
Genetic Determinants of Plasma Low-Density Lipoprotein Cholesterol Levels: Monogenicity, Polygenicity, and “Missing” Heritability


Abstract
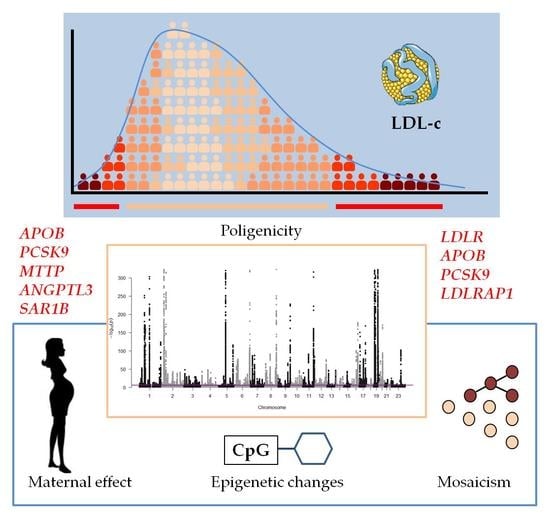
1. Introduction
2. Cholesterol Metabolism
2.1. The Endogenous Pathway
2.2. The Exogenous Pathway
3. Genetic Determinants of Plasma LDL Cholesterol Levels in the Pregenomic Era
3.1. Monogenic Forms of Hypercholesterolemia
- FHCL1 (OMIM #143890) or defective cellular LDL receptor (LDLR) is an autosomal dominant disorder due to loss-of-function LDLR gene variants [30]—the most common genetic defect in FH [31,32]. Defective LDLR results in reduced LDL-c uptake by hepatocytes, with a consequent increase in blood cholesterol.
- FHCL2 or familial ligand-defective hypercholesterolemia (OMIM #144010) is an autosomal dominant disorder due to missense APOB variants [33] (mainly p.Arg3527Gln). Since each LDL particle contains only one molecule of apoB100, a ligand-defective apoB results in an inability of LDL to bind to the LDLR, impairing its clearance from the blood. Mutations of the APOB gene account for 6–10% of ADH cases in Europeans [32].
- FHCL3 (OMIM #603776) is an autosomal dominant disorder due to gain-of-function variants of LDLR catabolic regulator proprotein convertase subtilisin/kexin type 9 (PCSK9) [34]. PCSK9 is an enzyme involved in the regulation of the degradation of LDLR in the lysosome, and gain-of-function mutants increase the degradation of LDLR, reducing its quantity on the hepatocyte surface [35].
- There are also recessive forms of phenotype HLP2A—referred to as autosomal recessive hypercholesterolemia (ARH or FHCL4) (OMIM #603813)—mainly due to protein-truncated mutations of the low-density lipoprotein receptor adaptor-protein 1 gene (LDLRAP1) [19]—a cytosolic protein that interacts with the cytoplasmatic tail of the LDLR.

