
Essential Role of Rho-Associated Kinase in ABO Immune Complex-Mediated Endothelial Barrier Disruption

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Research Subject Participation
2.2. Blood Sample Collection
2.3. Sample Processing and Protein Concentration
2.4. Measurement of Anti-A and Anti-B Titers
2.5. Immune Complex Formation
2.6. Endothelial Cell Culture
2.7. Measurement of Endothelial Cell Permeability by Transendothelial Electrical Resistance
2.8. Immunofluorescence and Confocal Microscopy
2.9. Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling (TUNEL) Assay
2.10. Scanning Electron Microscopy
2.11. Statistical Analysis
3. Results
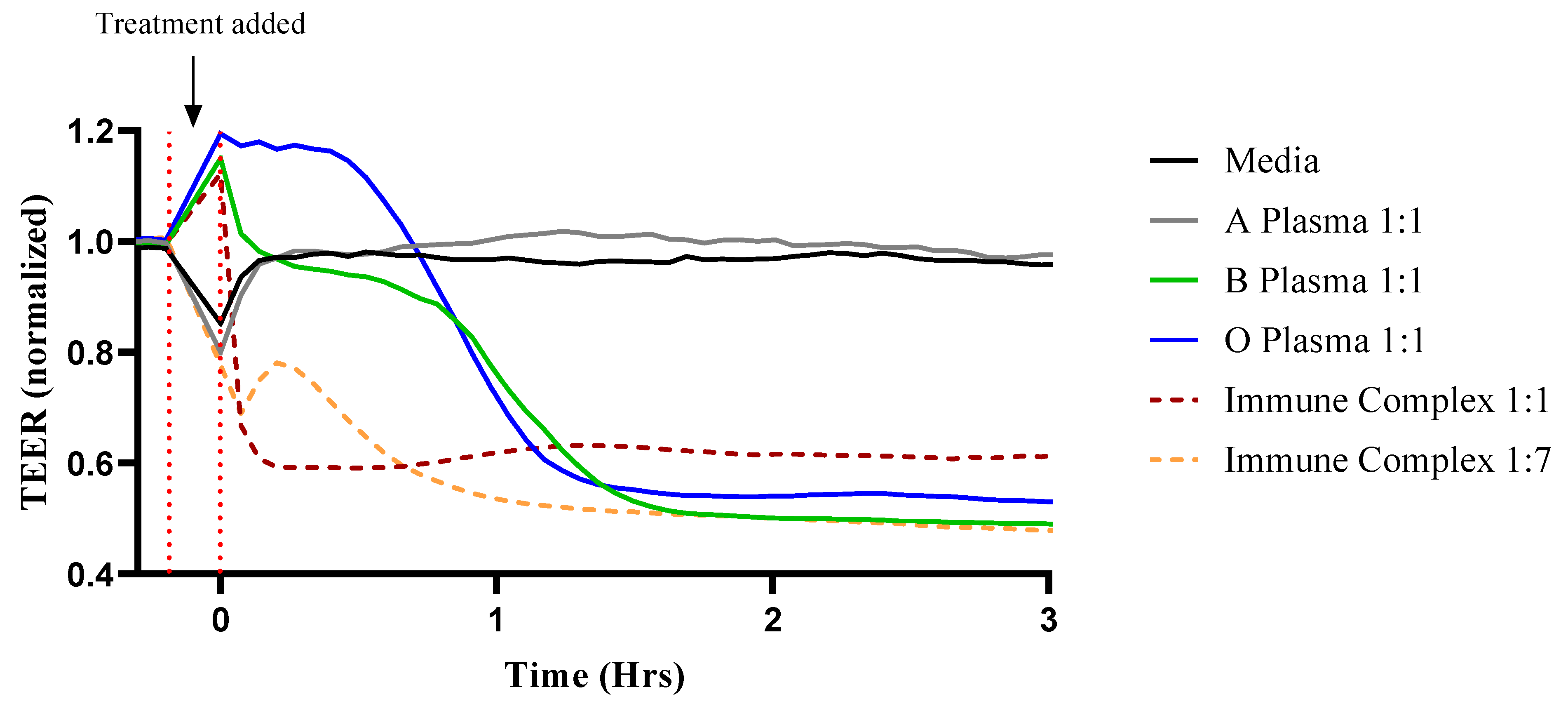
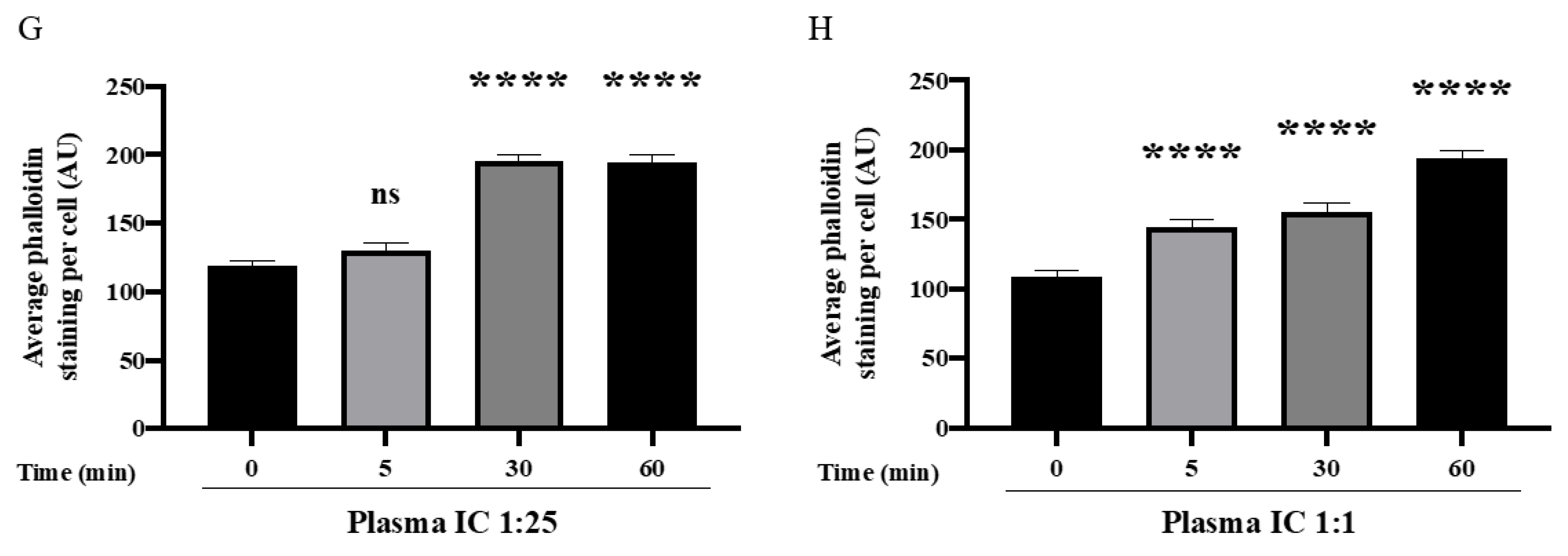
3.1. ABO Mismatched Plasma and IC Induce EC Barrier Disruption
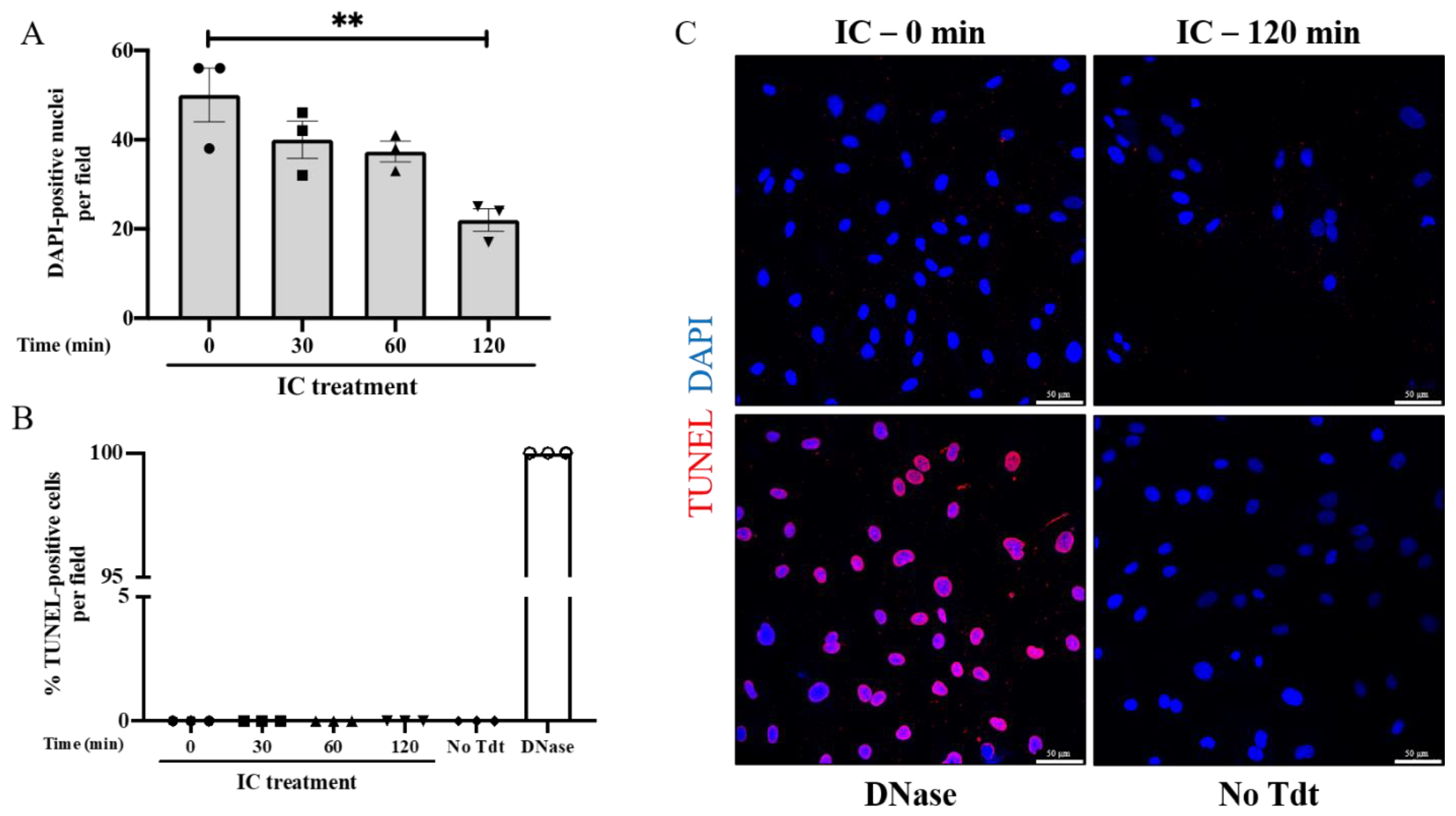
3.2. IC Induces EC Barrier Disruption without Causing Apoptosis
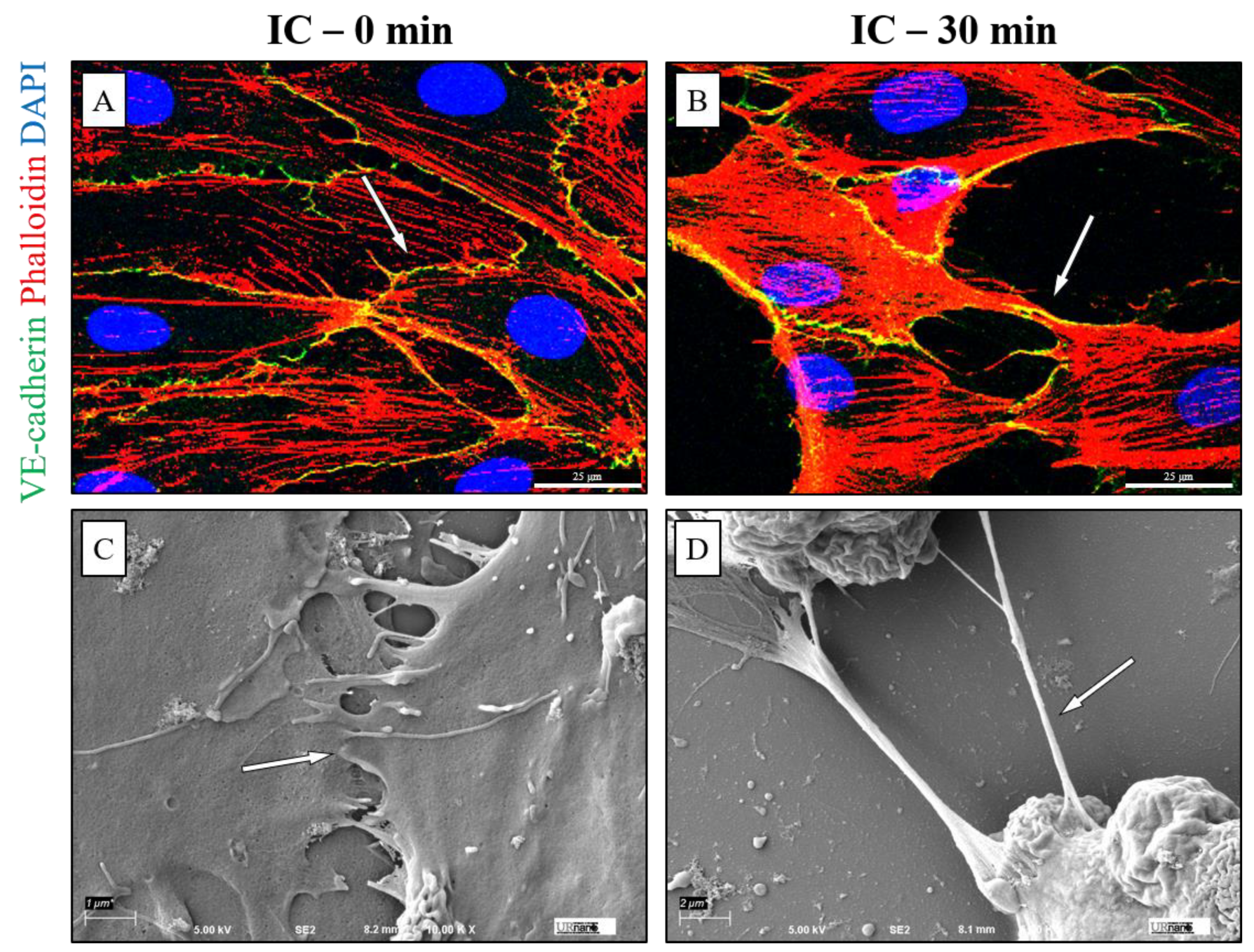
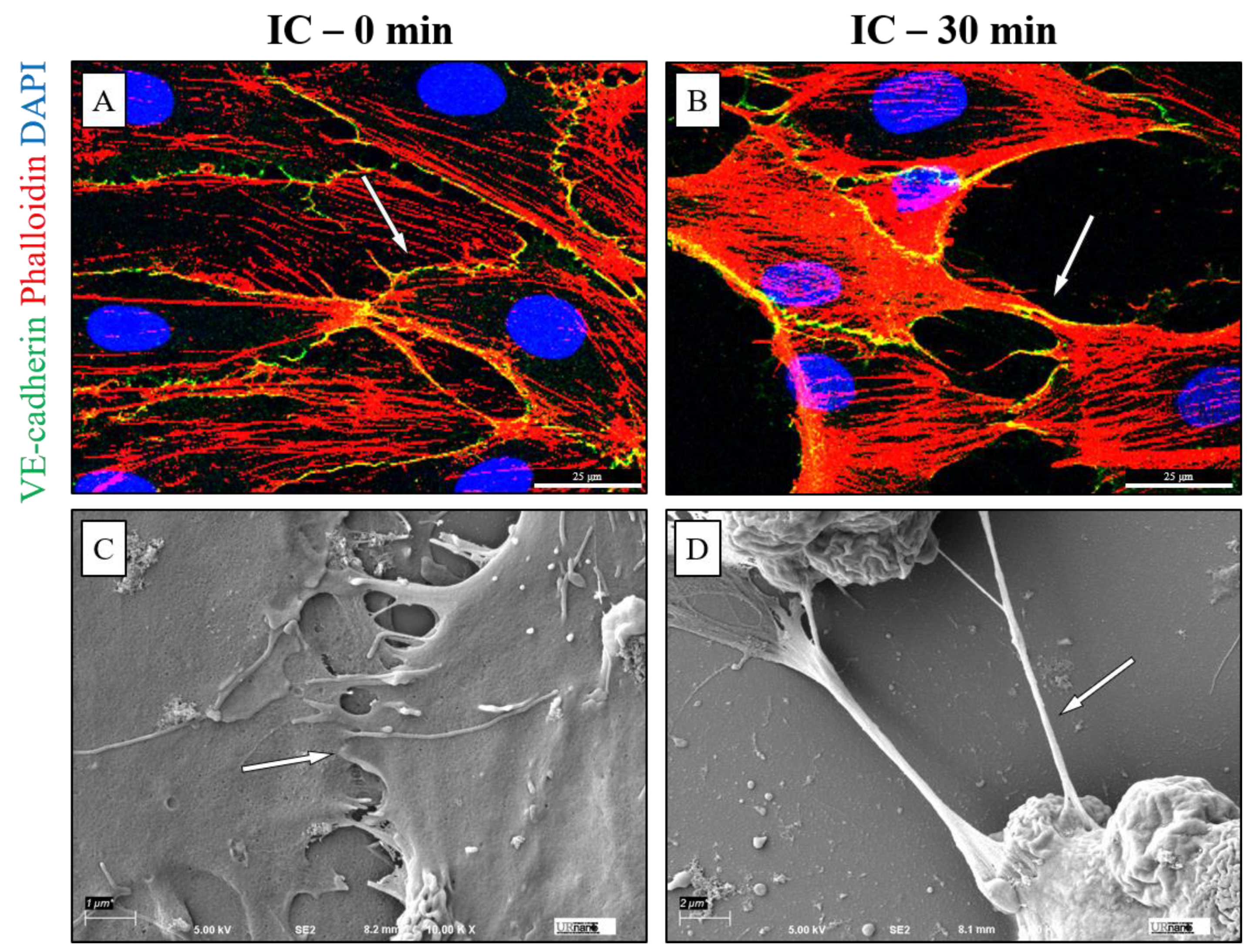
3.3. IC Treatment Induces Cell Contraction to Cause EC Barrier Disruption
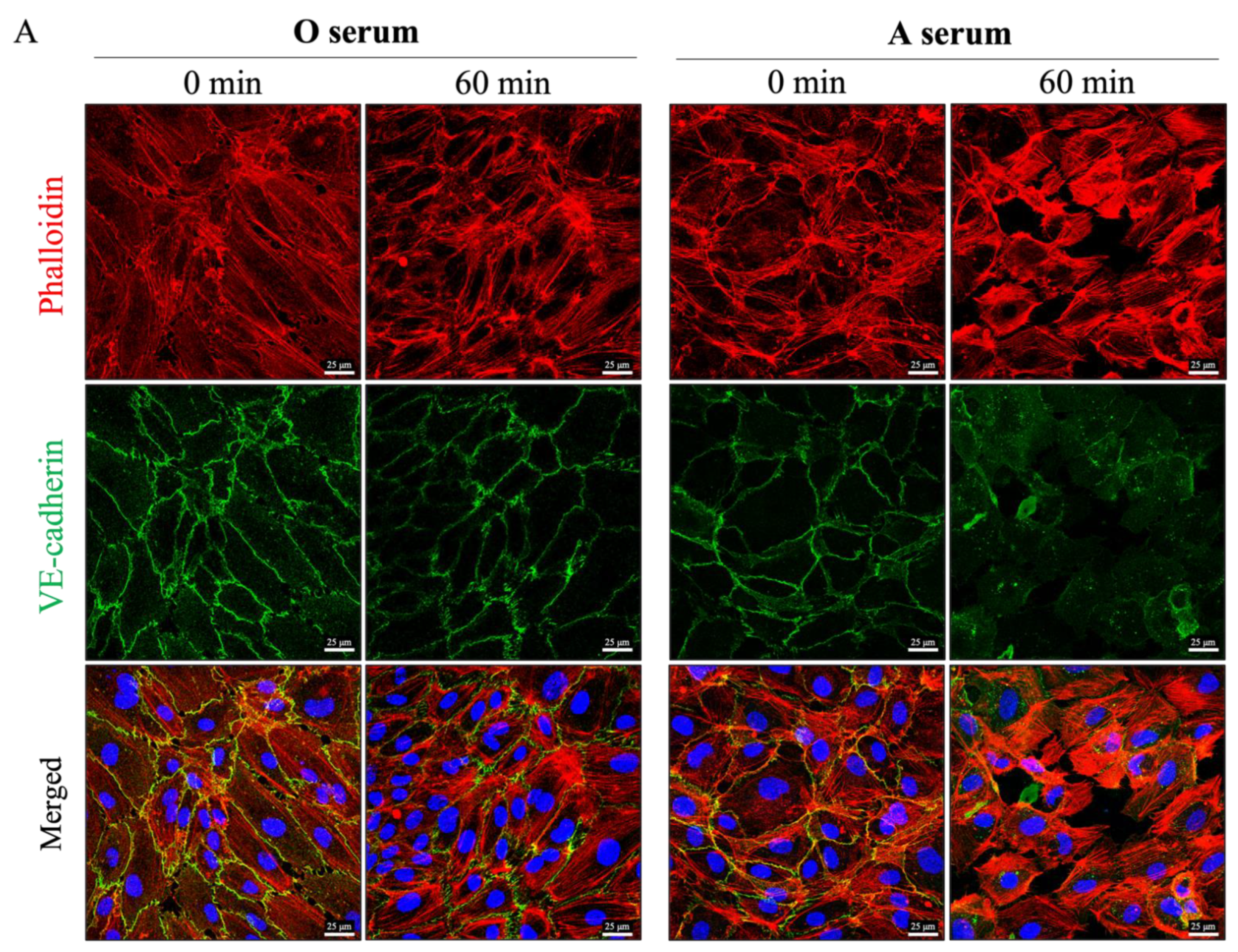
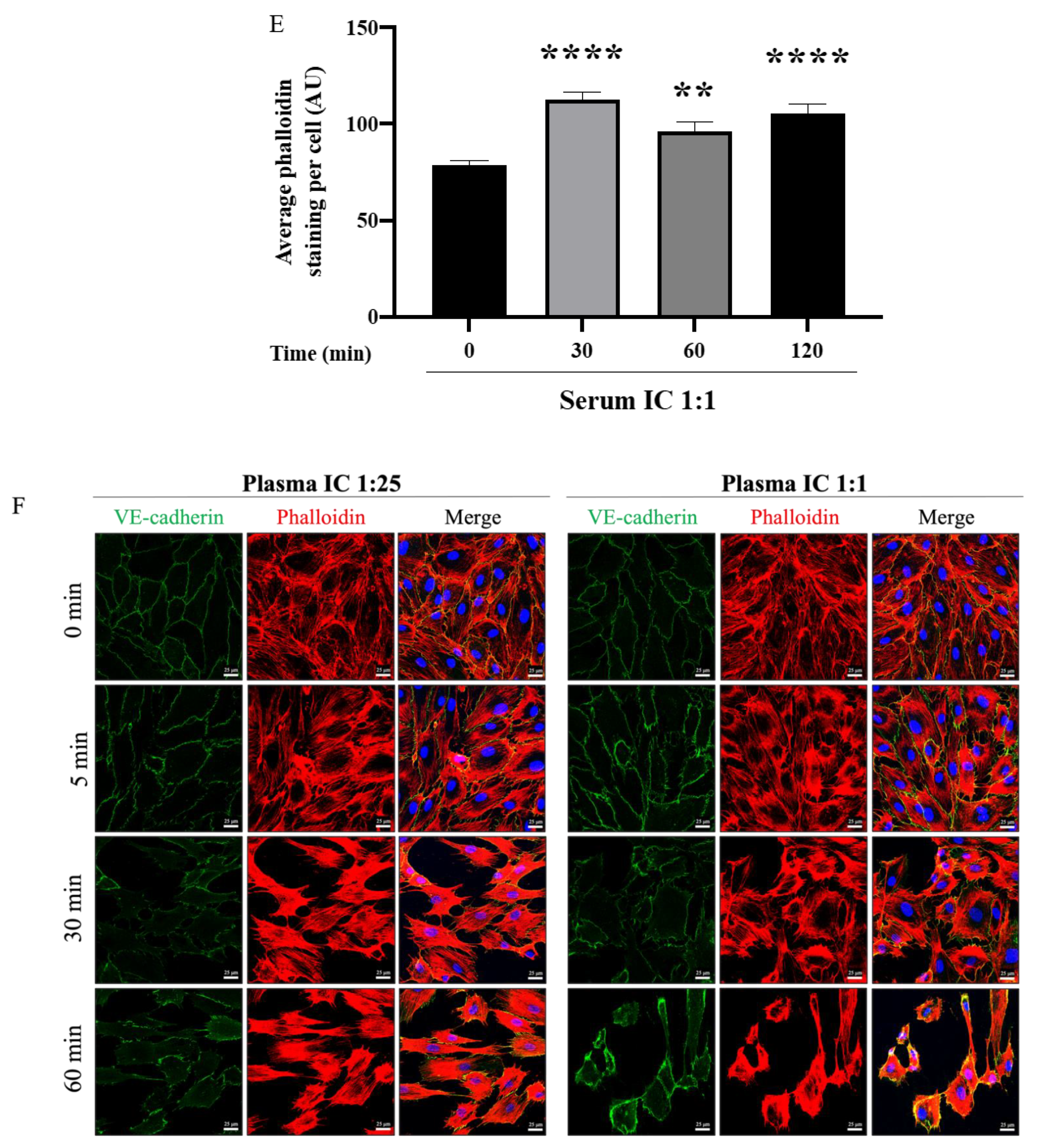
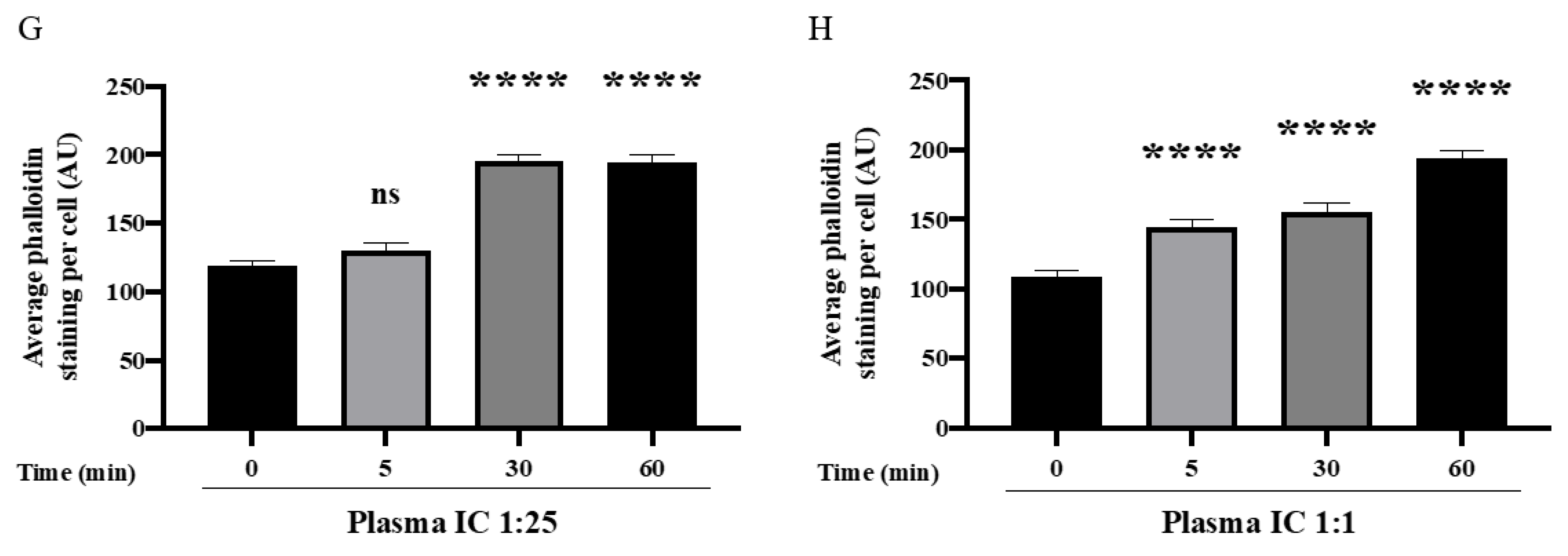
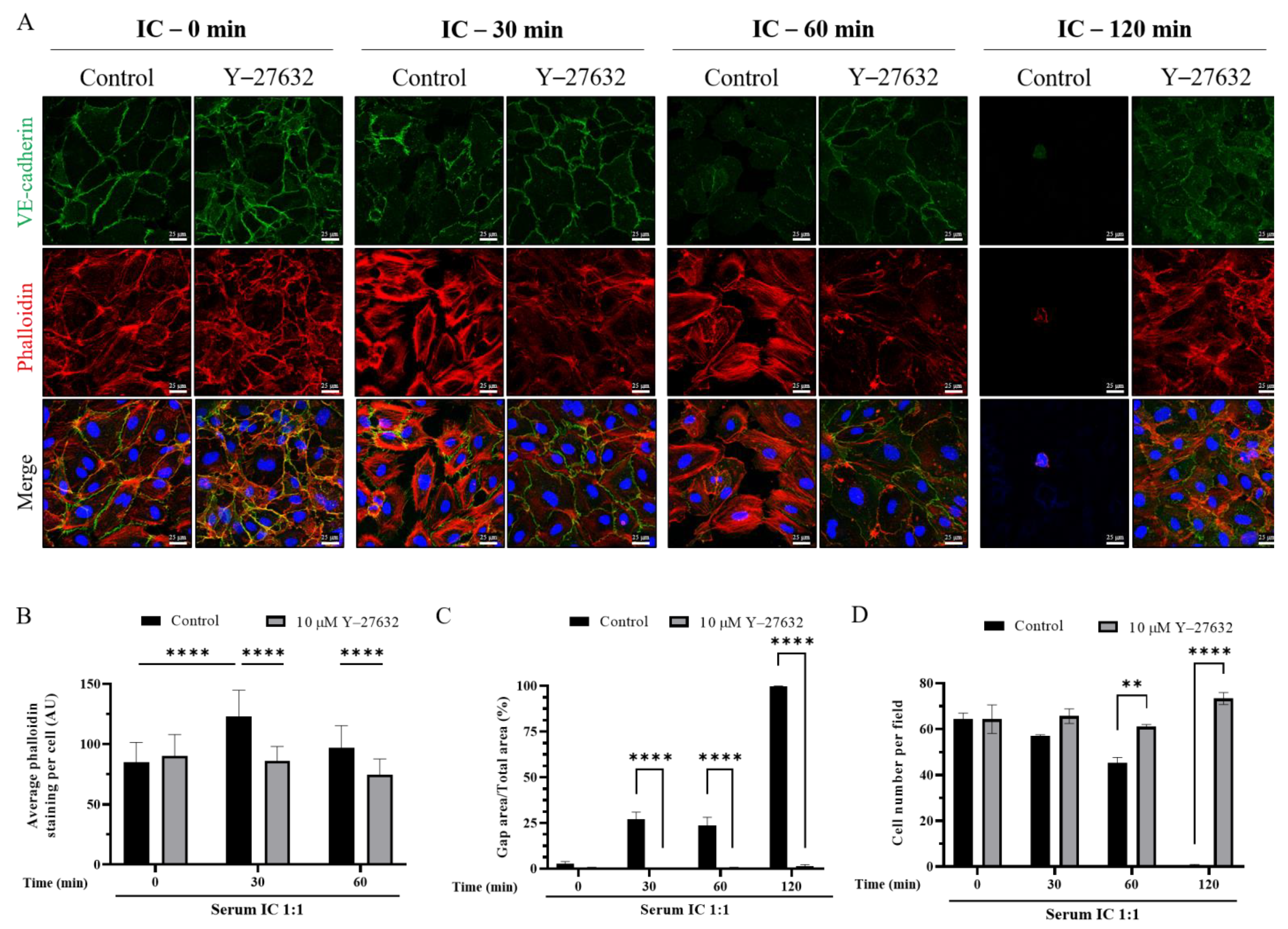
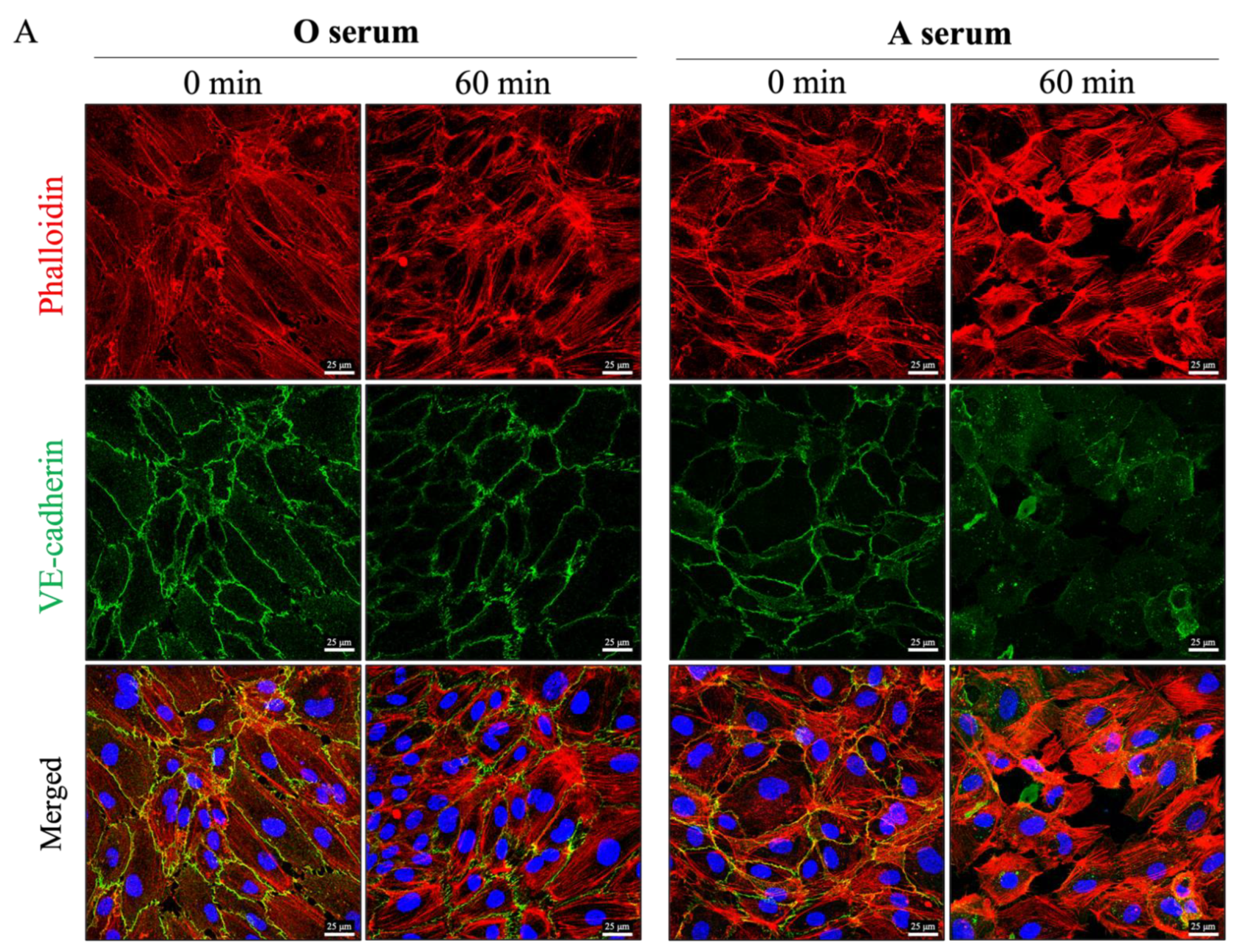
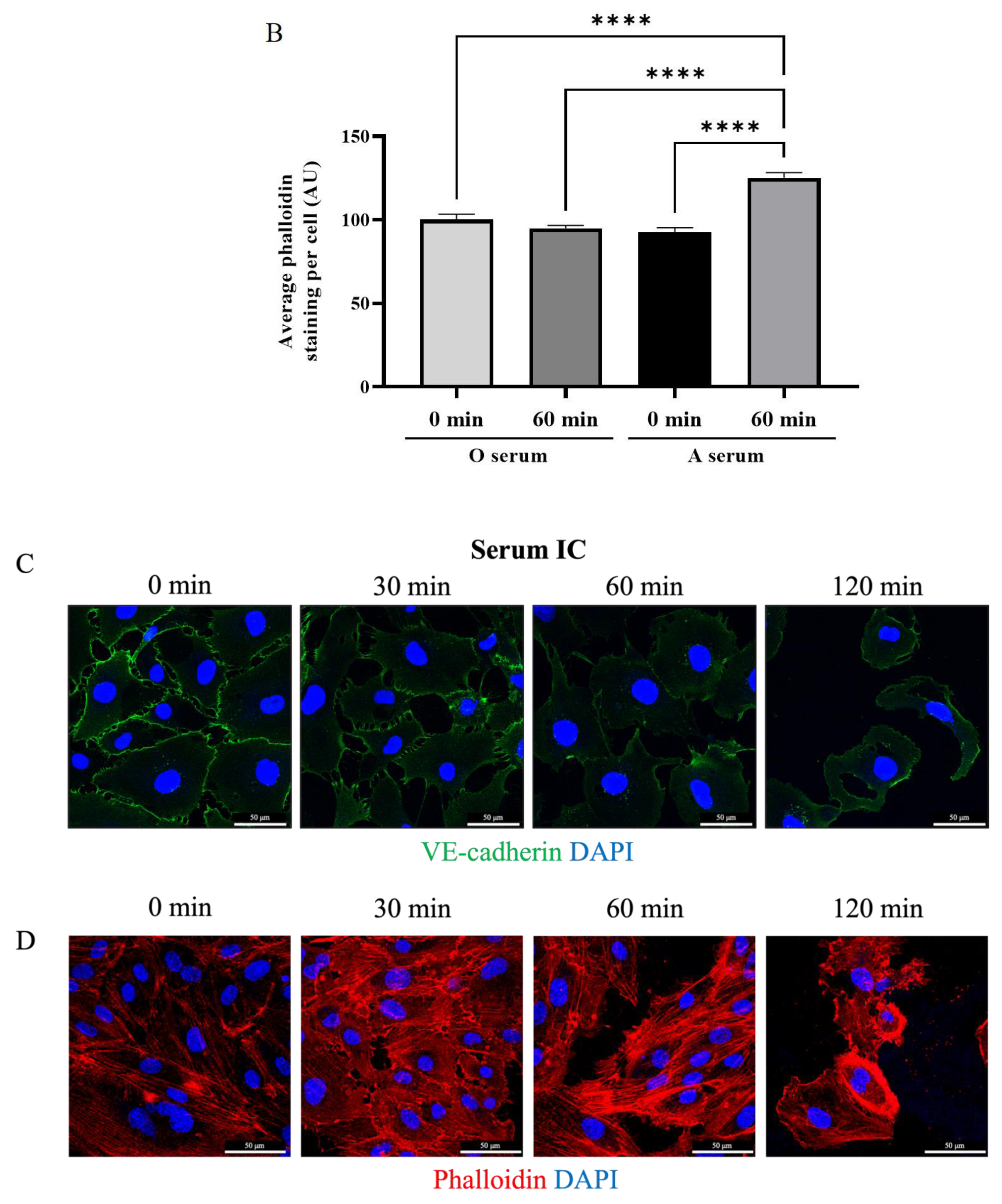
3.4. Mismatched ABO and IC Promotes VE-Cadherin Disassembly and Actin Stress Fiber Formation to Cause EC Barrier Disruption
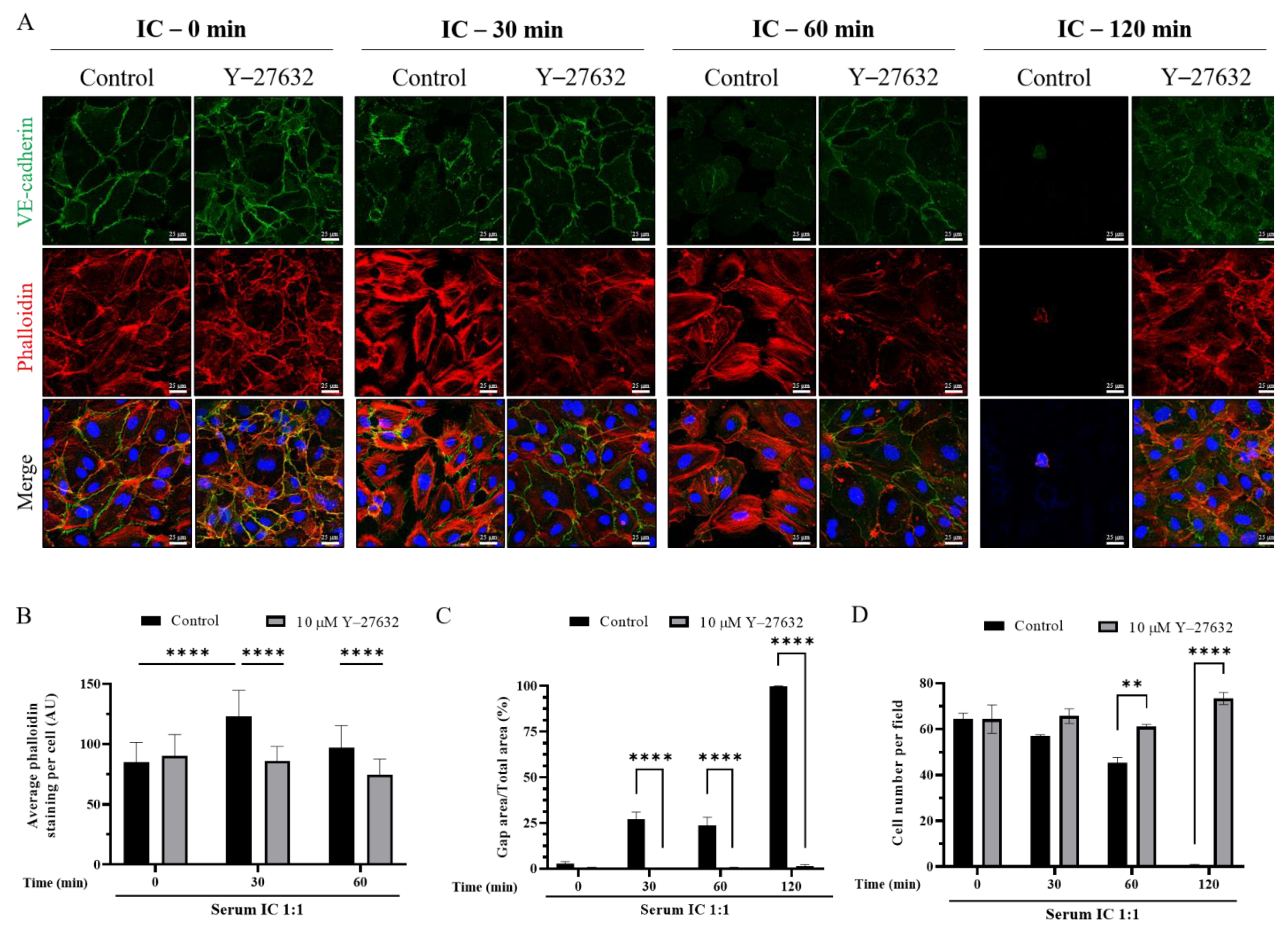
3.5. ROCK Inhibition Prevents IC-Induced EC Barrier Disruption
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blumberg, N.; Refaai, M.; Heal, J. ABO matching of platelet transfusions—”Start Making Sense”. “As we get older, and stop making sense…”—The Talking Heads (1984). Blood Transfus. 2015, 13, 347–350. [Google Scholar]
