activation after transient focal ischemia in rats. Eur. J. Neurosci. 2006, 24, 1653–1663. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Yang, T.; Liu, H.; Han, L.; Zhang, K.; Hu, X.; Zhang, X.; Yin, K.-J.; Gao, Y.; Bennett, M.V.L.; et al. Peroxisome proliferator-activated receptor γ (PPARγ): A master gatekeeper in CNS injury and repair. Prog. Neurobiol. 2018, 163–164, 27–58. [Google Scholar] [CrossRef] [PubMed]
- Shehata, A.H.F.; Ahmed, A.-S.F.; Abdelrehim, A.B.; Heeba, G.H. The impact of single and combined PPAR-α and PPAR-γ activation on the neurological outcomes following cerebral ischemia reperfusion. Life Sci. 2020, 252, 117679. [Google Scholar] [CrossRef]
- Mannan, A.; Garg, N.; Singh, T.G.; Kang, H.K. Peroxisome Proliferator-Activated Receptor-Gamma (PPAR-ɣ): Molecular Effects and Its Importance as a Novel Therapeutic Target for Cerebral Ischemic Injury. Neurochem. Res. 2021, 46, 2800–2831. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Ahmad, R.; Khare, S.K. Alzheimer’s disease and its treatment by different approaches: A review. Eur. J. Med. Chem. 2021, 216, 113320. [Google Scholar] [CrossRef] [PubMed]
- Inestrosa, N.C.; Godoy, J.A.; Quintanilla, R.A.; Koenig, C.S.; Bronfman, M. Peroxisome proliferator-activated receptor γ is expressed in hippocampal neurons and its activation prevents β-amyloid neurodegeneration: Role of Wnt signaling. Experiment. Cell Res. 2005, 304, 91–104. [Google Scholar] [CrossRef]
- Nenov, M.N.; Tempia, F.; Denner, L.; Dineley, K.T.; Laezza, F. Impaired firing properties of dentate granule neurons in an Alzheimer’s disease animal model are rescued by PPARγ agonism. J. Neurophysiol. 2015, 113, 1712–1726. [Google Scholar] [CrossRef]
- Wójtowicz, S.; Strosznajder, A.K.; Jeżyna, M.; Strosznajder, J.B. The Novel Role of PPAR Alpha in the Brain: Promising Target in Therapy of Alzheimer’s Disease and Other Neurodegenerative Disorders. Neurochem. Res. 2020, 45, 972–988. [Google Scholar] [CrossRef] [Green Version]
- Opara, J.; Małecki, A.; Małecka, E.; Socha, T. Motor assessment in Parkinsons disease. Ann. Agric. Environ. Med. 2017, 24, 411–415. [Google Scholar] [CrossRef]
- Tolosa, E.; Garrido, A.; Scholz, S.W.; Poewe, W. Challenges in the diagnosis of Parkinson’s disease. Lancet Neurol. 2021, 20, 385–397. [Google Scholar] [CrossRef]
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef] [Green Version]
- Dexter, D.T.; Jenner, P. Parkinson disease: From pathology to molecular disease mechanisms. Free Radic. Biol. Med. 2013, 62, 132–144. [Google Scholar] [CrossRef]
- Avarachan, J.; Augustine, A.; Shinde, P.M.; Gunasekaran, V. A Mechanistic approach of Peroxisome Proliferator-Activated Receptors and its subtypes on Clinical and preclinical model of Neurodegenerative disorders. Res. J. Pharm. Technol. 2021, 14, 3967–3975. [Google Scholar] [CrossRef]
- Upadhyay, A.; Amanullah, A.; Joshi, V.; Dhiman, R.; Prajapati, V.K.; Poluri, K.M.; Mishra, A. Ibuprofen-based advanced therapeutics: Breaking the inflammatory link in cancer, neurodegeneration, and diseases. Drug Metab. Rev. 2021, 53, 100–121. [Google Scholar] [CrossRef]
- Michael, J.; Zirknitzer, J.; Unger, M.S.; Poupardin, R.; Rieß, T.; Paiement, N.; Zerbe, H.; Hutter-Paier, B.; Reitsamer, H.; Aigner, L. The Leukotriene Receptor Antagonist Montelukast Attenuates Neuroinflammation and Affects Cognition in Transgenic 5xFAD Mice. Int. J. Mol. Sci. 2021, 22, 2782. [Google Scholar] [CrossRef] [PubMed]
- Mahalakshmi, B.; Maurya, N.; Lee, S.-D.; Bharath Kumar, V. Possible Neuroprotective Mechanisms of Physical Exercise in Neurodegeneration. Int. J. Mol. Sci. 2020, 21, 5895. [Google Scholar] [CrossRef] [PubMed]
- Behl, T.; Madaan, P.; Sehgal, A.; Singh, S.; Sharma, N.; Bhatia, S.; Al-Harrasi, A.; Chigurupati, S.; Alrashdi, I.; Bungau, S.G. Elucidating the Neuroprotective Role of PPARs in Parkinson’s Disease: A Neoteric and Prospective Target. Int. J. Mol. Sci. 2021, 22, 10161. [Google Scholar] [CrossRef]
- Gil, J.M.; Rego, A.C. Mechanisms of neurodegeneration in Huntington’s disease. Eur. J. Neurosci. 2008, 27, 2803–2820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, S.J.; Douglas, I.; Rawlins, M.D.; Wexler, N.S.; Tabrizi, S.J.; Smeeth, L. Prevalence of adult Huntington’s disease in the UK based on diagnoses recorded in general practice records. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1156–1160. [Google Scholar] [CrossRef] [Green Version]
- Túnez, I.; Tasset, I.; Santamaría, A. 3-Nitropropionic acid as a tool to study the mechanisms involved in Huntington’s disease: Past, present and future. Molecules 2010, 15, 878–916. [Google Scholar] [CrossRef] [Green Version]
- Chiang, M.-C.; Chen, C.-M.; Lee, M.-R.; Chen, H.-W.; Chen, H.-M.; Wu, Y.-S.; Hung, C.-H.; Kang, J.-J.; Chang, C.-P.; Chang, C.; et al. Modulation of energy deficiency in Huntington’s disease via activation of the peroxisome proliferator-activated receptor gamma. Hum. Mol. Genet. 2010, 19, 4043–4058. [Google Scholar] [CrossRef] [Green Version]
- Quintanilla, R.A.; Jin, Y.N.; Fuenzalida, K.; Bronfman, M.; Johnson, G.V.W. Rosiglitazone Treatment Prevents Mitochondrial Dysfunction in Mutant Huntingtin-expressing Cells: Possible role of peroxisome proliferator-activated receptor-γ (pparγ) in the pathogenesis of huntington disease. J. Biol. Chem. 2008, 283, 25628–25637. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Albertz, J.; Guo, Z.; Peng, Q.; Rudow, G.; Troncoso, J.C.; Ross, C.A.; Duan, W. Neuroprotective effects of PPAR-γ agonist rosiglitazone in N171-82Q mouse model of Huntington’s disease. J. Neurochem. 2013, 125, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Steinman, L. Immunology of relapse and remission in multiple sclerosis. Annu. Rev. Immunol. 2014, 32, 257–281. [Google Scholar] [CrossRef] [PubMed]
- Sospedra, M.; Martin, R. Immunology of multiple sclerosis. Annu. Rev. Immunol. 2005, 23, 683–747. [Google Scholar] [CrossRef] [Green Version]
- Walton, C.; King, R.; Rechtman, L.; Kaye, W.; Leray, E.; Marrie, R.A.; Robertson, N.; La Rocca, N.; Uitdehaag, B.; van der Mei, I. Rising prevalence of multiple sclerosis worldwide: Insights from the Atlas of MS. Mult. Scler. J. 2020, 26, 1816–1821. [Google Scholar] [CrossRef] [PubMed]
- Friese, M.A.; Schattling, B.; Fugger, L. Mechanisms of neurodegeneration and axonal dysfunction in multiple sclerosis. Nat. Rev. Neurol. 2014, 10, 225–238. [Google Scholar] [CrossRef]
- Ferret-Sena, V.; Capela, C.; Sena, A. Metabolic Dysfunction and Peroxisome Proliferator-Activated Receptors (PPAR) in Multiple Sclerosis. Int. J. Mol. Sci. 2018, 19, 1639. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Zhong, M.; He, L.; Li, W.; Huang, Y.; Liu, J.; Chen, Y.; Xiao, Z. Epidemiology of antibody-positive autoimmune encephalitis in Southwest China: A multicenter study. Front. Immunol. 2019, 10, 2611. [Google Scholar] [CrossRef] [Green Version]
- Giacoppo, S.; Galuppo, M.; Montaut, S.; Iori, R.; Rollin, P.; Bramanti, P.; Mazzon, E. An overview on neuroprotective effects of isothiocyanates for the treatment of neurodegenerative diseases. Fitoterapia 2015, 106, 12–21. [Google Scholar] [CrossRef]
- Galuppo, M.; Giacoppo, S.; De Nicola, G.R.; Iori, R.; Navarra, M.; Lombardo, G.E.; Bramanti, P.; Mazzon, E. Antiinflammatory activity of glucomoringin isothiocyanate in a mouse model of experimental autoimmune encephalomyelitis. Fitoterapia 2014, 95, 160–174. [Google Scholar] [CrossRef]
- Giacoppo, S.; Soundara Rajan, T.; De Nicola, G.R.; Iori, R.; Bramanti, P.; Mazzon, E. Moringin activates Wnt canonical pathway by inhibiting GSK3β in a mouse model of experimental autoimmune encephalomyelitis. Drug Des. Dev. Ther. 2016, 10, 3291–3304. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Li, X.; Ciric, B.; Curtis, M.T.; Chen, W.-J.; Rostami, A.; Zhang, G.-X. A dual effect of ursolic acid to the treatment of multiple sclerosis through both immunomodulation and direct remyelination. Proc. Natl. Acad. Sci. USA 2020, 117, 9082–9093. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, J.; Luo, S.; Zhan, Y.; Lu, Q. The roles of PPARγ and its agonists in autoimmune diseases: A comprehensive review. J. Autoimmun. 2020, 113, 102510. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Zhang, Y.; Zhu, S.; Luo, Y.; Xu, P.; Huang, Z. PPAR-mediated toxicology and applied pharmacology. Cells 2020, 9, 352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, S.; Yadav, A.; Chaturvedi, R.K. Peroxisome proliferator-activated receptors (PPARs) as therapeutic target in neurodegenerative disorders. Biochem. Biophys. Res. Commun. 2017, 483, 1166–1177. [Google Scholar] [CrossRef] [PubMed]
