
Pharmacological Inhibition of NOX4 Improves Mitochondrial Function and Survival in Human Beta-Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. EndoC-βH1 Cell and Human Islet Culture
2.3. Oxygen Consumption and Extracellular Acidification Rates
2.4. Mitochondrial Membrane Potential
2.5. Mitochondrial ROS
2.6. ATP and ADP
2.7. EndoC-βH1 Insulin Release
2.8. Single Islet Insulin Release and Survival Assessment after Gluco-Lipotoxic Stress
2.9. Statistical Analysis
3. Results
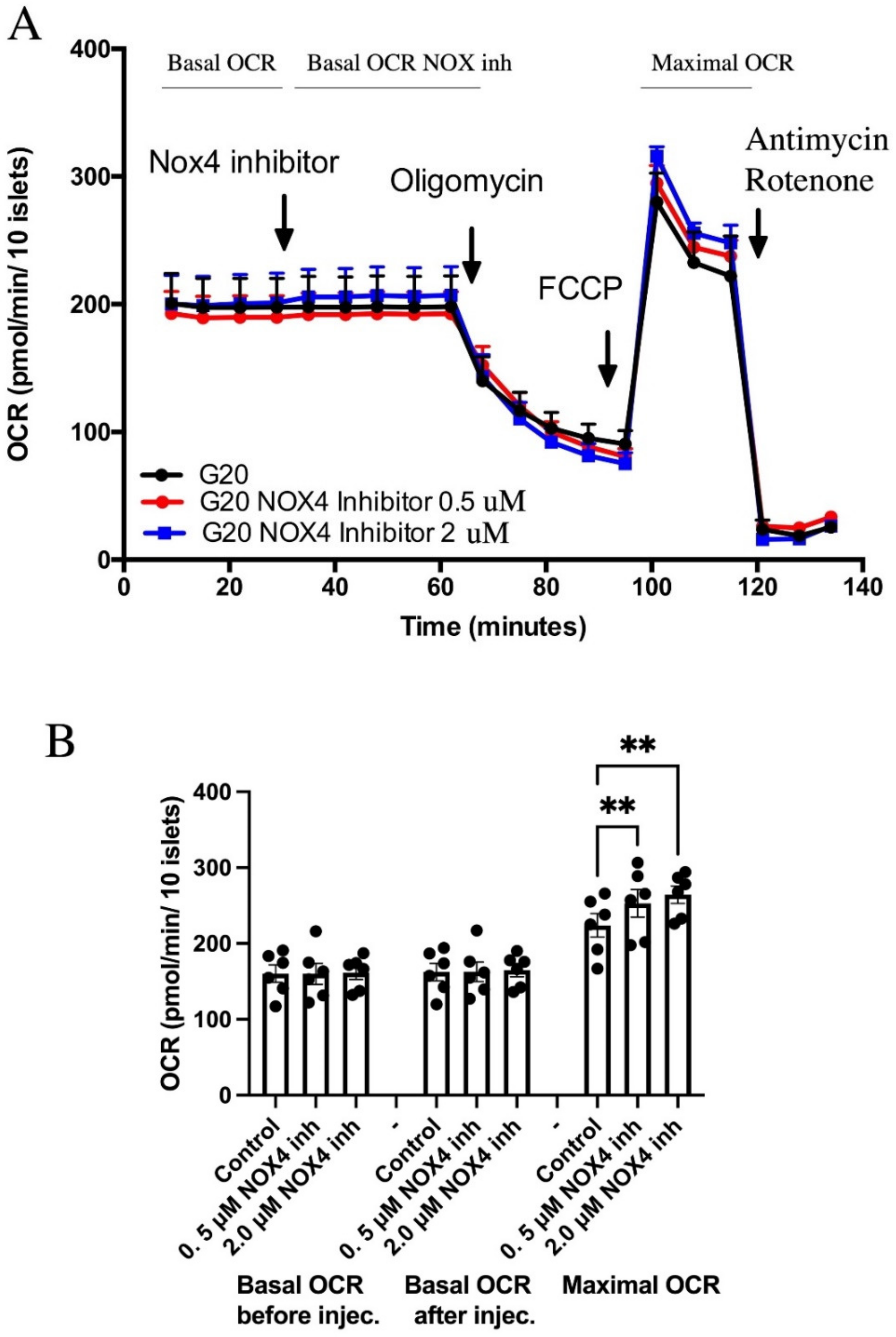
3.1. GLX7013114-Mediated NOX4 Inhibition Increased Maximal Oxygen Consumption Rates in Human Islets
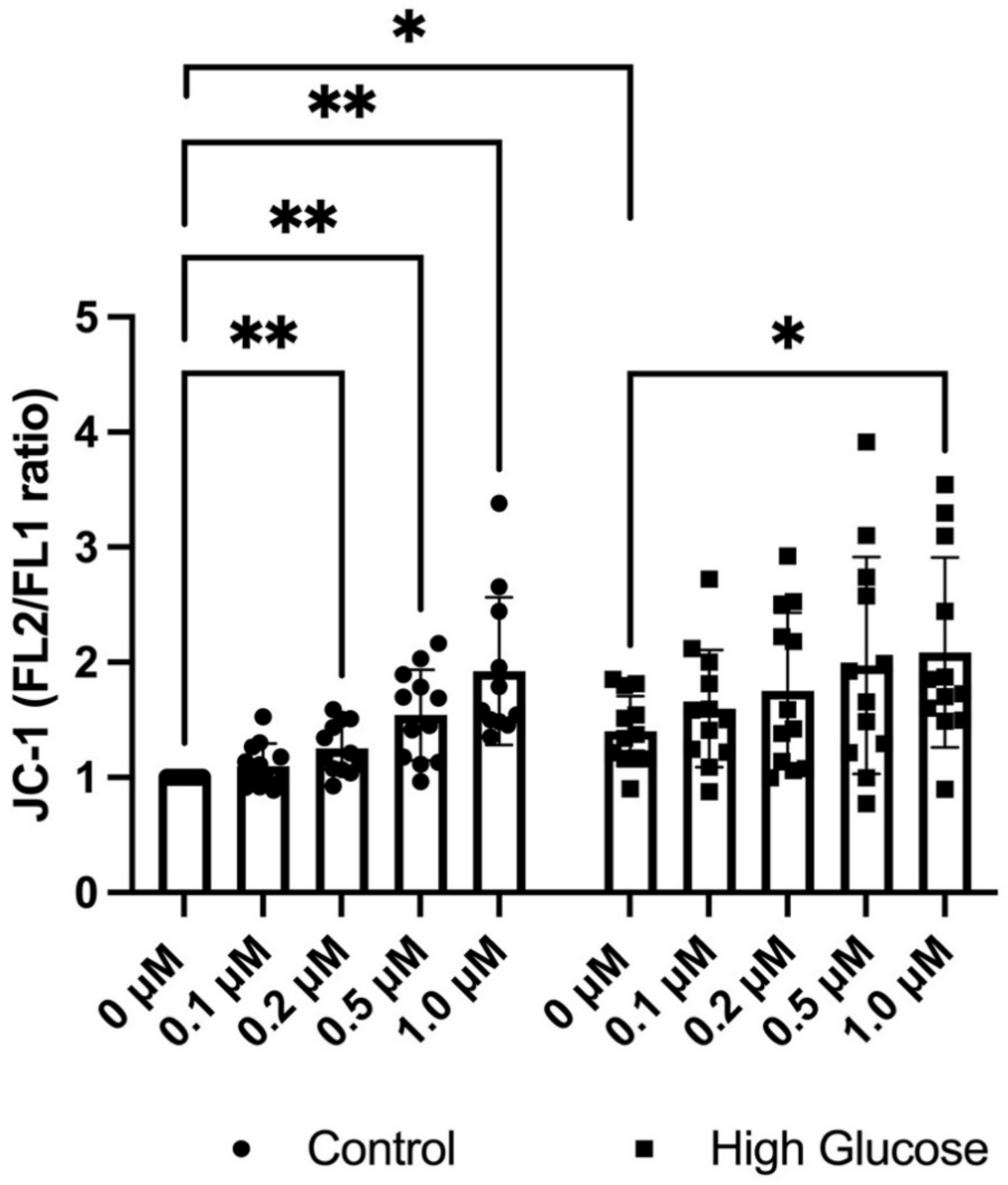
3.2. GLX7013114 Increased the Mitochondrial Membrane Potential in EndoC-βH1 Cells
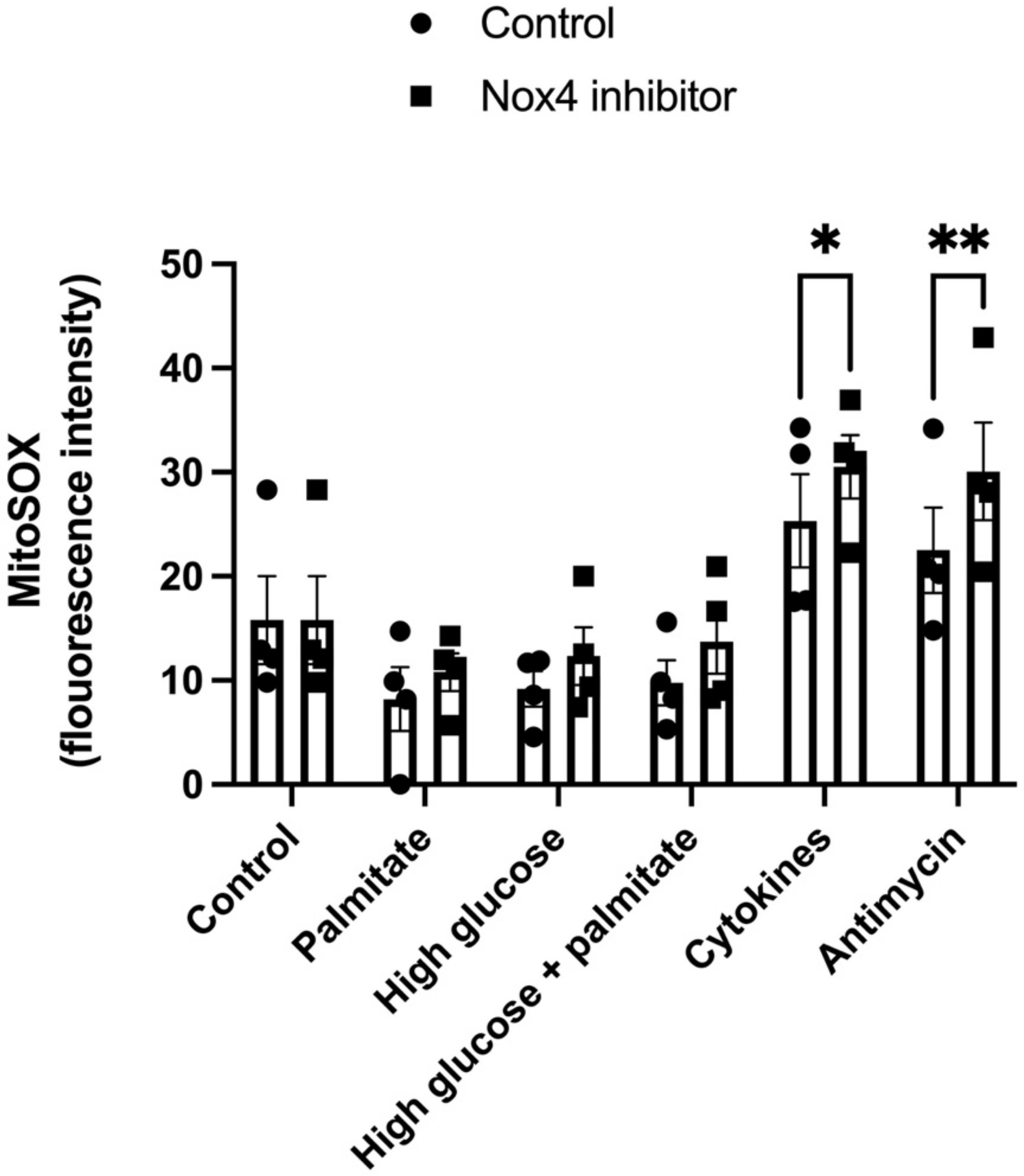
3.3. GLX7013114 Increased Mitochondrial ROS Production in EndoC-βH1 Cells
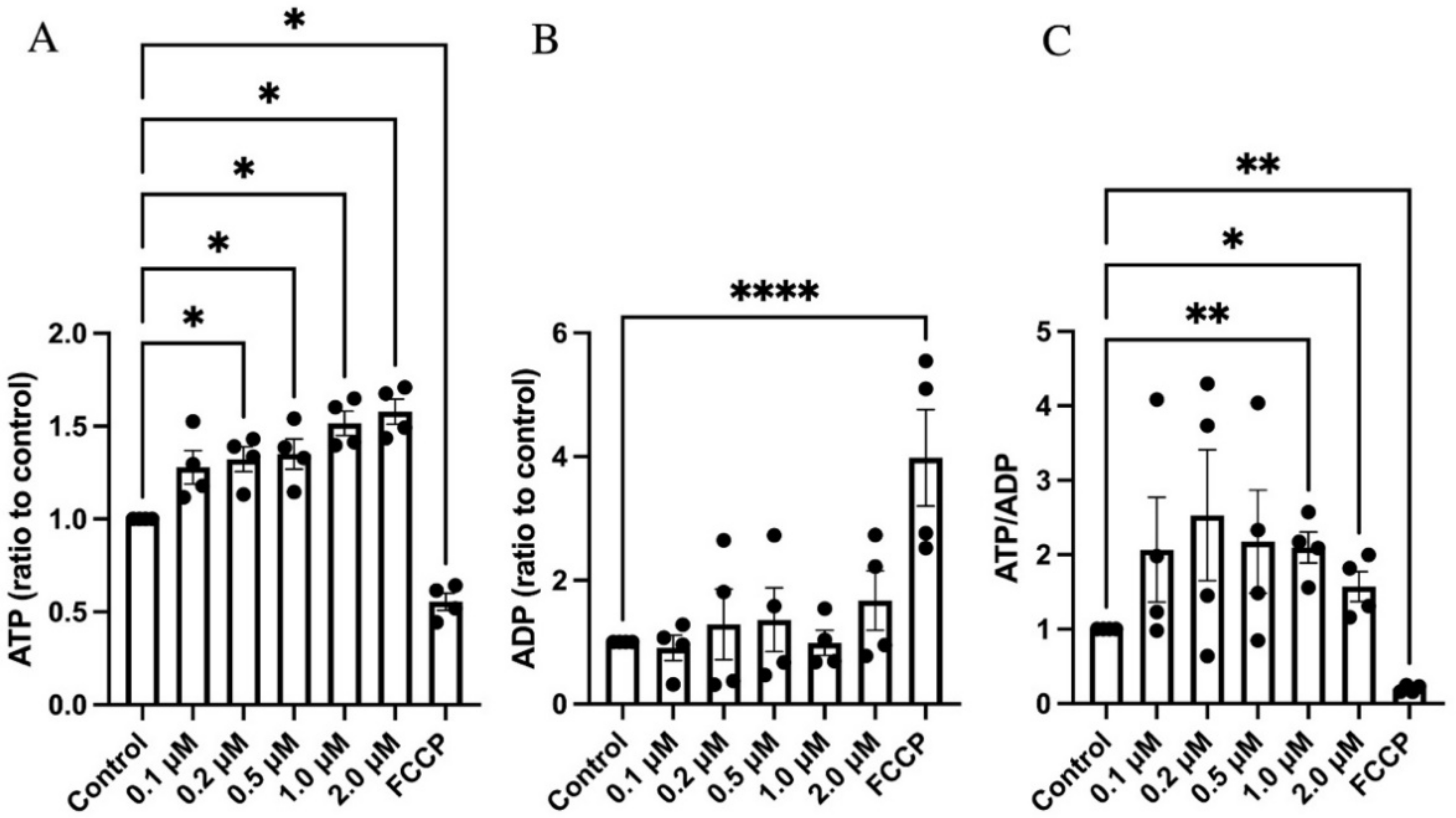
3.4. GLX7013114 Increased EndoC-βH1 Cell ATP and ATP/ADP
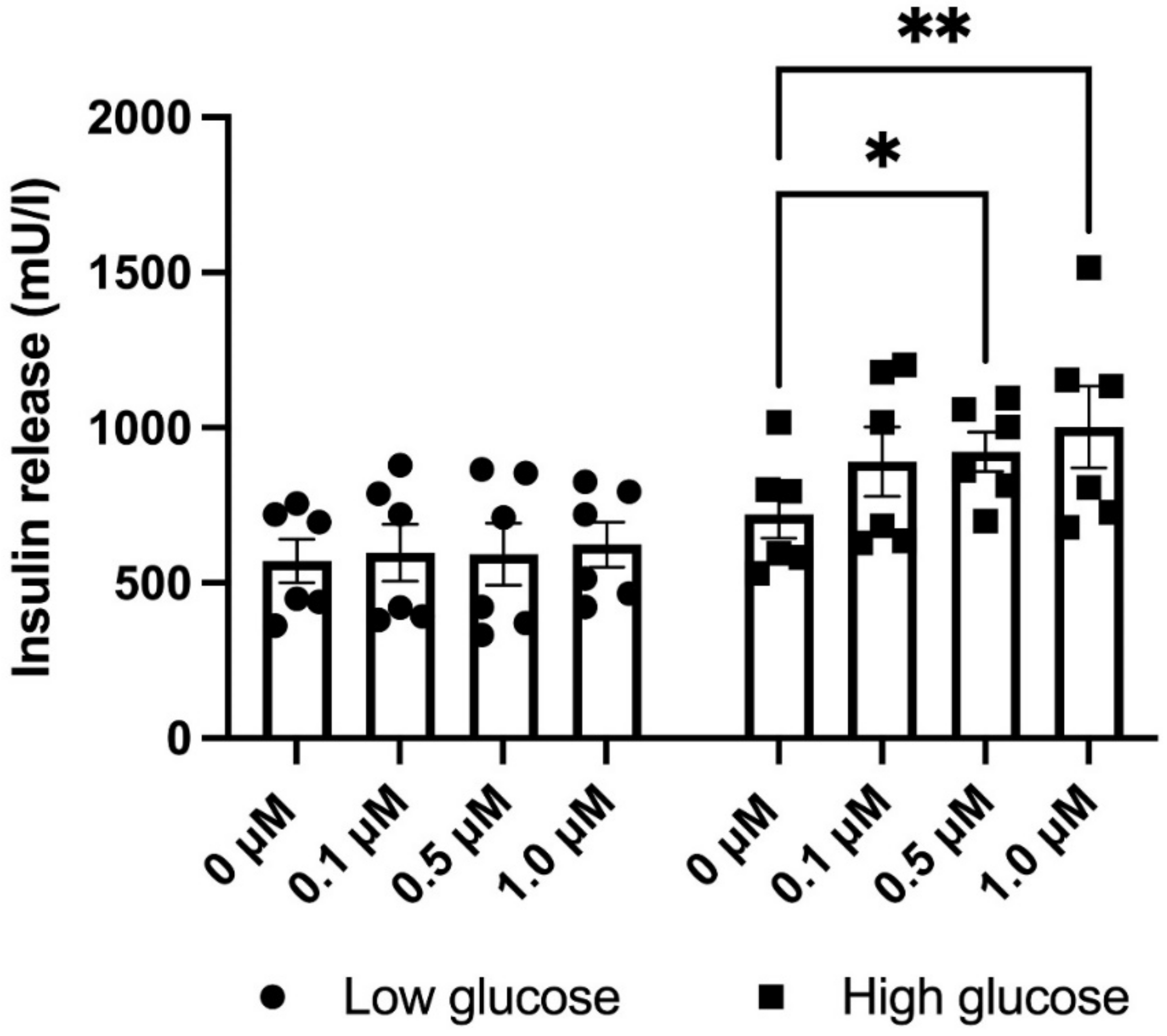
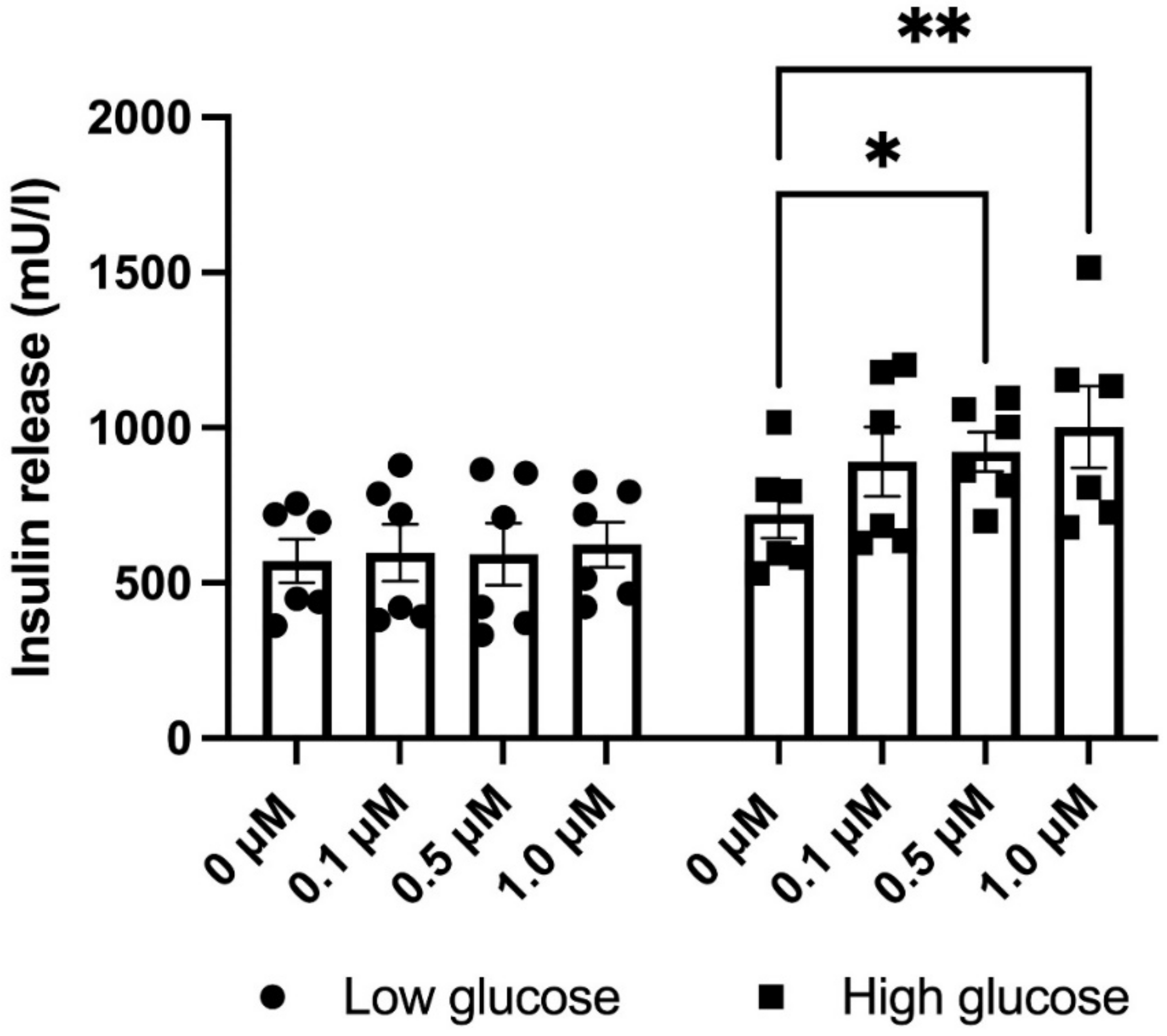
3.5. GLX7013114 Increased EndoC-βH1 Cell Glucose-Stimulated Insulin Release
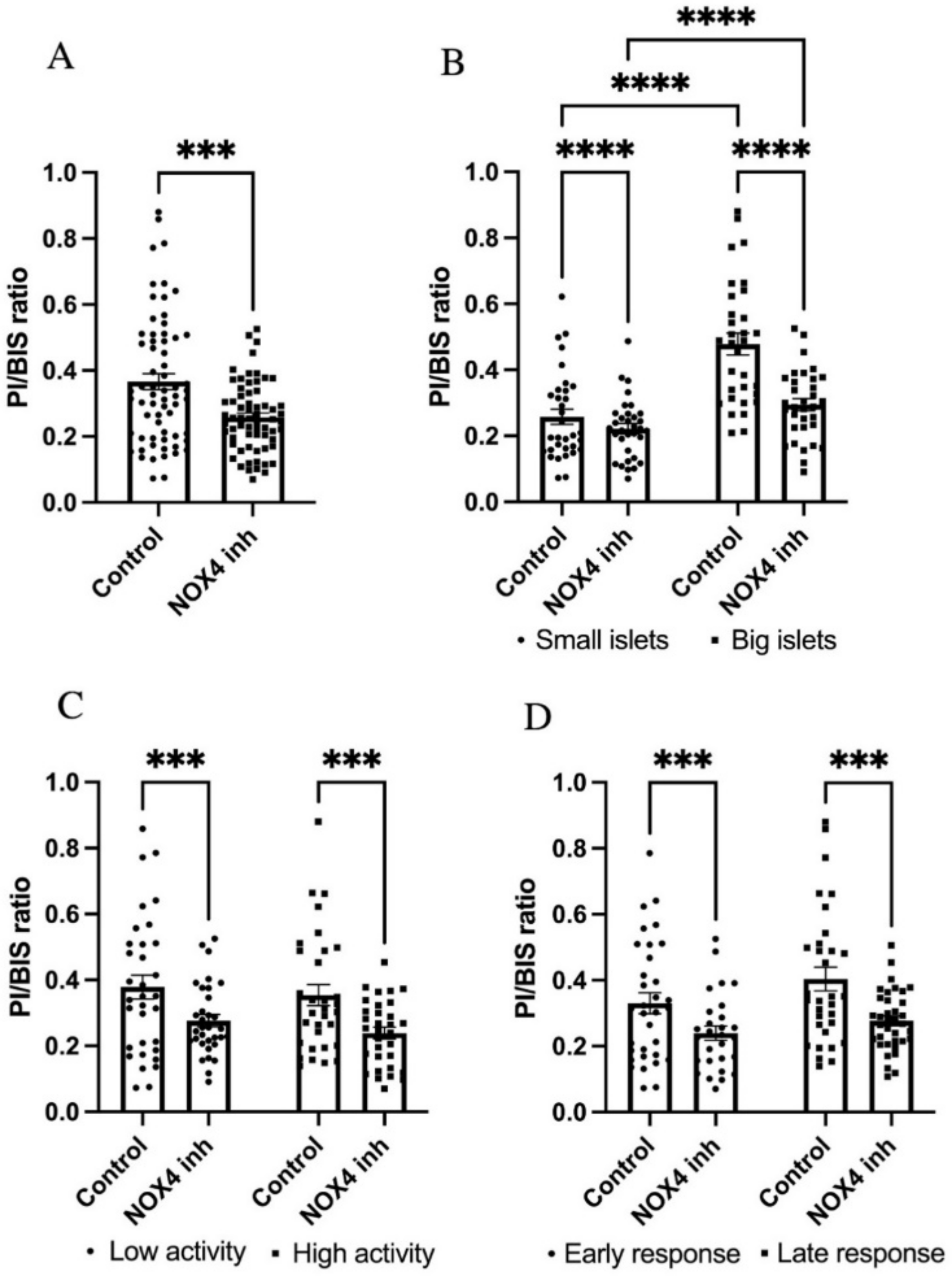
3.6. GLX7013114 Protected Human Islets against High Glucose + Palmitate-Induced Cell Death Independently of Size, Activity, and Glucose Responsiveness