- McManigal, S.; Sims, K.L. Intravascular hemolysis secondary to ABO incompatible platelet products. An underrecognized transfusion reaction. Am. J. Clin. Pathol. 1999, 111, 202–206. [Google Scholar] [CrossRef] [Green Version]
- Sadani, D.T.; Urbaniak, S.J.; Bruce, M.; Tighe, J.E. Repeat ABO-incompatible platelet transfusions leading to haemolytic transfusion reaction. Transfus. Med. 2006, 16, 375–379. [Google Scholar] [CrossRef]
- Zaffuto, B.J.; Conley, G.W.; Connolly, G.C.; Henrichs, K.F.; Francis, C.W.; Heal, J.M.; Blumberg, N.; Refaai, M.A. ABO-immune complex formation and impact on platelet function, red cell structural integrity and haemostasis: An in vitro model of ABO non-identical transfusion. Vox Sang. 2016, 110, 219–226. [Google Scholar] [CrossRef]
- Shanwell, A.; Andersson, T.M.-L.; Rostgaard, K.; Edgren, G.; Hjalgrim, H.; Norda, R.; Melbye, M.; Nyrén, O.; Reilly, M. Post-transfusion mortality among recipients of ABO-compatible but non-identical plasma. Vox Sang. 2009, 96, 316–323. [Google Scholar] [CrossRef]
- Inaba, K.; Branco, B.C.; Rhee, P.; Holcomb, J.B.; Blackbourne, L.H.; Shulman, I.; Nelson, J.; Demetriades, D. Impact of ABO-Identical vs ABO-Compatible Nonidentical Plasma Transfusion in Trauma Patients. Arch. Surg. 2010, 145, 899–906. [Google Scholar] [CrossRef] [Green Version]
- Magid-Bernstein, J.; Beaman, C.B.; Carvalho-Poyraz, F.; Boehme, A.; Hod, E.A.; Francis, R.O.; Elkind, M.S.V.; Agarwal, S.; Park, S.; Claassen, J.; et al. Impacts of ABO Incompatible Platelet Transfusions on Platelet Recovery and Outcomes after Intracerebral Hemorrhage. Blood 2021, 137, 2699–2703. [Google Scholar] [CrossRef]