- Panche, A.; Diwan, A.; Chandra, S. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, e47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rupasinghe, H. Special issue flavonoids and their disease prevention and treatment potential: Recent advances and future perspectives. Molecules 2020, 25, 4746. [Google Scholar] [CrossRef] [PubMed]
- Devi, S.; Kumar, V.; Singh, S.K.; Dubey, A.K.; Kim, J.-J. Flavonoids: Potential candidates for the treatment of neurodegenerative disorders. Biomedicines 2021, 9, 99. [Google Scholar] [CrossRef] [PubMed]
- Sarfraz, A.; Javeed, M.; Shah, M.A.; Hussain, G.; Shafiq, N.; Sarfraz, I.; Riaz, A.; Sadiqa, A.; Zara, R.; Zafar, S. Biochanin A: A novel bioactive multifunctional compound from nature. Sci. Total Environ. 2020, 722, 137907. [Google Scholar] [CrossRef]
- Wang, W.; Tang, L.; Li, Y.; Wang, Y. Biochanin A protects against focal cerebral ischemia/reperfusion in rats via inhibition of p38-mediated inflammatory responses. J. Neurol. Sci. 2015, 348, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Wang, X.; Chen, H.; Yan, Z.; Wang, M.; Li, Y. Neurochemical and behavior deficits in rats with iron and rotenone co-treatment: Role of redox imbalance and neuroprotection by biochanin A. Front. Neurosci. 2017, 11, 657. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, W.-a. Biochanin A inhibits lipopolysaccharide-induced inflammatory cytokines and mediators production in BV2 microglia. Neurochem. Res. 2015, 40, 165–171. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Wang, Z.; Shi, J. Pharmacological effects of icariin. Adv. Pharmacol. 2020, 87, 179–203. [Google Scholar]
- Xiong, D.; Deng, Y.; Huang, B.; Yin, C.; Liu, B.; Shi, J.; Gong, Q. Icariin attenuates cerebral ischemia–reperfusion injury through inhibition of inflammatory response mediated by NF-κB, PPARα and PPARγ in rats. Int. Immunopharmacol. 2016, 30, 157–162. [Google Scholar] [CrossRef]
- Siniscalchi, A.; Gallelli, L.; Malferrari, G.; Pirritano, D.; Serra, R.; Santangelo, E.; De Sarro, G. Cerebral stroke injury: The role of cytokines and brain inflammation. J. Basic Clin. Physiol. Pharmacol. 2014, 25, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Tobin, M.K.; Bonds, J.A.; Minshall, R.D.; Pelligrino, D.A.; Testai, F.D.; Lazarov, O. Neurogenesis and inflammation after ischemic stroke: What is known and where we go from here. J. Cereb. Blood Flow Metab. 2014, 34, 1573–1584. [Google Scholar] [CrossRef] [Green Version]
- Maddahi, A.; Edvinsson, L. Cerebral ischemia induces microvascular pro-inflammatory cytokine expression via the MEK/ERK pathway. J. Neuroinflamm. 2010, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Wan, F.; Lenardo, M.J. The nuclear signaling of NF-κB: Current knowledge, new insights, and future perspectives. Cell Res. 2010, 20, 24–33. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Zhou, Q.-x.; Shi, J.-s. Protective effects of icariin on neurons injured by cerebral ischemia/reperfusion. Chin. Med. J. 2005, 118, 1637–1643. [Google Scholar]
- Li, F.; Gong, Q.-H.; Wu, Q.; Lu, Y.-F.; Shi, J.-S. Icariin isolated from Epimedium brevicornum Maxim attenuates learning and memory deficits induced by d-galactose in rats. Pharmacol. Biochem. Behav. 2010, 96, 301–305. [Google Scholar] [CrossRef]
- Guo, J.; Li, F.; Wu, Q.; Gong, Q.; Lu, Y.; Shi, J. Protective effects of icariin on brain dysfunction induced by lipopolysaccharide in rats. Phytomedicine 2010, 17, 950–955. [Google Scholar] [CrossRef]
- Zhu, H.-R.; Wang, Z.-Y.; Zhu, X.-L.; Wu, X.-X.; Li, E.-g.; Xu, Y. Icariin protects against brain injury by enhancing SIRT1-dependent PGC-1α expression in experimental stroke. Neuropharmacology 2010, 59, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhu, T.; Wang, M.; Zhang, F.; Zhang, G.; Zhao, J.; Zhang, Y.; Wu, E.; Li, X. Icariin attenuates M1 activation of microglia and Aβ plaque accumulation in the hippocampus and prefrontal cortex by up-regulating PPARγ in restraint/isolation-stressed APP/PS1 mice. Front. Neurosci. 2019, 13, 291. [Google Scholar] [CrossRef] [PubMed]
- Dai, M.; Chen, B.; Wang, X.; Gao, C.; Yu, H. Icariin enhance mild hypothermia-induced neuroprotection via inhibiting the activation of NF-κB in experimental ischemic stroke. Metab. Brain Dis. 2021, 36, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Tian, Z.; Wang, M.; Kou, J.; Wang, C.; Rong, X.; Li, J.; Xie, X.; Pang, X. Luteoloside attenuates neuroinflammation in focal cerebral ischemia in rats via regulation of the PPARγ/Nrf2/NF-κB signaling pathway. Int. Immunopharmacol. 2019, 66, 309–316. [Google Scholar] [CrossRef]
- Tang, Y.; Tong, X.; Li, Y.; Jiang, G.; Yu, M.; Chen, Y.; Dong, S. JAK2/STAT3 pathway is involved in the protective effects of epidermal growth factor receptor activation against cerebral ischemia/reperfusion injury in rats. Neurosci. Lett. 2018, 662, 219–226. [Google Scholar] [CrossRef]
- Deng, Y.; Xiong, D.; Yin, C.; Liu, B.; Shi, J.; Gong, Q. Icariside II protects against cerebral ischemia–reperfusion injury in rats via nuclear factor-κB inhibition and peroxisome proliferator-activated receptor up-regulation. Neurochem. Int. 2016, 96, 56–61. [Google Scholar] [CrossRef]
- Yin, C.; Deng, Y.; Liu, Y.; Gao, J.; Yan, L.; Gong, Q. Icariside II ameliorates cognitive impairments induced by chronic cerebral hypoperfusion by inhibiting the amyloidogenic pathway: Involvement of BDNF/TrkB/CREB signaling and up-regulation of PPARα and PPARγ in rats. Front. Pharmacol. 2018, 9, 1211. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Shi, L.; Liu, Y.; Li, P.; Jiang, G.; Gao, X.; Zhang, Y.; Jiang, C.; Zhu, W.; Han, H. Activation of PPARγ mediates icaritin-induced cell cycle arrest and apoptosis in glioblastoma multiforme. Biomed. Pharmacother. 2018, 100, 358–366. [Google Scholar] [CrossRef]
- Wang, Z.; Zeng, M.; Wang, Z.; Qin, F.; Chen, J.; He, Z. Dietary Luteolin: A Narrative Review Focusing on Its Pharmacokinetic Properties and Effects on Glycolipid Metabolism. J. Agric. Food Chem. 2021, 69, 1441–1454. [Google Scholar] [CrossRef] [PubMed]
- Salehi, B.; Fokou, P.V.T.; Sharifi-Rad, M.; Zucca, P.; Pezzani, R.; Martins, N.; Sharifi-Rad, J. The therapeutic potential of naringenin: A review of clinical trials. Pharmaceuticals 2019, 12, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghofrani, S.; Joghataei, M.-T.; Mohseni, S.; Baluchnejadmojarad, T.; Bagheri, M.; Khamse, S.; Roghani, M. Naringenin improves learning and memory in an Alzheimer’s disease rat model: Insights into the underlying mechanisms. Eur. J. Pharmacol. 2015, 764, 195–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Ma, J.; Liu, Z.; Lu, Y.; Hu, B.; Yu, H. Effect of naringenin on brain insulin signaling and cognitive functions in ICV-STZ induced dementia model of rats. Neurol. Sci. 2014, 35, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Wang, G.; Kandhare, A.D.; Mukherjee-Kandhare, A.A.; Bodhankar, S.L. Neuroprotective effect of naringin, a flavone glycoside in quinolinic acid-induced neurotoxicity: Possible role of PPAR-γ, Bax/Bcl-2, and caspase-3. Food Chem. Toxicol. 2018, 121, 95–108. [Google Scholar] [CrossRef]
- Hoyer, S. Causes and consequences of disturbances of cerebral glucose metabolism in sporadic Alzheimer disease: Therapeutic implications. Front. Neurosci. 2004, 541, 135–152. [Google Scholar]
- Solano, D.C.; Sironi, M.; Bonfini, C.; Solerte, S.B.; Govoni, S.; Racchi, M. Insulin regulates soluble amyloid precursor protein release via phosphatidyl inositol 3 kinase-dependent pathway. FASEB J. 2000, 14, 1015–1022. [Google Scholar] [CrossRef]
- Rad, S.K.; Arya, A.; Karimian, H.; Madhavan, P.; Rizwan, F.; Koshy, S.; Prabhu, G. Mechanism involved in insulin resistance via accumulation of β-amyloid and neurofibrillary tangles: Link between type 2 diabetes and Alzheimer’s disease. Drug Des. Dev. Ther. 2018, 12, 3999–4021. [Google Scholar]
- Zhang, Y.; Huang, N.-q.; Yan, F.; Jin, H.; Zhou, S.-y.; Shi, J.-s.; Jin, F. Diabetes mellitus and Alzheimer’s disease: GSK-3β as a potential link. Behav. Brain Res. 2018, 339, 57–65. [Google Scholar] [CrossRef]
- Fu, H.; Liu, B.; Li, L.; Lemere, C.A. Microglia Do Not Take Up Soluble Amyloid-beta Peptides, But Partially Degrade Them by Secreting Insulin-degrading Enzyme. Neuroscience 2020, 443, 30–43. [Google Scholar] [CrossRef]