{kind=link}
{kind=link}
{kind=link}
| Gene | Chromosome | Phenotype 1 | Type | Inheritance 2 | OMIM |
|---|---|---|---|---|---|
| high LDL-c | |||||
| LDLR | 19p13.2 | FHCL1 | loss-of-function | AD | #143890 |
| APOB | 2p24.1 | FHCL2 | missense | AD | #144010 |
| PCSK9 | 1p32.3 | FHCL3 | gain-of-funtion | AD | #603776 |
| LDLRAP1 | 1p36.11 | FHCL4 | protein-truncated | AR | #603813 |
| Phenocopies | |||||
| ABCG5/8 | 2p21 | sitosterolemia | loss-of-function | AR | #618666/#210250 |
| APOE | 19q13.32 | FCHL/dysB | p.Leu167del | #617347 | |
| CYP7A1 | 2q35 | CTX | loss-of-function | AR | #213700 |
| LIPA | 10q23.21 | CESD/WD | loss-of-function | AR | #278000 |
| LPA | 6q25-q26 | AD | #618807 | ||
| low LDL-c | |||||
| APOB | 2p24.1 | FHBL | protein-truncated | AD | #615558 |
| PCSK9 | 1p32.3 | FHBL | loss-of-function | AD | #615558 |
| ANGPTL3 | 1p31.3 | FHBL2 | loss-of-function | AR | #605019 |
| MTTP | 4q23 | ABL | loss-of-function | AR | #200100 |
| SAR1B | 5q31.1 | CMRD | loss-of-function | AR | #246700 |
| Other genes | |||||
| NPC1L1 | 7p13 | ↓LDL-c | loss-of-function | #617966 | |
| MYLIP | 6p22.3 | ↓LDL-c | *610082 | ||
| SREBF1 | 17p11.2 | CHL | AD | *184756 |
3.2. Monogenic Forms of Hypocholesterolemia
- FHBL2 or familial hypobetalipoproteinemia type 2 (OMIM #605019)—also known as familial combined hypolipidemia—is an autosomal recessive disorder caused by loss-of-function mutations of the angiopoietin-like 3 (ANGPTL3) gene [73]. ANGPTL3 is an inhibitor of the lipases LPL and LIPG (endothelial lipase), reducing the clearance of triglyceride-rich particles [74];
- ABL or abetalipoproteinemia (OMIM #200100) is an autosomal recessive disorder caused by mutations of the microsomal triglyceride transfer protein (MTTP) gene [75]. MTTP is a chaperone and the major lipid transfer protein of triglyceride, cholesteryl esters, and phospholipid to nascent apoB-containing lipoproteins [76];
- Chylomicron retention disease (CMRD; OMIM #246700)—also known as Anderson’s disease—is an autosomal recessive disorder caused by mutations of the secretion-associated Ras-related GTPase 1B (SAR1B) gene [77]. SAR1B plays a central role in the specific prechylomicron transport vesicles within the Golgi apparatus, as a component of coat protein complex II [78].
4. Genetics of LDL Cholesterol in the Big-Data Era
4.1. Genes Related to LDL Cholesterol Metabolism

4.1.1. Apolipoproteins and Lipoprotein Receptors
4.1.2. Transporters
4.1.3. Enzymes
4.1.4. Transcription Factors and Transcriptional Modulators
4.1.5. Other Genes Related to LDL-c Metabolism
4.2. Genes Not Related to LDL Cholesterol Metabolism
4.3. Polygenicity and Polygenic Risk Scores
5. Other Causes of High Plasma LDL Cholesterol Levels
5.1. Somatic Mutations/Mosaicism
5.2. Maternal Effect
5.3. Epigenetic Modifications
6. The Future: The Era of System Genetics
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kannel, W.B.; Dawber, T.R.; Friedman, G.D.; Glennon, W.E.; McNamara, P.M. Risk Factors in Coronary Heart Disease. An Evaluation of Several Serum Lipids as Predictors of Coronary Heart Disease; the Framingham Study. Ann. Intern. Med. 1964, 61, 888–899. [Google Scholar] [CrossRef] [PubMed]
- Ference, B.A.; Graham, I.; Tokgozoglu, L.; Catapano, A.L. Impact of Lipids on Cardiovascular Health: JACC Health Promotion Series. J. Am. Coll. Cardiol. 2018, 72, 1141–1156. [Google Scholar] [CrossRef] [PubMed]
- Ivanovic, B.; Tadic, M. Hypercholesterolemia and Hypertension: Two Sides of the Same Coin. Am. J. Cardiovasc. Drugs 2015, 15, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Falcone, G.J.; Kirsch, E.; Acosta, J.N.; Noche, R.B.; Leasure, A.; Marini, S.; Chung, J.; Selim, M.; Meschia, J.F.; Brown, D.L.; et al. Genetically Elevated LDL Associates with Lower Risk of Intracerebral Hemorrhage. Ann. Neurol. 2020, 88, 56–66. [Google Scholar] [CrossRef]
- White, J.; Swerdlow, D.I.; Preiss, D.; Fairhurst-Hunter, Z.; Keating, B.J.; Asselbergs, F.W.; Sattar, N.; Humphries, S.E.; Hingorani, A.D.; Holmes, M.V. Association of Lipid Fractions with Risks for Coronary Artery Disease and Diabetes. JAMA Cardiol. 2016, 1, 692–699. [Google Scholar] [CrossRef]
- Higuchi, S.; Izquierdo, M.C.; Haeusler, R.A. Unexplained reciprocal regulation of diabetes and lipoproteins. Curr. Opin. Lipidol. 2018, 29, 186–193. [Google Scholar] [CrossRef]
- Cohain, A.T.; Barrington, W.T.; Jordan, D.M.; Beckmann, N.D.; Argmann, C.A.; Houten, S.M.; Charney, A.W.; Ermel, R.; Sukhavasi, K.; Franzen, O.; et al. An integrative multiomic network model links lipid metabolism to glucose regulation in coronary artery disease. Nat. Commun. 2021, 12, 547. [Google Scholar] [CrossRef]
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update from the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef]
- INE. Defunciones Según la Causa de Muerte. 2018. Available online: https://www.ine.es/prensa/edcm_2018.pdf (accessed on 6 August 2021).