- Heal, J.; Masel, D.; Blumberg, N. Platelets (PLT) coated with ABO immune-complexes (cic) from refractory patients are associated with increased adhesion to monocytes. In Transfusion; Amer Assoc Blood Banks 8101 Glenbrook Rd: Bethesda, MD, USA, 1993; pp. 20814–22749. [Google Scholar]
- Heal, J.M.; Masel, D.; Rowe, J.M.; Blumberg, N. Circulating immune complexes involving the ABO system after platelet transfusion. Br. J. Haematol. 1993, 85, 566–572. [Google Scholar] [CrossRef]
- Heal, J.M.; Rowe, J.M.; Blumberg, N. ABO and platelet transfusion revisited. Ann. Hematol. 1993, 66, 309–314. [Google Scholar]
- Heal, J.M.; Masel, D.; Blumberg, N. Interaction of platelet fc and complement receptors with circulating immune complexes involving the AB0 system. Vox Sang. 1996, 71, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Heal, J.M.; Rowe, J.M.; McMican, A.; Masel, D.; Finke, C.; Blumberg, N. The role of ABO matching in platelet transfusion. Eur. J. Haematol. 1993, 50, 110–117. [Google Scholar] [CrossRef]
- Blumberg, N.; Heal, J.M.; Hicks, G.L., Jr.; Risher, W.H. Association of ABO-mismatched platelet transfusions with morbidity and mortality in cardiac surgery. Transfusion 2001, 41, 790–793. [Google Scholar] [CrossRef]
- Goerge, T.; Ho-Tin-Noe, B.; Carbo, C.; Benarafa, C.; Remold-O’Donnell, E.; Zhao, B.-Q.; Cifuni, S.M.; Wagner, D.D. Inflammation induces hemorrhage in thrombocytopenia. Blood 2008, 111, 4958–4964. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, M.; Ishikawa, Y.; Satake, M. Activation of polymorphonuclear neutrophils by immune complex: Possible involvement in development of transfusion-related acute lung injury. Transfus. Med. 2004, 14, 359–367. [Google Scholar] [CrossRef]