- Du, J.; Zhang, L.; Liu, S.; Zhang, C.; Huang, X.; Li, J.; Zhao, N.; Wang, Z. PPARγ transcriptionally regulates the expression of insulin-degrading enzyme in primary neurons. Biochem. Biophys. Res. Commun. 2009, 383, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Kalonia, H.; Kumar, P.; Kumar, A. Pioglitazone ameliorates behavioral, biochemical and cellular alterations in quinolinic acid induced neurotoxicity: Possible role of peroxisome proliferator activated receptor-ϒ (PPARϒ) in Huntington’s disease. Pharmacol. Biochem. Behav. 2010, 96, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Chaudhary, T.; Mishra, J. Minocycline modulates neuroprotective effect of hesperidin against quinolinic acid induced Huntington’s disease like symptoms in rats: Behavioral, biochemical, cellular and histological evidences. Eur. J. Pharmacol. 2013, 720, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Dünschede, F.; Tybl, E.; Kiemer, A.K.; Dutkowski, P.; Erbes, K.; Kircher, A.; Gockel, I.; Zechner, U.; Schad, A.; Lang, H. Bcl-2 upregulation after 3-nitropropionic acid preconditioning in warm rat liver ischemia. Shock 2008, 30, 699–704. [Google Scholar] [CrossRef] [Green Version]
- Mani, R.; Natesan, V. Chrysin: Sources, beneficial pharmacological activities, and molecular mechanism of action. Phytochemistry 2018, 145, 187–196. [Google Scholar] [CrossRef]
- Bae, Y.; Lee, S.; Kim, S.-H. Chrysin suppresses mast cell-mediated allergic inflammation: Involvement of calcium, caspase-1 and nuclear factor-κB. Toxicol. Appl. Pharmacol. 2011, 254, 56–64. [Google Scholar] [CrossRef]
- Xiao, J.; Zhai, H.; Yao, Y.; Wang, C.; Jiang, W.; Zhang, C.; Simard, A.; Zhang, R.; Hao, J. Chrysin attenuates experimental autoimmune neuritis by suppressing immuno-inflammatory responses. Neuroscience 2014, 262, 156–164. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, J.; Liu, F.; Tong, L.; Chen, Z.; Chen, J.; He, H.; Xu, R.; Ma, Y.; Huang, C. Neuroprotective effects of anthocyanins and its major component cyanidin-3-O-glucoside (C3G) in the central nervous system: An outlined review. Eur. J. Pharmacol. 2019, 858, 172500. [Google Scholar] [CrossRef]
- Song, N.; Zhang, L.; Chen, W.; Zhu, H.; Deng, W.; Han, Y.; Guo, J.; Qin, C. Cyanidin 3-O-β-glucopyranoside activates peroxisome proliferator-activated receptor-γ and alleviates cognitive impairment in the APPswe/PS1ΔE9 mouse model. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 1786–1800. [Google Scholar] [CrossRef]
- Olivas-Aguirre, F.J.; Rodrigo-García, J.; Martínez-Ruiz, N.d.R.; Cárdenas-Robles, A.I.; Mendoza-Díaz, S.O.; Álvarez-Parrilla, E.; González-Aguilar, G.A.; De la Rosa, L.A.; Ramos-Jiménez, A.; Wall-Medrano, A. Cyanidin-3-O-glucoside: Physical-chemistry, foodomics and health effects. Molecules 2016, 21, 1264. [Google Scholar] [CrossRef] [Green Version]
- Fang, D.; Xiong, Z.; Xu, J.; Yin, J.; Luo, R. Chemopreventive mechanisms of galangin against hepatocellular carcinoma: A review. Biomed. Pharmacother. 2019, 109, 2054–2061. [Google Scholar] [CrossRef]
- Kong, Y.; Feng, Z.; Chen, A.; Qi, Q.; Han, M.; Wang, S.; Zhang, Y.; Zhang, X.; Yang, N.; Wang, J. The natural flavonoid galangin elicits apoptosis, pyroptosis, and autophagy in glioblastoma. Front. Oncol. 2019, 9, 942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mak, K.-K.; Tan, J.-J.; Marappan, P.; Balijepalli, M.K.; Choudhury, H.; Ramamurthy, S.; Pichika, M.R. Galangin’s potential as a functional food ingredient. J. Funct. Foods 2018, 46, 490–503. [Google Scholar] [CrossRef]
- Choi, M.-J.; Lee, E.-J.; Park, J.-S.; Kim, S.-N.; Park, E.-M.; Kim, H.-S. Anti-inflammatory mechanism of galangin in lipopolysaccharide-stimulated microglia: Critical role of PPAR-γ signaling pathway. Biochem. Pharmacol. 2017, 144, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.L.; Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuadrado, A.; Rojo, A.I. Heme oxygenase-1 as a therapeutic target in neurodegenerative diseases and brain infections. Curr. Pharm. Des. 2008, 14, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.-J.; Park, J.-S.; Park, J.-E.; Kim, H.S.; Kim, H.-S. Galangin suppresses pro-inflammatory gene expression in polyinosinic-polycytidylic acid-stimulated microglial cells. Biomol. Ther. 2017, 25, 641–647. [Google Scholar] [CrossRef] [Green Version]
- Calder, P.C. Fatty acids and inflammation: The cutting edge between food and pharma. Eur. J. Pharmacol. 2011, 668, S50–S58. [Google Scholar] [CrossRef]
- Shahidi, F.; Ambigaipalan, P. Omega-3 polyunsaturated fatty acids and their health benefits. Annu. Rev. Food Sci. Technol. 2018, 9, 345–381. [Google Scholar] [CrossRef]
- Kumar, N.G.; Contaifer, D.; Madurantakam, P.; Carbone, S.; Price, E.T.; Van Tassell, B.; Brophy, D.F.; Wijesinghe, D.S. Dietary bioactive fatty acids as modulators of immune function: Implications on human health. Nutrients 2019, 11, 2974. [Google Scholar] [CrossRef] [Green Version]
- Sokoła-Wysoczańska, E.; Wysoczański, T.; Wagner, J.; Czyż, K.; Bodkowski, R.; Lochyński, S.; Patkowska-Sokoła, B. Polyunsaturated fatty acids and their potential therapeutic role in cardiovascular system disorders—A review. Nutrients 2018, 10, 1561. [Google Scholar] [CrossRef] [Green Version]
- Calder, P.C. Marine omega-3 fatty acids and inflammatory processes: Effects, mechanisms and clinical relevance. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2015, 1851, 469–484. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C. Omega-3 fatty acids and inflammatory processes: From molecules to man. Biochem. Soc. Trans. 2017, 45, 1105–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braverman, N.E.; Moser, A.B. Functions of plasmalogen lipids in health and disease. Biochim. Biophys. Acta Mol. Basis Dis. 2012, 1822, 1442–1452. [Google Scholar] [CrossRef] [Green Version]
- Echeverría, F.; Valenzuela, R.; Hernandez-Rodas, M.C.; Valenzuela, A. Docosahexaenoic acid (DHA), a fundamental fatty acid for the brain: New dietary sources. Prostagland. Leukot. Essent. Fatty Acids 2017, 124, 1–10. [Google Scholar] [CrossRef]
- Zock, P.L.; Blom, W.A.; Nettleton, J.A.; Hornstra, G. Progressing insights into the role of dietary fats in the prevention of cardiovascular disease. Curr. Cardiol. Rep. 2016, 18, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Bozzatello, P.; Brignolo, E.; De Grandi, E.; Bellino, S. Supplementation with omega-3 fatty acids in psychiatric disorders: A review of literature data. J. Clin. Med. 2016, 5, 67. [Google Scholar] [CrossRef]
- Bazan, N.G.; Molina, M.F.; Gordon, W.C. Docosahexaenoic acid signalolipidomics in nutrition: Significance in aging, neuroinflammation, macular degeneration, Alzheimer’s, and other neurodegenerative diseases. Annu. Rev. Nutr. 2011, 31, 321–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, W.; Yen, J.-H.; Ganea, D. Docosahexaenoic acid prevents dendritic cell maturation, inhibits antigen-specific Th1/Th17 differentiation and suppresses experimental autoimmune encephalomyelitis. Brain Behav. Immun. 2011, 25, 872–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kliewer, S.A.; Sundseth, S.S.; Jones, S.A.; Brown, P.J.; Wisely, G.B.; Koble, C.S.; Devchand, P.; Wahli, W.; Willson, T.M.; Lenhard, J.M. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors α and γ. Proc. Natl. Acad. Sci. USA 1997, 94, 4318–4323. [Google Scholar] [CrossRef] [Green Version]
- Yu, K.; Bayona, W.; Kallen, C.B.; Harding, H.P.; Ravera, C.P.; McMahon, G.; Brown, M.; Lazar, M.A. Differential Activation of Peroxisome Proliferator-activated Receptors by Eicosanoids. J. Biol. Chem. 1995, 270, 23975–23983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Combs, C.K.; Johnson, D.E.; Karlo, J.C.; Cannady, S.B.; Landreth, G.E. Inflammatory mechanisms in Alzheimer’s disease: Inhibition of β-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARγ agonists. J. Neurosci. Res. 2000, 20, 558–567. [Google Scholar] [CrossRef]
- Niemoller, T.D.; Bazan, N.G. Docosahexaenoic acid neurolipidomics. Prostagland. Other Lipid Mediat. 2010, 91, 85–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonietta Ajmone-Cat, M.; Lavinia Salvatori, M.; De Simone, R.; Mancini, M.; Biagioni, S.; Bernardo, A.; Cacci, E.; Minghetti, L. Docosahexaenoic acid modulates inflammatory and antineurogenic functions of activated microglial cells. J. Neurosci. Res. 2012, 90, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, A.; Giammarco, M.; De Nuccio, C.; Ajmone-Cat, M.; Visentin, S.; De Simone, R.; Minghetti, L. Docosahexaenoic acid promotes oligodendrocyte differentiation via PPAR-γ signalling and prevents tumor necrosis factor-α-dependent maturational arrest. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1013–1023. [Google Scholar] [CrossRef]
- Guardiola-Diaz, H.M.; Ishii, A.; Bansal, R. Erk1/2 MAPK and mTOR signaling sequentially regulates progression through distinct stages of oligodendrocyte differentiation. Glia 2012, 60, 476–486. [Google Scholar] [CrossRef] [Green Version]