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guichard, C.; Moreau, R.; Pessayre, D.; Epperson, T.K.; Krause, K.H. NOX family NADPH oxidases in liver and in pancreatic islets: A role in the metabolic syndrome and diabetes? Biochem. Soc. Trans. 2008, 36, 920–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nisimoto, Y.; Diebold, B.A.; Cosentino-Gomes, D.; Lambeth, J.D. Nox4: A hydrogen peroxide-generating oxygen sensor. Biochemistry 2014, 53, 5111–5120. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Murugesan, P.; Huang, K.; Cai, H. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: Novel therapeutic targets. Nat. Rev. Cardiol. 2020, 17, 170–194. [Google Scholar] [CrossRef]
- Hsu, J.; Wang, C.H.; Huang, S.C.; Chen, Y.W.; Yu, S.; Hwang, J.J.; Lin, J.W.; Ma, M.C.; Chen, Y.S. Novel application of amino-acid buffered solution for neuroprotection against ischemia/reperfusion injury. PLoS ONE 2019, 14, e0221039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyle, A.N.; Deshpande, N.N.; Taniyama, Y.; Seidel-Rogol, B.; Pounkova, L.; Du, P.; Papaharalambus, C.; Lassègue, B.; Griendling, K.K. Poldip2, a novel regulator of Nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circ. Res. 2009, 105, 249–259. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Craige, S.E.; Keaney, J.F., Jr. Downstream targets and intracellular compartmentalization in Nox signaling. Antioxid. Redox Signal. 2009, 11, 2467–2480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hecker, L.; Vittal, R.; Jones, T.; Jagirdar, R.; Luckhardt, T.R.; Horowitz, J.C.; Pennathur, S.; Martinez, F.J.; Thannickal, V.J. NADPH Oxidase-4 Mediates Myofibroblast Activation and Fibrogenic Responses to Lung Injury. Nat. Med. 2009, 15, 1077–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, T.; Ramachandrarao, S.P.; Siva, S.; Valancius, C.; Zhu, Y.; Mahadev, K.; Toh, I.; Goldstein, B.J.; Woolkalis, M.; Sharma, K. Reactive oxygen species production via NADPH oxidase mediates TGF-beta-induced cytoskeletal alterations in endothelial cells. Am. J. Physiol.-Renal Phys. 2005, 289, F816–F825. [Google Scholar] [CrossRef] [Green Version]
- Mistry, R.K.; Murray, T.; Prysyazhna, O.; Martin, D.; Burgoyne, J.R.; Santos, C.; Eaton, P.; Shah, A.M.; Brewer, A.C. Transcriptional Regulation of Cystathionine-γ-Lyase in Endothelial Cells by NADPH Oxidase 4-Dependent Signaling. J. Biol. Chem. 2016, 291, 1774–1788. [Google Scholar] [CrossRef] [Green Version]