- Laborde, E.A.; Vanzulli, S.; Beigier-Bompadre, M.; Isturiz, M.A.; Ruggiero, R.A.; Fourcade, M.G.; Pellet, A.C.C.; Sozzani, S.; Vulcano, M. Immune complexes inhibit differentiation, maturation, and function of human monocyte-derived dendritic cells. J. Immunol. 2007, 179, 673–681. [Google Scholar] [CrossRef] [Green Version]
- Awad, H.A.; Tantawy, A.A.; El-Farrash, R.A.; Ismail, E.A.; Youssif, N.M. CD144+ endothelial microparticles as a marker of endothelial injury in neonatal ABO blood group incompatibility. Blood Transfus. 2014, 12, 250–259. [Google Scholar]
- Agre, P.; Cartron, J.P. Molecular biology of the Rh antigens. Blood 1991, 78, 551–563. [Google Scholar] [CrossRef] [Green Version]
- Calhoun, L.; Petz, L. Williams Hematology, 8th ed.; McGraw-Hill Education: New York, NY, USA, 2010. [Google Scholar]
- Gimbrone, M.A., Jr.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [Green Version]
- Goligorsky, M.S. Endothelial cell dysfunction: Can’t live with it, how to live without it. Am. J. Physiol. -Ren. Physiol. 2005, 288, F871–F880. [Google Scholar] [CrossRef] [Green Version]
- Avogaro, A.; Albiero, M.; Menegazzo, L.; de Kreutzenberg, S.; Fadini, G.P. Endothelial Dysfunction in Diabetes. Role Reparatory Mech. 2011, 34 (Suppl. 2), S285–S290. [Google Scholar]
- Roback, J.D.; Grossman, B.J.; Harris, T.; Hillyer, C.D. Technial Manual, 17th ed.; AABB Press: Bethesda, MD, USA, 2011. [Google Scholar]
- John Judd, W.; Johnson, S.T.; Storry, J.R. Judd’s Methods in Immunohematology, 3rd ed.; AABB Press: Bethesda, MD, USA, 2008. [Google Scholar]