- Bonora, M.; De Marchi, E.; Patergnani, S.; Suski, J.; Celsi, F.; Bononi, A.; Giorgi, C.; Marchi, S.; Rimessi, A.; Duszyński, J. Tumor necrosis factor-α impairs oligodendroglial differentiation through a mitochondria-dependent process. Cell Death Differ. 2014, 21, 1198–1208. [Google Scholar] [CrossRef] [Green Version]
- De Nuccio, C.; Bernardo, A.; Cruciani, C.; De Simone, R.; Visentin, S.; Minghetti, L. Peroxisome proliferator activated receptor-γ agonists protect oligodendrocyte progenitors against tumor necrosis factor-alpha-induced damage: Effects on mitochondrial functions and differentiation. Exp. Neurol. 2015, 271, 506–514. [Google Scholar] [CrossRef]
- Mancera, P.; Wappenhans, B.; Cordobilla, B.; Virgili, N.; Pugliese, M.; Rueda, F.; Espinosa-Parrilla, J.F.; Domingo, J.C. Natural docosahexaenoic acid in the triglyceride form attenuates in vitro microglial activation and ameliorates autoimmune encephalomyelitis in mice. Nutrients 2017, 9, 681. [Google Scholar] [CrossRef]
- Newell, M.; Baker, K.; Postovit, L.M.; Field, C.J. A critical review on the effect of docosahexaenoic acid (DHA) on cancer cell cycle progression. Int. J. Mol. Sci. 2017, 18, 1784. [Google Scholar] [CrossRef] [Green Version]
- Zapata-Gonzalez, F.; Rueda, F.; Petriz, J.; Domingo, P.; Villarroya, F.; Diaz-Delfin, J.; de Madariaga, M.A.; Domingo, J.C. Human dendritic cell activities are modulated by the omega-3 fatty acid, docosahexaenoic acid, mainly through PPARγ: RXR heterodimers: Comparison with other polyunsaturated fatty acids. J. Leukoc. Biol. 2008, 84, 1172–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storer, P.D.; Xu, J.; Chavis, J.; Drew, P.D. Peroxisome proliferator-activated receptor-gamma agonists inhibit the activation of microglia and astrocytes: Implications for multiple sclerosis. J. Neuroimmunol. 2005, 161, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Innes, J.K.; Calder, P.C. The differential effects of eicosapentaenoic acid and docosahexaenoic acid on cardiometabolic risk factors: A systematic review. Int. J. Mol. Sci. 2018, 19, 532. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez, S.; Svahn, S.L.; Johansson, M.E. Effects of omega-3 fatty acids on immune cells. Int. J. Mol. Sci. 2019, 20, 5028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calder, P.C. n− 3 Polyunsaturated fatty acids, inflammation, and inflammatory diseases. Am. J. Clin. Nutr. 2006, 83, 1505S–1519S. [Google Scholar] [CrossRef]
- Fatel, E.C.; Rosa, F.T.; Alfieri, D.F.; Flauzino, T.; Scavuzzi, B.M.; Lozovoy, M.A.; Iriyoda, T.M.; Simão, A.N.; Dichi, I. Beneficial effects of fish oil and cranberry juice on disease activity and inflammatory biomarkers in people with rheumatoid arthritis. Nutrition 2021, 86, 111183. [Google Scholar] [CrossRef]
- Lynch, A.M.; Loane, D.J.; Minogue, A.M.; Clarke, R.M.; Kilroy, D.; Nally, R.E.; Roche, Ó.J.; O’Connell, F.; Lynch, M.A. Eicosapentaenoic acid confers neuroprotection in the amyloid-β challenged aged hippocampus. Neurobiol. Aging 2007, 28, 845–855. [Google Scholar] [CrossRef]
- Kapadia, R.; Yi, J.-H.; Vemuganti, R. Mechanisms of anti-inflammatory and neuroprotective actions of PPAR-gamma agonists. Front. Biosci. 2008, 13, 1813. [Google Scholar] [CrossRef] [Green Version]
- Kawashima, A.; Harada, T.; Imada, K.; Yano, T.; Mizuguchi, K. Eicosapentaenoic acid inhibits interleukin-6 production in interleukin-1β-stimulated C6 glioma cells through peroxisome proliferator-activated receptor-gamma. Prostagland. Leukot. Essent. Fatty Acids 2008, 79, 59–65. [Google Scholar] [CrossRef]
- Unoda, K.; Doi, Y.; Nakajima, H.; Yamane, K.; Hosokawa, T.; Ishida, S.; Kimura, F.; Hanafusa, T. Eicosapentaenoic acid (EPA) induces peroxisome proliferator-activated receptors and ameliorates experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2013, 256, 7–12. [Google Scholar] [CrossRef]
- Satyanarayanan, S.K.; Shih, Y.-H.; Chien, Y.-C.; Huang, S.-Y.; Gałecki, P.; Kasper, S.; Chang, J.P.-C.; Su, K.-P. Anti-oxidative effects of melatonin receptor agonist and omega-3 polyunsaturated fatty acids in neuronal SH-SY5Y cells: Deciphering synergic effects on anti-depressant mechanisms. Mol. Neurobiol. 2018, 55, 7271–7284. [Google Scholar] [CrossRef]
- Corsi, L.; Momo Dongmo, B.; Avallone, R. Supplementation of omega 3 fatty acids improves oxidative stress in activated BV2 microglial cell line. Int. J. Food Sci. Nutr. 2015, 66, 293–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.-C.; Chen, Q.; Wan, X.-Z.; Yang, X.-L.; Liu, X.; Zhong, L. Effects of conjugated linoleic acid on cleavage of amyloid precursor protein via PPARγ. Neurol. Sci. 2011, 32, 1095–1101. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-J.; Liang, C.-L.; Li, G.-M.; Yu, C.-Y.; Yin, M. Neuroprotective effects of arachidonic acid against oxidative stress on rat hippocampal slices. Chem. Biol. 2006, 163, 207–217. [Google Scholar] [CrossRef]
- Wang, Z.-j.; Li, G.-m.; Tang, W.-l.; Yin, M. Neuroprotective effects of stearic acid against toxicity of oxygen/glucose deprivation or glutamate on rat cortical or hippocampal slices. Acta Pharmacol. Sin. 2006, 27, 145–150. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.-j.; Liang, C.-l.; Li, G.-m.; Yu, C.-y.; Yin, M. Stearic acid protects primary cultured cortical neurons against oxidative stress 4. Acta Pharmacol. Sin. 2007, 28, 315–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.; Kim, Y.-S.; Lee, D.H.; Lee, S.H.; Park, H.J.; Lee, D.; Kim, H. Neuroprotective effects of oleic acid in rodent models of cerebral ischaemia. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassett, C.M.; McCullough, R.S.; Edel, A.L.; Patenaude, A.; LaVallee, R.K.; Pierce, G.N. The α-linolenic acid content of flaxseed can prevent the atherogenic effects of dietary trans fat. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2220–H2226. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, V.; Abidi, A.; Markerb, S.; Bilal, S. Linseed and linseed oil: Health benefits-a review. Int. J. Pharm. Biol. Sci. 2013, 3, 434–442. [Google Scholar]
- Ramaprasad, T.; Baskaran, V.; Krishnakantha, T.; Lokesh, B. Modulation of antioxidant enzyme activities, platelet aggregation and serum prostaglandins in rats fed spray-dried milk containing n-3 fatty acid. Mol. Cell. Biochem. 2005, 280, 9–16. [Google Scholar] [CrossRef]
- Rao, Y.P.C.; Lokesh, B. Down-regulation of NF-κB expression by n-3 fatty acid-rich linseed oil is modulated by PPARγ activation, eicosanoid cascade and secretion of cytokines by macrophages in rats fed partially hydrogenated vegetable fat. Eur. J. Nutr. 2017, 56, 1135–1147. [Google Scholar] [CrossRef] [PubMed]
- Reeves, P.G.; Nielsen, F.H.; Fahey, G.C., Jr. AIN-93 purified diets for laboratory rodents: Final report of the American Institute of Nutrition ad hoc writing committee on the reformulation of the AIN-76A rodent diet. J. Nutr. 1993, 123, 1939–1951. [Google Scholar] [CrossRef] [PubMed]
- Magarkar, A.; Dhawan, V.; Kallinteri, P.; Viitala, T.; Elmowafy, M.; Róg, T.; Bunker, A. Cholesterol level affects surface charge of lipid membranes in saline solution. Sci. Rep. 2014, 4, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Pablo, M.A.; De Cienfuegos, G.Á. Modulatory effects of dietary lipids on immune system functions. Immunol. Cell Biol. 2000, 78, 31–39. [Google Scholar] [CrossRef]
- Mensink, R.P.; Katan, M.B. Effect of dietary trans fatty acids on high-density and low-density lipoprotein cholesterol levels in healthy subjects. N. Eng. J. Med. 1990, 323, 439–445. [Google Scholar] [CrossRef] [Green Version]
- Zhi, T.; Yanhong, L.; Rui, L.; Jian, S.; Jinping, L.; Junzhu, W. Trans fatty acids influence the oxidation of LDL in ECV304 cells. Eur. J. Lipid Sci. Technol. 2012, 114, 880–888. [Google Scholar] [CrossRef]
- Estadella, D.; Da Penha Oller do Nascimento, C.; Oyama, L.M.; Ribeiro, E.B.; Damaso, A.R.; de Piano, A. Lipotoxicity: Effects of dietary saturated and transfatty acids. Mediat. Inflamm. 2013, 2013, 137579. [Google Scholar] [CrossRef] [Green Version]
- Nagy, L.; Szanto, A.; Szatmari, I.; Széles, L. Nuclear hormone receptors enable macrophages and dendritic cells to sense their lipid environment and shape their immune response. Physiol. Rev. 2012, 92, 739–789. [Google Scholar] [CrossRef] [Green Version]
- Cassano, T.; Villani, R.; Pace, L.; Carbone, A.; Bukke, V.N.; Orkisz, S.; Avolio, C.; Serviddio, G. From Cannabis sativa to cannabidiol: Promising therapeutic candidate for the treatment of neurodegenerative diseases. Front. Pharmacol. 2020, 11, 124. [Google Scholar] [CrossRef] [Green Version]
- Carroll, C.; Zeissler, M.L.; Hanemann, C.; Zajicek, J. Δ9-tetrahydrocannabinol (Δ9-THC) exerts a direct neuroprotective effect in a human cell culture model of Parkinson’s disease. Neuropathol. Appl. Neurobiol. 2012, 38, 535–547. [Google Scholar] [CrossRef]