- Boudreau, H.E.; Casterline, B.W.; Rada, B.; Korzeniowska, A.; Leto, T.L. Nox4 involvement in TGF-beta and SMAD3-driven induction of the epithelial-to-mesenchymal transition and migration of breast epithelial cells. Free Radic. Biol. Med. 2012, 53, 1489–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beretta, M.; Santos, C.X.; Molenaar, C.; Hafstad, A.D.; Miller, C.C.; Revazian, A.; Betteridge, K.; Schröder, K.; Streckfuß-Bömeke, K.; Doroshow, J.H.; et al. Nox4 regulates InsP3 receptor-dependent Ca2+ release into mitochondria to promote cell survival. EMBO J. 2020, 39, e103530. [Google Scholar] [CrossRef] [PubMed]
- Block, K.; Gorin, Y.; Abboud, H.E. Subcellular localization of Nox4 and regulation in diabetes. Proc. Natl. Acad. Sci. USA 2009, 106, 14385–14390. [Google Scholar] [CrossRef] [Green Version]
- Shanmugasundaram, K.; Nayak, B.K.; Friedrichs, W.E.; Kaushik, D.; Rodriguez, R.; Block, K. NOX4 functions as a mitochondrial energetic sensor coupling cancer metabolic reprogramming to drug resistance. Nat. Commun. 2017, 8, 997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veith, C.; Boots, A.W.; Idris, M.; van Schooten, F.J.; van der Vliet, A. Redox Imbalance in Idiopathic Pulmonary Fibrosis: A Role for Oxidant Cross-Talk Between NADPH Oxidase Enzymes and Mitochondria. Antioxid. Redox Signal. 2019, 31, 1092–1115. [Google Scholar] [CrossRef] [PubMed]