- Cid, J.; Harm, S.K.; Yazer, M.H. Platelet Transfusion—The Art and Science of Compromise. Transfus. Med. Hemotherapy 2013, 40, 160–171. [Google Scholar] [CrossRef] [Green Version]
- Minhajuddin, M.; Bijli, K.M.; Fazal, F.; Sassano, A.; Nakayama, K.I.; Hay, N.; Platanias, L.C.; Rahman, A. Protein kinase C-delta and phosphatidylinositol 3-kinase/Akt activate mammalian target of rapamycin to modulate NF-kappaB activation and intercellular adhesion molecule-1 (ICAM-1) expression in endothelial cells. J. Biol. Chem. 2009, 284, 4052–4061. [Google Scholar] [CrossRef] [Green Version]
- Slavin, S.A.; Leonard, A.; Grose, V.; Fazal, F.; Rahman, A. Autophagy inhibitor 3-methyladenine protects against endothelial cell barrier dysfunction in acute lung injury. Am. J. Physiol. Lung Cell Mol Physiol. 2018, 314, L388–l396. [Google Scholar] [CrossRef] [Green Version]
- Bijli, K.M.; Fazal, F.; Slavin, S.A.; Leonard, A.; Grose, V.; Alexander, W.B.; Smrcka, A.V.; Rahman, A.S. Phospholipase C-epsilon signaling mediates endothelial cell inflammation and barrier disruption in acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L517–L524. [Google Scholar] [CrossRef] [Green Version]
- Dudek, S.M.; Garcia, J.G.N. Cytoskeletal regulation of pulmonary vascular permeability. J. Appl. Physiol. 2001, 91, 1487–1500. [Google Scholar] [CrossRef] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Leonard, A.; Millar, M.W.; Slavin, S.A.; Bijli, K.M.; Santos, D.A.D.; Dean, D.A.; Fazal, F.; Rahman, A. Critical role of autophagy regulator Beclin1 in endothelial cell inflammation and barrier disruption. Cell. Signal. 2019, 61, 120–129. [Google Scholar] [CrossRef]