- Zeissler, M.-L.; Eastwood, J.; McCorry, K.; Hanemann, C.O.; Zajicek, J.P.; Carroll, C.B. Delta-9-tetrahydrocannabinol protects against MPP+ toxicity in SH-SY5Y cells by restoring proteins involved in mitochondrial biogenesis. Oncotarget 2016, 7, 46603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadal, X.; Del Río, C.; Casano, S.; Palomares, B.; Ferreiro-Vera, C.; Navarrete, C.; Sánchez-Carnerero, C.; Cantarero, I.; Bellido, M.L.; Meyer, S. Tetrahydrocannabinolic acid is a potent PPARγ agonist with neuroprotective activity. Br. J. Pharmacol. 2017, 174, 4263–4276. [Google Scholar] [CrossRef] [Green Version]
- Roura, X.N. Methods of purifying cannabinoids, compositions and kits thereof. U.S. Patent US9765000B2, 19 September 2017. [Google Scholar]
- Chiang, M.-C.; Cheng, Y.-C.; Nicol, C.J.; Lin, K.-H.; Yen, C.-H.; Chen, S.-J.; Huang, R.-N. Rosiglitazone activation of PPARγ-dependent signaling is neuroprotective in mutant huntingtin expressing cells. Exp. Cell Res. 2015, 338, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Valdeolivas, S.; Navarrete, C.; Cantarero, I.; Bellido, M.L.; Muñoz, E.; Sagredo, O. Neuroprotective properties of cannabigerol in Huntington’s disease: Studies in R6/2 mice and 3-nitropropionate-lesioned mice. Neurotherapeutics 2015, 12, 185–199. [Google Scholar] [CrossRef] [Green Version]
- Esposito, G.; Scuderi, C.; Valenza, M.; Togna, G.I.; Latina, V.; De Filippis, D.; Cipriano, M.; Carratù, M.R.; Iuvone, T.; Steardo, L. Cannabidiol reduces Aβ-induced neuroinflammation and promotes hippocampal neurogenesis through PPARγ involvement. PLoS ONE 2011, 6, e28668. [Google Scholar] [CrossRef] [PubMed]
- Scuderi, C.; Steardo, L.; Esposito, G. Cannabidiol promotes amyloid precursor protein ubiquitination and reduction of beta amyloid expression in SHSY5YAPP+ cells through PPARγ involvement. Phytother. Res. 2014, 28, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- Dos-Santos-Pereira, M.; da-Silva, C.A.; Guimaraes, F.S.; Del-Bel, E. Co-administration of cannabidiol and capsazepine reduces L-DOPA-induced dyskinesia in mice: Possible mechanism of action. Neurobiol. Dis. 2016, 94, 179–195. [Google Scholar] [CrossRef] [PubMed]
- Hind, W.H.; England, T.J.; O’Sullivan, S.E. Cannabidiol protects an in vitro model of the blood–brain barrier from oxygen-glucose deprivation via PPARγ and 5-HT1A receptors. Br. J. Pharmacol. 2016, 173, 815–825. [Google Scholar] [CrossRef] [Green Version]
- Giacoppo, S.; Pollastro, F.; Grassi, G.; Bramanti, P.; Mazzon, E. Target regulation of PI3K/Akt/mTOR pathway by cannabidiol in treatment of experimental multiple sclerosis. Fitoterapia 2017, 116, 77–84. [Google Scholar] [CrossRef]
- Sadeghian, M.; Rahmani, S.; Jamialahmadi, T.; Johnston, T.P.; Sahebkar, A. The effect of oral curcumin supplementation on health-related quality of life: A systematic review and meta-analysis of randomized controlled trials. J. Affect. Disord. 2020, 278, 627–636. [Google Scholar] [CrossRef]
- Chearwae, W.; Bright, J.J. 15-Deoxy-Δ 12, 14-prostaglandin J 2 and curcumin modulate the expression of toll-like receptors 4 and 9 in autoimmune T lymphocyte. J. Clin. Immunol. 2008, 28, 558–570. [Google Scholar] [CrossRef]
- Forman, B.M.; Tontonoz, P.; Chen, J.; Brun, R.P.; Spiegelman, B.M.; Evans, R.M. 15-deoxy-Δ12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPARγ. Cell 1995, 83, 803–812. [Google Scholar] [CrossRef] [Green Version]
- Natarajan, C.; Bright, J.J. Curcumin inhibits experimental allergic encephalomyelitis by blocking IL-12 signaling through Janus kinase-STAT pathway in T lymphocytes. J. Immunol. Res. 2002, 168, 6506–6513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natarajan, C.; Bright, J. Peroxisome proliferator-activated receptor-gamma agonists inhibit experimental allergic encephalomyelitis by blocking IL-12 production, IL-12 signaling and Th1 differentiation. Genes Immun. 2002, 3, 59–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bright, J.J.; Natarajan, C.; Muthian, G.; Barak, Y.; Evans, R.M. Peroxisome proliferator-activated receptor-γ-deficient heterozygous mice develop an exacerbated neural antigen-induced Th1 response and experimental allergic encephalomyelitis. J. Immunol. Res. 2003, 171, 5743–5750. [Google Scholar] [CrossRef] [Green Version]
- Kanakasabai, S.; Casalini, E.; Walline, C.C.; Mo, C.; Chearwae, W.; Bright, J.J. Differential regulation of CD4+ T helper cell responses by curcumin in experimental autoimmune encephalomyelitis. J. Nutr. Biochem. 2012, 23, 1498–1507. [Google Scholar] [CrossRef] [PubMed]
- Rinwa, P.; Kaur, B.; Jaggi, A.S.; Singh, N. Involvement of PPAR-gamma in curcumin-mediated beneficial effects in experimental dementia. Naunyn Schmiedebergs Arch. Pharmacol. 2010, 381, 529–539. [Google Scholar] [CrossRef]
- Liu, Z.-J.; Liu, W.; Liu, L.; Xiao, C.; Wang, Y.; Jiao, J.-S. Curcumin protects neuron against cerebral ischemia-induced inflammation through improving PPAR-gamma function. Evid.-Based Complement. Altern. Med. 2013, 2013, 470975. [Google Scholar] [CrossRef] [Green Version]
- Chin, D.; Hagl, S.; Hoehn, A.; Huebbe, P.; Pallauf, K.; Grune, T.; Frank, J.; Eckert, G.P.; Rimbach, G. Adenosine triphosphate concentrations are higher in the brain of APOE3-compared to APOE4-targeted replacement mice and can be modulated by curcumin. Genes Nutr. 2014, 9, 397. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.-J.; Li, Z.-H.; Liu, L.; Tang, W.-X.; Wang, Y.; Dong, M.-R.; Xiao, C. Curcumin attenuates beta-amyloid-induced neuroinflammation via activation of peroxisome proliferator-activated receptor-gamma function in a rat model of Alzheimer’s disease. Front. Pharmacol. 2016, 7, 261. [Google Scholar] [CrossRef] [Green Version]
- Bernardo, A.; Plumitallo, C.; De Nuccio, C.; Visentin, S.; Minghetti, L. Curcumin promotes oligodendrocyte differentiation and their protection against TNF-α through the activation of the nuclear receptor PPAR-γ. Sci. Rep. 2021, 11, 1–13. [Google Scholar]
- Szoka, L.; Palka, J. Capsaicin up-regulates pro-apoptotic activity of thiazolidinediones in glioblastoma cell line. Biomed. Pharmacother. 2020, 132, 110741. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sun, B.-L.; Xiang, Y.; Tian, D.-Y.; Zhu, C.; Li, W.-W.; Liu, Y.-H.; Bu, X.-L.; Shen, L.-L.; Jin, W.-S. Capsaicin consumption reduces brain amyloid-beta generation and attenuates Alzheimer’s disease-type pathology and cognitive deficits in APP/PS1 mice. Transl. Psychiatry 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Shahbazi, S.; Zakerali, T.; Frycz, B.A.; Kaur, J. The critical role of piperamide derivative D4 in the regulation of inflammatory response by the microglia and astrocytic glial cells. Biomed. Pharmacother. 2020, 132, 110895. [Google Scholar] [CrossRef] [PubMed]
- Valles, S.L.; Dolz-Gaiton, P.; Gambini, J.; Borras, C.; LLoret, A.; Pallardo, F.V.; Viña, J. Estradiol or genistein prevent Alzheimer’s disease-associated inflammation correlating with an increase PPARγ expression in cultured astrocytes. Brain Res. 2010, 1312, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Yu, S.; Zheng, W.; Feng, G.; Luo, G.; Wang, L.; Zhao, Y. Curcumin improves outcomes and attenuates focal cerebral ischemic injury via antiapoptotic mechanisms in rats. Neurochem. Res. 2010, 35, 374–379. [Google Scholar] [CrossRef]
- Strittmatter, W.J.; Roses, A.D. Apolipoprotein E and Alzheimer’s disease. Annu. Rev. Neurosci. 1996, 19, 53–77. [Google Scholar] [CrossRef]
- Jofre-Monseny, L.; Minihane, A.M.; Rimbach, G. Impact of apoE genotype on oxidative stress, inflammation and disease risk. Mol. Nutr. Food Res. 2008, 52, 131–145. [Google Scholar] [CrossRef]
- Hauptmann, S.; Scherping, I.; Dröse, S.; Brandt, U.; Schulz, K.; Jendrach, M.; Leuner, K.; Eckert, A.; Müller, W. Mitochondrial dysfunction: An early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol. Aging 2009, 30, 1574–1586. [Google Scholar] [CrossRef]
- Basith, S.; Cui, M.; Hong, S.; Choi, S. Harnessing the therapeutic potential of capsaicin and its analogues in pain and other diseases. Molecules 2016, 21, 966. [Google Scholar] [CrossRef] [Green Version]
- Caterina, M.J.; Schumacher, M.A.; Tominaga, M.; Rosen, T.A.; Levine, J.D.; Julius, D. The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature 1997, 389, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Szallasi, A.; Blumberg, P.M. Vanilloid (Capsaicin) receptors and mechanisms. Pharmacol. Rev. 1999, 51, 159–212. [Google Scholar]