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. A century of cholesterol and coronaries: From plaques to genes to statins. Cell 2015, 161, 161–172. [Google Scholar] [CrossRef]
- McLean, K.J.; Hans, M.; Munro, A.W. Cholesterol, an essential molecule: Diverse roles involving cytochrome P450 enzymes. Biochem. Soc. Trans. 2012, 40, 587–593. [Google Scholar] [CrossRef]
- Schade, D.S.; Shey, L.; Eaton, R.P. Cholesterol Review: A Metabolically Important Molecule. Endocr. Pract. 2020, 26, 1514–1523. [Google Scholar] [CrossRef]
- Simons, K.; Sampaio, J.L. Membrane organization and lipid rafts. Cold Spring Harb. Perspect. Biol. 2011, 3, a004697. [Google Scholar] [CrossRef]
- Goedeke, L.; Fernández-Hernando, C. Regulation of cholesterol homeostasis. Cell Mol. Life Sci. 2012, 69, 915–930. [Google Scholar] [CrossRef]
- Shapiro, D.J.; Rodwell, V.W. Regulation of hepatic 3-hydroxy-3-methylglutaryl coenzyme A reductase and cholesterol synthesis. J. Biol. Chem. 1971, 246, 3210–3216. [Google Scholar] [CrossRef]
- Chang, T.Y.; Chang, C.C.; Cheng, D. Acyl-coenzyme A:cholesterol acyltransferase. Annu. Rev. Biochem. 1997, 66, 613–638. [Google Scholar] [CrossRef]
- Tall, A. Plasma lipid transfer proteins. Annu. Rev. Biochem. 1995, 64, 235–257.e246. [Google Scholar] [CrossRef]
- Garcia, C.K.; Wilund, K.; Arca, M.; Zuliani, G.; Fellin, R.; Maioli, M.; Calandra, S.; Bertolini, S.; Cossu, F.; Grishin, N.; et al. Autosomal recessive hypercholesterolemia caused by mutations in a putative LDL receptor adaptor protein. Science 2001, 292, 1394–1398. [Google Scholar] [CrossRef]
- Warner, T.G.; Dambach, L.M.; Shin, J.H.; O’Brien, J.S. Separation and characterization of the acid lipase and neutral esterases from human liver. Am. J. Hum. Genet. 1980, 32, 869–879. [Google Scholar]
- Sleat, D.E.; Wiseman, J.A.; El-Banna, M.; Price, S.M.; Verot, L.; Shen, M.M.; Tint, G.S.; Vanier, M.T.; Walkley, S.U.; Lobel, P. Genetic evidence for nonredundant functional cooperativity between NPC1 and NPC2 in lipid transport. Proc. Natl. Acad. Sci. USA 2004, 101, 5886–5891. [Google Scholar] [CrossRef]
- Schmidt, R.J.; Beyer, T.P.; Bensch, W.R.; Qian, Y.W.; Lin, A.; Kowala, M.; Alborn, W.E.; Konrad, R.J.; Cao, G. Secreted proprotein convertase subtilisin/kexin type 9 reduces both hepatic and extrahepatic low-density lipoprotein receptors in vivo. Biochem. Biophys. Res. Commun. 2008, 370, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Altmann, S.W.; Davis, H.R., Jr.; Zhu, L.J.; Yao, X.; Hoos, L.M.; Tetzloff, G.; Iyer, S.P.; Maguire, M.; Golovko, A.; Zeng, M.; et al. Niemann-Pick C1 Like 1 protein is critical for intestinal cholesterol absorption. Science 2004, 303, 1201–1204. [Google Scholar] [CrossRef] [PubMed]