- Shadab, M.; Millar, M.W.; Slavin, S.A.; Leonard, A.; Fazal, F.; Rahman, A. Autophagy protein ATG7 is a critical regulator of endothelial cell inflammation and permeability. Sci. Rep. 2020, 10, 13708. [Google Scholar] [CrossRef]
- Vandenbroucke St Amant, E.; Tauseef, M.; Vogel, S.M.; Gao, X.P.; Mehta, D.; Komarova, Y.A.; Malik, A.B. PKCα; Activation of p120-Catenin Serine 879 Phospho-Switch Disassembles VE-Cadherin Junctions and Disrupts Vascular Integrity. Circ. Res. 2012, 111, 739–749. [Google Scholar] [CrossRef] [Green Version]
- Mehta, D.; Rahman, A.; Malik, A.B. Protein kinase C-alpha signals rho-guanine nucleotide dissociation inhibitor phosphorylation and rho activation and regulates the endothelial cell barrier function. J. Biol. Chem. 2001, 276, 22614–22620. [Google Scholar] [CrossRef] [Green Version]
- Ge, M.; Tang, G.; Ryan, T.J.; Malik, A.B. Fibrinogen degradation product fragment D induces endothelial cell detachment by activation of cell-mediated fibrinolysis. J. Clin. Investig. 1992, 90, 2508–2516. [Google Scholar] [CrossRef] [Green Version]
- Bailey, K.E.; MacGowan, G.A.; Tual-Chalot, S.; Phillips, L.; Mohun, T.J.; Henderson, D.J. Disruption of embryonic ROCK signaling reproduces the sarcomeric phenotype of hypertrophic cardiomyopathy. JCI Insight 2019, 4, e146654. [Google Scholar] [CrossRef]
- Katoh, K.; Kano, Y.; Amano, M.; Kaibuchi, K.; Fujiwara, K. Stress fiber organization regulated by MLCK and Rho-kinase in cultured human fibroblasts. Am. J. Physiol. -Cell Physiol. 2001, 280, C1669–C1679. [Google Scholar] [CrossRef] [Green Version]
- Pipparelli, A.; Arsenijevic, Y.; Thuret, G.; Gain, P.; Nicolas, M.; Majo, F. ROCK inhibitor enhances adhesion and wound healing of human corneal endothelial cells. PLoS ONE 2013, 8, e62095. [Google Scholar]