- Grüter, T.; Blusch, A.; Motte, J.; Sgodzai, M.; Bachir, H.; Klimas, R.; Ambrosius, B.; Gold, R.; Ellrichmann, G.; Pitarokoili, K. Immunomodulatory and anti-oxidative effect of the direct TRPV1 receptor agonist capsaicin on Schwann cells. J. Neuroinflamm. 2020, 17, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Strakova, N.; Ehrmann, J.; Dzubak, P.; Bouchal, J.; Kolar, Z. The synthetic ligand of peroxisome proliferator-activated receptor-γ ciglitazone affects human glioblastoma cell lines. J. Pharmacol. Exp. Ther. 2004, 309, 1239–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strakova, N.; Ehrmann, J.; Bartos, J.; Malikova, J.; Dolezel, J.; Kolar, Z. Peroxisome proliferator-activated receptors (PPAR) agonists affect cell viability, apoptosis and expression of cell cycle related proteins in cell lines of glial brain tumors. Neoplasma 2005, 52, 126–136. [Google Scholar]
- Kumar, N.; Misra, P.; Dube, A.; Bhattacharya, S.; Dikshit, M.; Ranade, S. Piper betle Linn. A maligned Pan-Asiatic plant with an array of pharmacological activities and prospects for drug discovery. Curr. Sci. 2010, 99, 922–932. [Google Scholar]
- Zarai, Z.; Boujelbene, E.; Salem, N.B.; Gargouri, Y.; Sayari, A. Antioxidant and antimicrobial activities of various solvent extracts, piperine and piperic acid from Piper nigrum. LWT—Food Sci. Technol. 2013, 50, 634–641. [Google Scholar] [CrossRef]
- Thangavel, P.; Puga-Olguín, A.; Rodríguez-Landa, J.F.; Zepeda, R.C. Genistein as potential therapeutic candidate for menopausal symptoms and other related diseases. Molecules 2019, 24, 3892. [Google Scholar] [CrossRef] [Green Version]
- Tan, J.W.; Tham, C.L.; Israf, D.A.; Lee, S.H.; Kim, M.K. Neuroprotective effects of biochanin A against glutamate-induced cytotoxicity in PC12 cells via apoptosis inhibition. Neurochem. Res. 2013, 38, 512–518. [Google Scholar] [CrossRef]
- Chen, H.-Q.; Jin, Z.-Y.; Li, G.-H. Biochanin A protects dopaminergic neurons against lipopolysaccharide-induced damage through inhibition of microglia activation and proinflammatory factors generation. Neurosci. Lett. 2007, 417, 112–117. [Google Scholar] [CrossRef]
- Berköz, M.; Krośniak, M.; Özkan-Yılmaz, F.; Özlüer-Hunt, A. Prophylactic effect of Biochanin A in lipopolysaccharide-stimulated BV2 microglial cells. Immunopharmacol. Immunotoxicol. 2020, 42, 330–339. [Google Scholar] [CrossRef] [PubMed]
- El-Sherbeeny, N.A.; Soliman, N.; Youssef, A.M.; Abd El-Fadeal, N.M.; El-Abaseri, T.B.; Hashish, A.A.; Abdelbasset, W.K.; Batiha, G.E.-S.; Zaitone, S.A. The protective effect of biochanin A against rotenone-induced neurotoxicity in mice involves enhancing of PI3K/Akt/mTOR signaling and beclin-1 production. Ecotoxicol. Environ. Saf. 2020, 205, 111344. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Zhu, G.; He, J.; Wang, G.; Li, D.; Zhang, F. Icariin targets Nrf2 signaling to inhibit microglia-mediated neuroinflammation. Int. Immunopharmacol. 2019, 73, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Feng, X.; Fu, Y.; Zheng, Y.; Ma, M.; Wang, C.; Zhang, Y. Icariin attenuates amyloid-β (Aβ)-Induced neuronal insulin resistance through PTEN downregulation. Front. Pharmacol. 2020, 11, 880. [Google Scholar] [CrossRef]
- Zheng, J.; Hu, S.; Wang, J.; Zhang, X.; Yuan, D.; Zhang, C.; Liu, C.; Wang, T.; Zhou, Z. Icariin improves brain function decline in aging rats by enhancing neuronal autophagy through the AMPK/mTOR/ULK1 pathway. Pharmaceut. Biol. 2021, 59, 183–191. [Google Scholar] [CrossRef]
- Qin, L.; Chen, Z.; Yang, L.; Shi, H.; Wu, H.; Zhang, B.; Zhang, W.; Xu, Q.; Huang, F.; Wu, X. Luteolin-7-O-glucoside protects dopaminergic neurons by activating estrogen-receptor-mediated signaling pathway in MPTP-induced mice. Toxicology 2019, 426, 152256. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.H.; Fatima, M.; Ali, M.; Rizvi, M.A.; Mondal, A.C. Naringenin alleviates paraquat-induced dopaminergic neuronal loss in SH-SY5Y cells and a rat model of Parkinson’s disease. Neuropharmacology 2021, 201, 108831. [Google Scholar] [CrossRef]
- Ahsan, A.U.; Sharma, V.L.; Wani, A.; Chopra, M. Naringenin Upregulates AMPK-Mediated Autophagy to Rescue Neuronal Cells From β-Amyloid (1–42) Evoked Neurotoxicity. Mol. Neurobiol. 2020, 57, 3589–3602. [Google Scholar] [CrossRef]
- Goes, A.T.; Jesse, C.R.; Antunes, M.S.; Ladd, F.V.L.; Ladd, A.A.L.; Luchese, C.; Paroul, N.; Boeira, S.P. Protective role of chrysin on 6-hydroxydopamine-induced neurodegeneration a mouse model of Parkinson’s disease: Involvement of neuroinflammation and neurotrophins. Chem.-Biol. Interact. 2018, 279, 111–120. [Google Scholar] [CrossRef]
- Angelopoulou, E.; Pyrgelis, E.-S.; Piperi, C. Neuroprotective potential of chrysin in Parkinson’s disease: Molecular mechanisms and clinical implications. Neurochem. Int. 2020, 132, 104612. [Google Scholar] [CrossRef]
- Campos, H.M.; da Costa, M.; da Silva Moreira, L.K.; da Silva Neri, H.F.; da Silva, C.R.B.; Pruccoli, L.; Dos Santos, F.C.A.; Costa, E.A.; Tarozzi, A.; Ghedini, P.C. Protective Effects of Chrysin against the Neurotoxicity Induced by Aluminium: In Vitro and In Vivo Studies. Toxicology 2021, 465, 153033. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Mathew, S.; Nair, P.; Ramadan, W.S.; Vazhappilly, C.G. Health benefits of cyanidin-3-glucoside as a potent modulator of Nrf2-mediated oxidative stress. Inflammopharmacology 2021, 29, 907–923. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-C.; Hsiao, L.-D.; Yang, C.-M. Galangin Inhibits LPS-Induced MMP-9 Expression via Suppressing Protein Kinase-Dependent AP-1 and FoxO1 Activation in Rat Brain Astrocytes. J. Inflamm. Res. 2020, 13, 945. [Google Scholar] [CrossRef] [PubMed]
- Sarparast, M.; Dattmore, D.; Alan, J.; Lee, K.S.S. Cytochrome P450 Metabolism of Polyunsaturated Fatty Acids and Neurodegeneration. Nutrients 2020, 12, 3523. [Google Scholar] [CrossRef] [PubMed]
- Scheinman, S.B.; Sugasini, D.; Zayed, M.; Yalagala, P.C.; Marottoli, F.M.; Subbaiah, P.V.; Tai, L.M. LPC-DHA/EPA-enriched diets increase brain DHA and modulate behavior in mice that express human APOE4. Front. Neurosci. 2021, 15, 767. [Google Scholar] [CrossRef]
- Alam, S.-I.; Kim, M.-W.; Shah, F.A.; Saeed, K.; Ullah, R.; Kim, M.-O. Alpha-Linolenic Acid Impedes Cadmium-Induced Oxidative Stress, Neuroinflammation, and Neurodegeneration in Mouse Brain. Cells 2021, 10, 2274. [Google Scholar] [CrossRef]
- Litwiniuk, A.; Domańska, A.; Chmielowska, M.; Martyńska, L.; Bik, W.; Kalisz, M. The effects of alpha-linolenic acid on the secretory activity of astrocytes and β amyloid-associated neurodegeneration in differentiated SH-SY5Y cells: Alpha-linolenic acid protects the SH-SY5Y cells against β amyloid toxicity. Oxid. Med. Cell. Longev. 2020, 2020, 8908901. [Google Scholar] [CrossRef]
- Calina, D.; Buga, A.M.; Mitroi, M.; Buha, A.; Caruntu, C.; Scheau, C.; Bouyahya, A.; El Omari, N.; El Menyiy, N.; Docea, A.O. The Treatment of Cognitive, Behavioural and Motor Impairments from Brain Injury and Neurodegenerative Diseases through Cannabinoid System Modulation—Evidence from In Vivo Studies. J. Clin. Med. 2020, 9, 2395. [Google Scholar] [CrossRef]
- Chayasirisobhon, S. The Role of Cannabidiol in Neurological Disorders. Perm. J. 2021, 25. [Google Scholar] [CrossRef]
- Abrahams, S.; Haylett, W.L.; Johnson, G.; Carr, J.A.; Bardien, S. Antioxidant effects of curcumin in models of neurodegeneration, aging, oxidative and nitrosative stress: A review. Neuroscience 2019, 406, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Salam, O.M.; Sleem, A.A.; Sayed, M.A.E.B.M.; Youness, E.R.; Shaffie, N. Capsaicin exerts anti-convulsant and neuroprotective effects in pentylenetetrazole-induced seizures. Neurochem. Res. 2020, 45, 1045–1061. [Google Scholar] [CrossRef]
- Shahbazi, S.; Zakerali, T.; Frycz, B.; Kaur, J. Impact of novel N-aryl substituted piperamide on NF-kappa B translocation as a potent anti-neuroinflammatory agent. Biomed. Pharmacother. 2020, 127, 110199. [Google Scholar] [CrossRef] [PubMed]
- Ngo, Q.T.; Tran, P.T.; Tran, M.H.; Kim, J.A.; Rho, S.S.; Lim, C.H.; Kim, J.C.; Woo, M.H.; Choi, J.S.; Lee, J.H. Alkaloids from Piper nigrum exhibit antiinflammatory activity via activating the Nrf2/HO1 pathway. Phytother. Res. 2017, 31, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Mousavi Nasl-khameneh, A.; Mirshafiey, A.; Naser Moghadasi, A.; Chahardoli, R.; Mahmoudi, M.; Parastouei, K.; Yekaninejad, M.S.; Saboor-Yaraghi, A.A. Combination treatment of docosahexaenoic acid (DHA) and all-trans-retinoic acid (ATRA) inhibit IL-17 and RORγt gene expression in PBMCs of patients with relapsing-remitting multiple sclerosis. Neurol. Res. 2018, 40, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Brucklacher-Waldert, V.; Stuerner, K.; Kolster, M.; Wolthausen, J.; Tolosa, E. Phenotypical and functional characterization of T helper 17 cells in multiple sclerosis. Brain 2009, 132, 3329–3341. [Google Scholar] [CrossRef] [PubMed]