- Kozieł, R.; Pircher, H.; Kratochwil, M.; Lener, B.; Hermann, M.; Dencher, N.A.; Jansen-Dürr, P. Mitochondrial respiratory chain complex I is inactivated by NADPH oxidase Nox4. Biochem. J. 2013, 452, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Bernard, K.; Logsdon, N.J.; Miguel, V.; Benavides, G.A.; Zhang, J.; Carter, A.B.; Darley-Usmar, V.M.; Thannickal, V.J. NADPH Oxidase 4 (Nox4) Suppresses Mitochondrial Biogenesis and Bioenergetics in Lung Fibroblasts via a Nuclear Factor Erythroid-derived 2-like 2 (Nrf2)-dependent Pathway. J. Biol. Chem. 2017, 292, 3029–3038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fex, M.; Nicholas, L.M.; Vishnu, N.; Medina, A.; Sharoyko, V.V.; Nicholls, D.G.; Spégel, P.; Mulder, H. The pathogenetic role of β-cell mitochondria in type 2 diabetes. J. Endocrinol. 2018, 236, R145–R159. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Elksnis, A.; Wikstrom, P.; Walum, E.; Welsh, N.; Carlsson, P.O. The novel NADPH oxidase 4 selective inhibitor GLX7013114 counteracts human islet cell death in vitro. PLoS ONE 2018, 13, e0204271. [Google Scholar] [CrossRef]