- Sakamoto, K.; Hori, M.; Izumi, M.; Oka, T.; Kohama, K.; Ozaki, H.; Karaki, H. Inhibition of High K+-Induced Contraction by the ROCKs Inhibitor Y-27632 in Vascular Smooth Muscle: Possible Involvement of ROCKs in a Signal Transduction Pathway. J. Pharmacol. Sci. 2003, 92, 56–69. [Google Scholar] [CrossRef] [Green Version]
- Giannotta, M.; Trani, M.; Dejana, E. VE-cadherin and endothelial adherens junctions: Active guardians of vascular integrity. Dev. Cell. 2013, 26, 441–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiruppathi, C.; Ahmmed, G.U.; Vogel, S.M.; Malik, A.B. Ca2+ signaling, TRP channels, and endothelial permeability. Microcirculation 2006, 13, 693–708. [Google Scholar] [CrossRef]
- Mehta, D.; Malik, A.B. Signaling mechanisms regulating endothelial permeability. Physiol. Rev. 2006, 86, 279–367. [Google Scholar] [CrossRef]
- Wojciak-Stothard BRidley, A.J. Rho GTPases and the regulation of endothelial permeability. Vascul. Pharmacol. 2002, 39, 187–199. [Google Scholar] [CrossRef]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | N | Endothelial Cell ABO Type(s) | Percent Change at 1 h (Mean ± SD) | Percent Change at 8 h (Mean ± SD) |
|---|---|---|---|---|
| Media (−) | 16 | A, B, O | 2 ± 5 | 3 ± 6 |
| Thrombin (+) | 6 | A, B, O | 20 ± 11 | −17 ± 17 |
| ABO-Identical Exposure | ||||
| A Plasma | 6 | A | 5 ± 6 | 12 ± 5 |
| B Plasma | 4 | B | 4 ± 7 | 10 ± 3 |
| O Plasma | 6 | O | 12 ± 12 | 13 ± 6 |
| ABO-Non-identical Exposure | ||||
| A Plasma | 4 | B | 33 ± 7 *† | 44 ± 8 *‡ |
| B Plasma | 6 | A | 38 ± 4 *§ | 51 ± 4 *§ |
| O Plasma | 10 | A, B | 36 ± 6 *§ | 49 ± 6 *‡ |