- Berge, K.E.; Tian, H.; Graf, G.A.; Yu, L.; Grishin, N.V.; Schultz, J.; Kwiterovich, P.; Shan, B.; Barnes, R.; Hobbs, H.H. Accumulation of dietary cholesterol in sitosterolemia caused by mutations in adjacent ABC transporters. Science 2000, 290, 1771–1775. [Google Scholar] [CrossRef] [PubMed]
- May, P.; Woldt, E.; Matz, R.L.; Boucher, P. The LDL receptor-related protein (LRP) family: An old family of proteins with new physiological functions. Ann. Med. 2007, 39, 219–228. [Google Scholar] [CrossRef]
- Kenny, D.J.; Plichta, D.R.; Shungin, D.; Koppel, N.; Hall, A.B.; Fu, B.; Vasan, R.S.; Shaw, S.Y.; Vlamakis, H.; Balskus, E.P.; et al. Cholesterol Metabolism by Uncultured Human Gut Bacteria Influences Host Cholesterol Level. Cell Host Microbe 2020, 28, 245–257. [Google Scholar] [CrossRef]
- Zhong, V.W.; Van Horn, L.; Cornelis, M.C.; Wilkins, J.T.; Ning, H.; Carnethon, M.R.; Greenland, P.; Mentz, R.J.; Tucker, K.L.; Zhao, L.; et al. Associations of Dietary Cholesterol or Egg Consumption with Incident Cardiovascular Disease and Mortality. JAMA 2019, 321, 1081–1095. [Google Scholar] [CrossRef]
- Fredrickson, D.S.; Lees, R.S. A System for Phenotyping Hyperlipoproteinemia. Circulation 1965, 31, 321–327. [Google Scholar] [CrossRef]
- Hegele, R.A. Plasma lipoproteins: Genetic influences and clinical implications. Nat. Rev. Genet. 2009, 10, 109–121. [Google Scholar] [CrossRef]
- Lehrman, M.A.; Schneider, W.J.; Südhof, T.C.; Brown, M.S.; Goldstein, J.L.; Russell, D.W. Mutation in LDL receptor: Alu-Alu recombination deletes exons encoding transmembrane and cytoplasmic domains. Science 1985, 227, 140–146. [Google Scholar] [CrossRef]
- Do, R.; Stitziel, N.O.; Won, H.H.; Jørgensen, A.B.; Duga, S.; Merlini, P.A.; Kiezun, A.; Farrall, M.; Goel, A.; Zuk, O.; et al. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature 2015, 518, 102–106. [Google Scholar] [CrossRef]
- Sharifi, M.; Futema, M.; Nair, D.; Humphries, S.E. Genetic Architecture of Familial Hypercholesterolaemia. Curr. Cardiol. Rep. 2017, 19, 44. [Google Scholar] [CrossRef]
- Innerarity, T.L.; Weisgraber, K.H.; Arnold, K.S.; Mahley, R.W.; Krauss, R.M.; Vega, G.L.; Grundy, S.M. Familial defective apolipoprotein B-100: Low density lipoproteins with abnormal receptor binding. Proc. Natl. Acad. Sci. USA 1987, 84, 6919–6923. [Google Scholar] [CrossRef]
- Abifadel, M.; Varret, M.; Rabès, J.P.; Allard, D.; Ouguerram, K.; Devillers, M.; Cruaud, C.; Benjannet, S.; Wickham, L.; Erlich, D.; et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 2003, 34, 154–156. [Google Scholar] [CrossRef]
- Lagace, T.A.; Curtis, D.E.; Garuti, R.; McNutt, M.C.; Park, S.W.; Prather, H.B.; Anderson, N.N.; Ho, Y.K.; Hammer, R.E.; Horton, J.D. Secreted PCSK9 decreases the number of LDL receptors in hepatocytes and in livers of parabiotic mice. J. Clin. Investig. 2006, 116, 2995–3005. [Google Scholar] [CrossRef]