- De Urquiza, A.M.; Liu, S.; Sjöberg, M.; Zetterström, R.H.; Griffiths, W.; Sjövall, J.; Perlmann, T. Docosahexaenoic acid, a ligand for the retinoid X receptor in mouse brain. Science 2000, 290, 2140–2144. [Google Scholar] [CrossRef]
- Dawson, M.I.; Xia, Z. The retinoid X receptors and their ligands. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2012, 1821, 21–56. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Zhen, J.; Lu, Z. Synergetic neuroprotective effect of docosahexaenoic acid and aspirin in SH-Y5Y by inhibiting miR-21 and activating RXRα and PPARα. DNA Cell Biol. 2017, 36, 482–489. [Google Scholar] [CrossRef]
- Pan, X.; Wang, Z.-X.; Wang, R. MicroRNA-21: A novel therapeutic target in human cancer. Cancer Biol. Ther. 2010, 10, 1224–1232. [Google Scholar] [CrossRef] [Green Version]
- Strickland, I.T.; Richards, L.; Holmes, F.E.; Wynick, D.; Uney, J.B.; Wong, L.-F. Axotomy-induced miR-21 promotes axon growth in adult dorsal root ganglion neurons. PLoS ONE 2011, 6, e23423. [Google Scholar] [CrossRef] [Green Version]
- Quinn, J.F.; Raman, R.; Thomas, R.G.; Yurko-Mauro, K.; Nelson, E.B.; Van Dyck, C.; Galvin, J.E.; Emond, J.; Jack, C.R.; Weiner, M.; et al. Docosahexaenoic Acid Supplementation and Cognitive Decline in Alzheimer Disease: A Randomized Trial. JAMA 2010, 304, 1903–1911. [Google Scholar] [CrossRef] [PubMed]
- Ringman, J.M.; Frautschy, S.A.; Teng, E.; Begum, A.N.; Bardens, J.; Beigi, M.; Gylys, K.H.; Badmaev, V.; Heath, D.D.; Apostolova, L.G.; et al. Oral curcumin for Alzheimer’s disease: Tolerability and efficacy in a 24-week randomized, double blind, placebo-controlled study. Alzheimer’s Res. Ther. 2012, 4, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Sendón Moreno, J.L.; García Caldentey, J.; Trigo Cubillo, P.; Ruiz Romero, C.; García Ribas, G.; Alonso Arias, M.A.A.; García de Yébenes, M.J.; Tolón, R.M.; Galve-Roperh, I.; Sagredo, O.; et al. A double-blind, randomized, cross-over, placebo-controlled, pilot trial with Sativex in Huntington’s disease. J. Neurol. 2016, 263, 1390–1400. [Google Scholar] [CrossRef]
- Filošević Vujnović, A.; Jović, K.; Pištan, E.; Andretić Waldowski, R. Influence of Dopamine on Fluorescent Advanced Glycation End Products Formation Using Drosophila melanogaster. Biomolecules 2021, 11, 453. [Google Scholar] [CrossRef]
- Prüßing, K.; Voigt, A.; Schulz, J.B. Drosophila melanogaster as a model organism for Alzheimer’s disease. Mol. Neurodegener. 2013, 8, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naz, F.; Siddique, Y.H. Drosophila melanogaster a versatile model of Parkinson’s Disease. CNS Neurol. Disord. Drug Targets 2021, 20, 487–530. [Google Scholar] [CrossRef] [PubMed]
- Joardar, A.; Menzl, J.; Podolsky, T.C.; Manzo, E.; Estes, P.S.; Ashford, S.; Zarnescu, D.C. PPAR gamma activation is neuroprotective in a Drosophila model of ALS based on TDP-43. Hum. Mol. Genet. 2015, 24, 1741–1754. [Google Scholar] [CrossRef] [Green Version]
- Westfall, S.; Lomis, N.; Prakash, S. A novel synbiotic delays Alzheimer’s disease onset via combinatorial gut-brain-axis signaling in Drosophila melanogaster. PLoS ONE 2019, 14, e0214985. [Google Scholar] [CrossRef]
- Haghighat, N.; Rajabi, S.; Mohammadshahi, M. Effect of synbiotic and probiotic supplementation on serum brain-derived neurotrophic factor level, depression and anxiety symptoms in hemodialysis patients: A randomized, double-blinded, clinical trial. Nutr. Neurosci. 2021, 24, 490–499. [Google Scholar] [CrossRef]
- Lee, Y.-M.; Sun, Y.H. Drosophila as a model to study the role of glia in neurodegeneration. J. Neurogenet. 2015, 29, 69–79. [Google Scholar] [CrossRef]

{kind=link}
| Compound Type | Neuro Model | Cell/Animal Type | Treatment | PPAR Type | Outcome | Ref. |
|---|---|---|---|---|---|---|
| Biochanin A | NI | BV2 | Pretreatment of Biochanin A (5, 10, 20 μM), 1 h+ LPS (0.5 μg/mL), 24 h | PPARγ | ↓iNOS, ↓PGE2, ↓TNF-α, ↓IL-1β, ↓NF-κB | [103] |
| Icariin | CI/R | SD rats | Pretreated with ICA (10, 30 mg/kg, twice a day), 3 days | PPARα PPARγ | ↓IL-1β, ↓TGF-β1, ↓NF-κB, ↑IκB-α, ↑PPARα, ↑PPARγ | [105] |
| Icariin | AD | APP/PS1 Mice | Control (ddH2O) + icariin group (60 mg/kg/day), 6 months | PPARγ | ↓IL-1β, ↓IL-6, ↓TNF-α, ↑ IL-4, ↑IL-10, ↑TGF-β1, ↓NF-κB, ↓Aβ42, ↓iNOS, ↓iNOS:Iba1, ↑PPARγ, ↓ memory impair | [115] |
| Icariin | Experimental ischemic stroke | SD rats | Sham (saline), MCAO + saline, MCAO + MH +saline, MCAO + MH + icariin (60 mg/kg/day), 28 days | PPARα PPARγ | ↓TNF-α, ↓IL-6, ↓Bax, ↓cleaved caspase-3, ↑Bcl-2, ↓NF-κB, ↑Nrf2, ↑PPARα, ↑PPARγ | [116] |
| Icariside II | CI/R | SD rats | Sham (saline), Vehicle (Saline), IC-II (L, H-10, 30 mg/kg), twice a day, 3 days | PPARα PPARγ | ↓IL-1β, ↑TGF-β1, ↓IκB-α degradation, ↓NF-κB, ↑PPARα, ↑PPARγ | [119] |
| Icariside II | VD | SD rats | Sham (saline), BCCAO, BCCAO + ICS II (4, 8, 16 mg/kg/day), 28 days | PPARα PPARγ | Improved learning and memory, ↓neuronal death, ↓Aβ oligomers, ↓APP, ↓BACE1, ↑ADAM10, ↑IDE | [120] |
| Icaritin | GBM | U87MG, T98G | Icaritin (10, 20 μM), 24 h, 48 h GW9662 (5 μM), 6 h + Icaritin (20 μM), 48 h | PPARγ | ↓cell growth, cell cycle arrest at G1/G0 phase, ↓cyclin D1, ↓CDK4, ↓CDK6, ↑apoptosis, ↑caspase-3, ↓Bcl-2, ↑Bax, ↑PPARγ, ↑p-AMPK | [121] |
| Luteoloside | CI/R | Male Rats | Luteoloside (20, 40, 80 mg/kg) + nimodipine (4 mg/kg) | PPARγ | ↑neural function, ↓cerebral edema, ↓IL-1β, ↓TNF-α, ↓iNOS, ↓COX-2, ↓ NF-κB, ↓ p- IκB-α, ↑nuclear Nrf2 | [117] |
| Naringenin | Dementia | SD rats | Control (5 μL saline), STZ (3 mg/kg), NAR-STZ (L-25, M-50, H-100 mg/kg/day), 21 days | PPARγ | Improved learning and memory, ↑INS, ↑INSR, ↓p-Tau, ↓GSK-3β, ↓Aβ levels, ↑IDE | [125] |
| HD | SD rats | Control (Saline, 4 μL) + QA (10 mL/kg), NAR + QA (20, 40, 80 mg/kg). Pio (40 mg/kg) + QA, Pio (40 mg/kg) + NAR (80 mg/kg) + QA, 28 days | PPARγ | ↓stress, ↓TNF-α, ↓IL-1β, ↓IL-6, ↓ NF-κB ↓Bax-Bcl-2, ↓caspase-3, ↑mitochondrial complex (I-IV) activity, ↑PPARγ | [126] | |
| Chrysin | EAN | Lewis rats | P0 peptide (180–199) 300 μL. Chrysin (50 mg/kg/day), 16 days | PPARγ | ↓iNOS, ↓COX-2, ↓NF-κB, ↓IL-1β, ↓IL-2, ↓IL-6, ↓IL-12, ↓IFNγ, ↓TNF-α, ↑IL-4 | [138] |
| Cy-3-G | AD | SH-SY5Y | Control, Aβ (10 μM) 24 h + Cy3G (25 μM), 24 h + GW9662 (20 μM), 3 h | PPARγ | ↓cytotoxicity, ↓ROS ↓Aβ (25–35) aggregation, ↑PPARγ | [140] |
| APP/PS1 Mice | Cy3G (5 mg/kg/day), GW9662 1 mg/kg/day + Cy3G 5 mg/kg, 2 months | Improved learning and memory | ||||
| Galangin | AD | BV2 | Pretreated with galangin (10, 30, 50 µM), 1 h followed by LPS treatment (100 ng/mL), 6 or 16h | PPARγ | ↓iNOS, ↓IL-1β,↓IL-6,↓TNF-α,↑IL-10,↓NO,↓COX-2 ↓MAPK & NF κB signaling, ↑Nrf2, ↑ CREB, ↑PPAR-γ, ↓NADPH oxidase subunits- p47and gp91, ↑ HO-1 | [145] |
| Galangin | NI | BV2 | Galangin pretreatment (0, 10, 30, 50 μM) 1 h + poly(I:C) (10 μg/mL), 6 and 16 h. | PPARγ | ↓NO, ↓iNOS, ↓IL-1β, ↓IL-6, ↓TNF-α, ↓ROS, ↓COX-2, ↑IL-10 | [148] |
| ICR mice | Pretreatment of galangin (50 mg/kg), 4 days + Poly(I:C) (12 mg/kg), 3 h | ↓IL-1β, ↓IL-6, ↓TNF-α, ↓iNOS, ↓MMP-8, ↓NF-κB, ↓p-Akt/Akt |
| Compound Type | Neuro Model | Cell/Animal Type | Treatment | PPAR Type | Outcome | Ref. |
|---|---|---|---|---|---|---|
| DHA | NI | Primary glial cultures | LPS (10 ng/mL) + DHA (10, 20 μM), 24 h. IFN-γ (200 U/mL) + DHA (10, 20 μM), 24 h | PPARγ | ↓NO, ↓iNOS. ↓TNF-α, ↓IL-6, ↑Arg1 activity, ↑IGF-1, ↓p-38 MAPK, ↑ PPARγ, ↑NPC survival ↑NPC differentiation | [165] |
| MS | OP | DHA (5, 10 μM), 24 h, PPARγ antagonist GW9662 (1 μM), 30 min | PPARγ | ↑OP maturation, ↓ maturational arrest, ↑p-ERK1/2 | [166] | |
| AE | BV2 | TG-DHA (1, 5, 10 and 20 μM) or EE-DHA (1, 5, 10, 20 μM), 30 min before LPS (100 ng/mL), 3h and IFN-γ (50 pg/mL), 24 h | PPARγ | ↑Cell viability, ↓NO2, ↓TNF-α, ↓IL-6 ↓splenocyte proliferation | [170] | |
| C57BL/6J mice | TG-DHA, (50, 250 mg/kg), or vehicle (0.3% DMSO in water), 56 days | ↓ disease severity, improved weight profile | ||||
| EPA | Tumor | C6 glioma cells | IL-1β (50 ng/mL), 24 h + EPA (12.5, 25, 50, 100 μM), 24 h | PPARγ | ↓IL-1β-induced IL-6 | [180] |
| EAE | C57BL/6 (B6) mice | Fish-oil-free diet with or without 5% (w/w) EPA ester, 2 weeks | PPARαPPARβPPARγ | ↓clinical EAE scores, ↓IFN-γ, ↓ IL-17, ↑PPARs | [181] | |
| Depression | SH-SY5Y cells | H2O2 (0.07, 0.15, 0.30, 0.62, 1.25, 2.5, 5, 10 μM), agonist (RMT; 10, 20 nM), fluoxetine (5, 10 μM), EPA (30 μM), 24 h | PPARγ | ↑cell viability, ↓NF-κB, ↓ROS, ↑cFos, ↑tyrosine hydroxylase ↑PPARγ | [182] | |
| PUFAs | NI | BV2 cells | PUFAs (SRP) 0.5 μg/mL (0.5 μM EPA+ 0.25 μM DHA) to 40 μg/mL (40 μM EPA + 20 μM DHA), 24 h + LPS (1 µg/mL) | PPARγ | ↑Cell Viability, ↓ROS, ↓NO, ↑PPARγ | [183] |