- Anvari, E.; Wikstrom, P.; Walum, E.; Welsh, N. The novel NADPH oxidase 4 inhibitor GLX351322 counteracts glucose intolerance in high-fat diet-treated C57BL/6 mice. Free Radic. Res. 2015, 49, 1308–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsuta, H.; Aguayo-Mazzucato, C.; Katsuta, R.; Akashi, T.; Hollister-Lock, J.; Sharma, A.J.; Bonner-Weir, S.; Weir, G.C. Subpopulations of GFP-marked mouse pancreatic β-cells differ in size, granularity, and insulin secretion. Endocrinology 2012, 153, 5180–5187. [Google Scholar] [CrossRef] [Green Version]
- Bader, E.; Migliorini, A.; Gegg, M.; Moruzzi, N.; Gerdes, J.; Roscioni, S.S.; Bakhti, M.; Brandl, E.; Irmler, M.; Beckers, J.; et al. Identification of proliferative and mature β-cells in the islets of Langerhans. Nature 2016, 535, 430–434. [Google Scholar] [CrossRef] [PubMed]
- Pipeleers, D.; Kiekens, R.; Ling, Z.; Wilikens, A.; Schuit, F. Physiologic relevance of heterogeneity in the pancreatic beta-cell population. Diabetologia 1994, 37 (Suppl. 2), S57–S64. [Google Scholar] [CrossRef] [PubMed]
- Ravassard, P.; Hazhouz, Y.; Pechberty, S.; Bricout-Neveu, E.; Armanet, M.; Czernichow, P.; Scharfmann, R. A genetically engineered human pancreatic beta cell line exhibiting glucose-inducible insulin secretion. J. Clin. Investig. 2011, 121, 3589–3597. [Google Scholar] [CrossRef]