| Treatment | N | Percent Change at 1 h (Mean ± SD) | Percent Change at 8 h (Mean ± SD) |
|---|---|---|---|
| Media (−) | 16 | 2 ± 5 | 3 ± 6 |
| Thrombin (+) | 6 | 20 ± 11 | −17 ± 17 |
| IC 1:1 | 26 | 50 ± 8 *‡ | 66 ± 9 *‡ |
| IC 1:3 | 20 | 44 ± 7 *‡ | 61 ± 7 *‡ |
| IC 1:7 | 20 | 42 ± 7 *‡ | 58 ± 7 *‡ |
| IC 1:10 | 6 | 32 ± 11 * | 69 ± 3 *§ |
| IC 1:20 | 6 | 31 ± 9 * | 65 ± 6 *§ |
| IC 1:25 | 6 | 27 ± 8 * | 64 ± 4 *§ |
| IC 1:30 | 6 | 29 ± 8 * | 64 ± 8 *§ |
| IC 1:50 | 6 | 22 ± 93 † | 62 ± 5 *§ |
| IC 1:100 | 6 | 11 ± 6 † | 51 ± 11 *§ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McRae, H.L.; Millar, M.W.; Slavin, S.A.; Blumberg, N.; Rahman, A.; Refaai, M.A. Essential Role of Rho-Associated Kinase in ABO Immune Complex-Mediated Endothelial Barrier Disruption. Biomedicines 2021, 9, 1851. https://doi.org/10.3390/biomedicines9121851
McRae HL, Millar MW, Slavin SA, Blumberg N, Rahman A, Refaai MA. Essential Role of Rho-Associated Kinase in ABO Immune Complex-Mediated Endothelial Barrier Disruption. Biomedicines. 2021; 9(12):1851. https://doi.org/10.3390/biomedicines9121851
Chicago/Turabian StyleMcRae, Hannah L., Michelle Warren Millar, Spencer A. Slavin, Neil Blumberg, Arshad Rahman, and Majed A. Refaai. 2021. "Essential Role of Rho-Associated Kinase in ABO Immune Complex-Mediated Endothelial Barrier Disruption" Biomedicines 9, no. 12: 1851. https://doi.org/10.3390/biomedicines9121851
APA StyleMcRae, H. L., Millar, M. W., Slavin, S. A., Blumberg, N., Rahman, A., & Refaai, M. A. (2021). Essential Role of Rho-Associated Kinase in ABO Immune Complex-Mediated Endothelial Barrier Disruption. Biomedicines, 9(12), 1851. https://doi.org/10.3390/biomedicines9121851