| SD rats | Control olive oil (2 g/day), SRP (2 g/day), 30 days | ↑PPARγ | ||||
| CLA | AD | SH-SY5Y cells | CLA (10, 20, 40, 60, 80 μmol/L), 48 h | PPARγ | ↓BACE1, ↑sAPPα, ↑PPARγ | [184] |
| AA | NI/OS | Hippocampal slices from SD rats | Control (artificial CSF immerged), 3 h, glutamate (1 mM), 30min, PUFA (1–10 μM), 30 min + glutamate, H2O2 (2 mM), 30 min, PUFA (1–10 μM), 30 min + H2O2, NaN3 (10 mM), 30 min, PUFAs (1–10 μM), 30 min + NaN3 | PPARγ | ↓ stress, improved Cu/Zn-SOD activity, ↑CAT | [185] |
| SA | NI/OS | Brain slices from SD rats | Control (ACSF), OGD (glucose-free ACSF), 2 h, SA (3, 10, 30 µmol/L), 30 min + OGD. Control (ACSF), glutamate (1 mmol/L), 30 min, SA (3, 10, 30 µmol/L), 30 min + glutamate (1 mmol/L), 30 min. Control, NaN3 (10 mmol/L), 30 min. SA (3, 10, 30 µmol/L), 30 min + NaN3 (10 mmol/L), 30min. BADGE (100 µmol/L), 3 h before | PPARγ | ↑ tissue activity | [186] |
| SA | NI/OS | Cortical neurons from SD rats | SA (3, 10, 30 μM/L), 24 h + glutamate (100 µmol/L), H2O2 (50 µmol/L), NaN3 (20 µmol/L), 24 h. BADGE (100 µmol/L), 1 h | PPARγ | ↑Cell viability, ↓lipid peroxidation, ↑SOD, ↑CAT, ↑GSH-Px, ↑PPARγ | [187] |
| OA | IC/R | SD rats C57BL/6 mice Wistar rats | MCAO model: OA (10, 30, 100 mg/kg), 90 min, edaravone (30 mg/kg), 90 min. Photothrombosis model: OA (20, 60, 200 mg/kg) 24 h after ischemia. 4-VO model: OA (10, 20, 100 mg/kg) 7 days after ischemia. Control, OA (30 mg/kg), GW9661 (4 mg/kg), OA (30 mg/kg) + GW9661 (4 mg/kg), 1 h before MCAO, 24 h. | PPARγ | ↓infarct volume, ↓functional deficit, ↓neuronal death, ↓COX-2, ↓iNOS, ↓TNF-α | [188] |
| LSO | NI | Wistar rats | Supplemented diet with lipids: GNO-10 wt%, PHVF-10 wt%, LSO-10 wt%, PHVF + LSO (2.5, 5.0, 7.5 wt%), 60 days | PPARγ | ↓Cholesterol level, ↑cell viability, ↑membrane fluidity, ↓PGE2, ↓6-keto PFA1α, ↓TXB2, ↓LTB4, ↓LTC4, ↓cPLA2, ↓COX-2, ↓5-LOX, ↑PPARγ, ↑NF-κB | [192] |
| Compound Type | Neuro model | Model | Treatment | PPAR type | Outcome | Ref. |
|---|---|---|---|---|---|---|
| Δ9-THC | PD | SH-SY5Y differentiated neuronal cells | MPP+ (5 mM), lactacystin (20 mM), and Paraquat (500 mM), 6, 12, 24, 48, Δ9-THC (0.1, 1, 5 ,10 μM), 24 h. PPARγ inhibitor (T0070907; 10 μM), 24 h | PPARγ | ↓cell death, ↓cleaved caspase-3, ↓ROS, ↑ PPARγ | [201] |
| Δ9-THC | PD | SH-SY5Y differentiated neuronal cells | MPP+ (7 mM) + Δ9-THC (10) µM /pio (5 µM), 48 h. PPARγ inhibitor (T0070907; 10 and 5 µM) | PPARγ | ↓cell death, ↑PPARγ, ↑PGC-1α, ↑TFAM, ↑mitochondrial DNA content | [202] |
| ∆9-THCA, CBDA, CBG | HD | HEK-293 T Neuro-2a STHdhQ7/Q7, STHdhQ111/Q111 cells C57BL/6 Mice | N2a cells (Δ9-THCA, 0, 0.5, and 1 μM), 48 h. HEK-293 T cells (∆9-THCA, CBDA, and CBGA, 0.1–15 μM), 6 h. STHdhQ7/Q7 cells (1–10 μM ∆9-THCA), 1 h/CB 20 mg/kg, 30 min before 3-NPA, every 24 h, 4 days | PPARγ | ↑ neuronal cell viability, ↑ mitochondrial mass, ↓TNF-α, ↓ iNOS, ↓COX-2, ↓IL-6, ↑PPAR-γ expression in HEK-293 T cellsImproved behavioral symptomatology ↓TNF-α, ↓iNOS, ↓COX-2, ↓IL-6 | [203] |
| CBG | HD | STHdhQ7/Q7 and STHdh Q111/Q111 cells * C57BL/6 and R6/2 mice | CBG (10 mg/kg), every 24 h, 6 weeks | PPARγ | CBG dose-dependently activated PPARγ ↓neuronal loss, improved motor activities, recovered catalase, SOD, GSH, ↓sgkl, ↓Cd44, ↓COX-2, ↓iNOS, ↓IL-6, ↓TNF-α | [206] |
| CBD | NI | Primary astrocytes of/and SD rats | Aβ (1 µg/mL), Aβ (1 µg/mL) + CBD (10−9–10−7 M), Aβ (1 µg/mL) + CBD (10−9–10−7 M) + GW9662 (9 nM), Aβ (1 µg/mL) + CBD (10−9–10−7 M) + PPARα (MK886; 3 µM), 24 h. Aβ (30 ng), Aβ (30 ng) + CBD (10 mg/kg), Aβ (30 ng) + CBD (10mg/kg) + MK886 (10 mg/kg, Aβ (30 ng) + CBD (10 mg/kg) + GW9662 (1 mg/kg), 15 days. | PPARα PPARγ | ↓NO, ↓iNOS, ↓TNF-α, ↓S100B, ↓IL-1β, ↑GFAP, ↓NF- κB ↓iNOS, ↓NF-κB, ↓S100B, ↑GFAP, ↓calinbindin, ↓gliosis, ↑neuronal survival, ↑neurogenesis | [207] |
| CBD | AD | SH-SY5Y | SH-SY5Yempty vector and SH-SY5YAPP+; CBD (10-9–10-6 M), CBD (10-9–10-6 M) + MK886 (3 μM), CBD (10-9–10-6 M) + GW9662 (9nM), 24 h | PPARα PPARγ | ↓APP, ↓Aβ, ↑APP ubiquitination, ↑ PPARγ, ↓apoptosis | [208] |
| CBD | PD | C57⁄BL6 mice | 6-Hydroxydopamine (2.5 mg/mL), L-DOPA (25 mg/kg), 2 injections/day, 21 days with benserazide (10 mg/kg), CBD (15, 30, 60 mg/kg), 3 days 15 min before L-DOPA. CPZ (5 mg/kg) + CBD (30 mg/kg)/L-DOPA and AM251 (1 mg/kg), GW9662 (4 mg/kg), 45 mins | PPARγ | ↓orofacial abnormal involuntary movements, ↓ p-ERK1/ 2, ↓p-AcH3, ↓COX-2, ↓NF-κB | [209] |
| CBD | Ischemia | HBMEC and human astrocyte co-cultures model | CBD (100 nM, 1 μM and 10 μM), 6, 8, 12, 16, 20, 24, 28, 32 h + OGD PPARγ inhibitor (100 nM), similar to CBD | PPARγ | ↓BBB permeability, ↓LDH, ↓VCAM-1 | [210] |
| CBD | Experimental MS | C57BL/6 mice | Naïve, EAE, EAE + CBD (10 mg/kg), daily, 14 days | PPARγ | ↑histological EAE score, ↑pPI3K/PI3K, ↑pAkt/Akt, ↑pmTOR/mTOR, ↑ pS6K/S6K, ↑BDNF, ↓IFN-γ, ↓IL-17, ↑PPARγ, ↓JNK, ↓ pp38/p38 | [211] |
| Compound Type | Neuro Model | Cell/Animal type | Treatment | PPAR Type | Outcome | Ref. |
|---|---|---|---|---|---|---|
| Curcumin/15d-PGJ2 | AE | C57BL/6 SJL/J mice | Immunized mice (MOGp35–55 antigen), curcumin or 15d-PGJ2 (100 μg in 25 μL DMSO) alternate day, 14 days | PPARγ | ↓TLR4, ↓TLR9 in CD4+ and CD8+ T cells | [213] |
| Curcumin | AE | C57BL/6 mice | Curcumin (100 µg) alternate day, 14 days. Immunized (MOGp35-55), 36 h. Spleen cells with curcumin (2.5, 5, 10, 25 µM) | PPARγ | ↓IFNγ, ↓IL-17, ↓IL-12, ↓IL-23, ↑IL-10, ↑PPARγ ↑ CD4CD25+Foxp3+ Treg cells | [218] |
| Curcumin | Experimental dementia | Swiss albino mice | Control (DW; 10 mL/kg, 30 min), artificial CSF control (25 mg/mL, 10 µL), STZ (3 mg/kg, 10 µL) alternate days, 14 days, STZ + curcumin (20 mg/kg), 14 days | PPARγ | Improved learning and memory, ↓AChE activity, ↓oxidative stress, ↑PPARγ | [219] |
| CI/R | SD rats | MCAO group, curcumin (200 mg/kg) + MCAO group, curcumin (200 mg/kg) + GW9662 (4 mg/kg) + MCAO group, MG132 + MCAO group, and MG132 alone group. Curcumin + PPARγ inhibitor GW96624 mg/kg, 3 days | PPARγ | ↑PPARγ, ↑PPARγ-PPRE binding activity, ↓ infarct volume, ↓neurological deficits, ↓neuronal damage ↓IL-1β, ↓TNF-α, ↓PGE2, ↓NO, ↓COX-2, ↓iNOS, ↓IκB degradation, ↓NF-κB | [220] | |
| AD | APOE3- and AOE4-targeted gene replacement mice | Two diet groups (control and 0.2 % curcumin-supplemented), 3 months | PPARγ | ↑ATP, ↑TFAM, ↑PPARγ, ↑PGC-1α, GABPa, ↑mitochondrial respiratory complex IV in APOE-3 | [221] | |
| AD | Neuronal/glial cells from APP/PS1 | Pretreatment curcumin (10 μM), 1 h + Aβ42 (25 μM), 24 h. GW9662 (1 μM) or PPARγ siRNA was transfected 1 h prior to Aβ42 treatment | PPARγ | Improved learning and memory ↑ChAT, ↑Ach ↓LDH, ↓TNF-α, ↓IL-1b, ↓COX-2, ↓NO, ↓GFAP, ↓Iba-1, ↓Mac-1, ↑PPAR-γ, ↑IkB-α expression, ↓ NF-κB p65 | [222] | |
| MS | OPs | OPs were treated with Curcumin (1, 5 μM), 24 h. Curcumin + GW9662 (1 μM pretreatment) 30 min. TNF-α (10 ng/mL), 24 h + curcumin (1 μM) | PPARγ | ↑OPs differentiation, ↓TNF-α induced maturation arrest, ↑p-ERK1/ERK2, ↑PGC1-α, ↑COX-1, ↑PPARγ | [223] | |
| Capsaicin | Tumor | LN-18 | Capsaicin (25, 50, 100, 200, 300, 400 µM), CPZ (2, 10, 20 µM), 24 and 48 h | PPARγ | ↑apoptosis, ↑caspase-9, -8 and -3, ↑PPARγ | [224] |
| AD | SH-SY5Y-APP695 cells | Capsaicin (0.1, 1, 5, 10, 50 μM), 24 h | PPARα | ↓Aβ40, ↓Aβ42, ↑CTF-α/CTF-β | [225] | |
| APP/PS1 mice | Capsaicin (30 mg/kg), 6 months | ↓cognitive impairment, ↓Aβ40, ↑CTF-α/CTF-β, ↑ADAM10, ↓TNF-α, ↓IFN-γ, ↓IL-6, ↑PSD98, ↑SYN1, ↑Map-2, ↑SNAP25, ↑VAMP1, ↑NeuN, ↑PPARα | ||||
| Piperine (D4) | AE | SVG, CHME3 | Pretreatment D4 (0.86 μM) and aspirin (50.51μM), 2 h + LPS (100 ng/ml), 24 h | PPARγ | ↑cell viability, ↓TNF-α, ↓IL-1β, ↓iNOS, ↓NF-κB, ↑ PPARγ | [226] |
| Estradiol | AD | Primary astrocytes | Pretreatment 17-βestradiol (0.2 nM) or genistein (0.5 µM), 48 h + 5 μM amyloid beta (Aβ), 24 h | PPARγ | ↓TNF-α, ↓IL-1β, ↓COX-2, ↓NO, ↑PPARγ | [227] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanjay; Sharma, A.; Lee, H.-J. Role of Phytoconstituents as PPAR Agonists: Implications for Neurodegenerative Disorders. Biomedicines 2021, 9, 1914. https://doi.org/10.3390/biomedicines9121914
Sanjay, Sharma A, Lee H-J. Role of Phytoconstituents as PPAR Agonists: Implications for Neurodegenerative Disorders. Biomedicines. 2021; 9(12):1914. https://doi.org/10.3390/biomedicines9121914
Chicago/Turabian StyleSanjay, Anshul Sharma, and Hae-Jeung Lee. 2021. "Role of Phytoconstituents as PPAR Agonists: Implications for Neurodegenerative Disorders" Biomedicines 9, no. 12: 1914. https://doi.org/10.3390/biomedicines9121914
APA StyleSanjay, Sharma, A., & Lee, H.-J. (2021). Role of Phytoconstituents as PPAR Agonists: Implications for Neurodegenerative Disorders. Biomedicines, 9(12), 1914. https://doi.org/10.3390/biomedicines9121914