- Krizhanovskii, C.; Kristinsson, H.; Elksnis, A.; Wang, X.; Gavali, H.; Bergsten, P.; Scharfmann, R.; Welsh, N. EndoC-βH1 cells display increased sensitivity to sodium palmitate when cultured in DMEM/F12 medium. Islets 2017, 9, e1296995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Misawa, R.; Zielinski, M.C.; Cowen, P.; Jo, J.; Periwal, V.; Ricordi, C.; Khan, A.; Szust, J.; Shen, J.; et al. Regional differences in islet distribution in the human pancreas--preferential beta-cell loss in the head region in patients with type 2 diabetes. PLoS ONE 2013, 8, e67454. [Google Scholar] [CrossRef] [PubMed]
- Ullsten, S.; Bohman, S.; Oskarsson, M.E.; Nilsson, K.P.R.; Westermark, G.T.; Carlsson, P.O. Islet amyloid deposits preferentially in the highly functional and most blood-perfused islets. Endocr. Connect. 2017, 6, 458–468. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Younis, S.; Cen, J.; Wang, Y.; Krizhanovskii, C.; Andersson, L.; Welsh, N. ZBED6 counteracts high-fat diet-induced glucose intolerance by maintaining beta cell area and reducing excess mitochondrial activation. Diabetologia 2021, 64, 2292–2305. [Google Scholar] [CrossRef] [PubMed]
- Chareyron, I.; Christen, S.; Moco, S.; Valsesia, A.; Lassueur, S.; Dayon, L.; Wollheim, C.B.; Domingo, J.S.; Wiederkehr, A. Augmented mitochondrial energy metabolism is an early response to chronic glucose stress in human pancreatic beta cells. Diabetologia 2020, 63, 2628–2640. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elksnis, A.; Cen, J.; Wikström, P.; Carlsson, P.-O.; Welsh, N. Pharmacological Inhibition of NOX4 Improves Mitochondrial Function and Survival in Human Beta-Cells. Biomedicines 2021, 9, 1865. https://doi.org/10.3390/biomedicines9121865
Elksnis A, Cen J, Wikström P, Carlsson P-O, Welsh N. Pharmacological Inhibition of NOX4 Improves Mitochondrial Function and Survival in Human Beta-Cells. Biomedicines. 2021; 9(12):1865. https://doi.org/10.3390/biomedicines9121865
Chicago/Turabian StyleElksnis, Andris, Jing Cen, Per Wikström, Per-Ola Carlsson, and Nils Welsh. 2021. "Pharmacological Inhibition of NOX4 Improves Mitochondrial Function and Survival in Human Beta-Cells" Biomedicines 9, no. 12: 1865. https://doi.org/10.3390/biomedicines9121865
APA StyleElksnis, A., Cen, J., Wikström, P., Carlsson, P.-O., & Welsh, N. (2021). Pharmacological Inhibition of NOX4 Improves Mitochondrial Function and Survival in Human Beta-Cells. Biomedicines, 9(12), 1865. https://doi.org/10.3390/biomedicines9121865