- Beheshti, S.O.; Madsen, C.M.; Varbo, A.; Nordestgaard, B.G. Worldwide Prevalence of Familial Hypercholesterolemia: Meta-Analyses of 11 Million Subjects. J. Am. Coll. Cardiol. 2020, 75, 2553–2566. [Google Scholar] [CrossRef]
- Vallejo-Vaz, A.J.; De Marco, M.; Stevens, C.A.T.; Akram, A.; Freiberger, T.; Hovingh, G.K.; Kastelein, J.J.P.; Mata, P.; Raal, F.J.; Santos, R.D.; et al. Overview of the current status of familial hypercholesterolaemia care in over 60 countries—The EAS Familial Hypercholesterolaemia Studies Collaboration (FHSC). Atherosclerosis 2018, 277, 234–255. [Google Scholar] [CrossRef]
- Zamora, A.; Masana, L.; Comas-Cufí, M.; Vila, À.; Plana, N.; García-Gil, M.; Alves-Cabratosa, L.; Marrugat, J.; Roman, I.; Ramos, R. Familial hypercholesterolemia in a European Mediterranean population-Prevalence and clinical data from 2.5 million primary care patients. J. Clin. Lipidol. 2017, 11, 1013–1022. [Google Scholar] [CrossRef]
- Benn, M.; Watts, G.F.; Tybjærg-Hansen, A.; Nordestgaard, B.G. Mutations causative of familial hypercholesterolaemia: Screening of 98 098 individuals from the Copenhagen General Population Study estimated a prevalence of 1 in 217. Eur. Heart J. 2016, 37, 1384–1394. [Google Scholar] [CrossRef]
- Lamiquiz-Moneo, I.; Civeira, F.; Mateo-Gallego, R.; Laclaustra, M.; Moreno-Franco, B.; Tejedor, M.T.; Palacios, L.; Martín, C.; Cenarro, A. Diagnostic yield of sequencing familial hypercholesterolemia genes in individuals with primary hypercholesterolemia. Rev. Esp. Cardiol. 2021, 74, 664–673. [Google Scholar] [CrossRef]
- Watts, G.F.; Gidding, S.; Wierzbicki, A.S.; Toth, P.P.; Alonso, R.; Brown, W.V.; Bruckert, E.; Defesche, J.; Lin, K.K.; Livingston, M.; et al. Integrated guidance on the care of familial hypercholesterolaemia from the international FH foundation. Int. J. Cardiol. 2014, 171, 309–325. [Google Scholar] [CrossRef]
- Austin, M.A.; Hutter, C.M.; Zimmern, R.L.; Humphries, S.E. Genetic causes of monogenic heterozygous familial hypercholesterolemia: A HuGE prevalence review. Am. J. Epidemiol. 2004, 160, 407–420. [Google Scholar] [CrossRef]
- Fouchier, S.W.; Dallinga-Thie, G.M.; Meijers, J.C.; Zelcer, N.; Kastelein, J.J.; Defesche, J.C.; Hovingh, G.K. Mutations in STAP1 are associated with autosomal dominant hypercholesterolemia. Circ. Res. 2014, 115, 552–555. [Google Scholar] [CrossRef]
- Blanco-Vaca, F.; Martín-Campos, J.M.; Pérez, A.; Fuentes-Prior, P. A rare STAP1 mutation incompletely associated with familial hypercholesterolemia. Clin. Chim. Acta 2018, 487, 270–274. [Google Scholar] [CrossRef]
- Lamiquiz-Moneo, I.; Restrepo-Córdoba, M.A.; Mateo-Gallego, R.; Bea, A.M.; del Pino Alberiche-Ruano, M.; García-Pavía, P.; Cenarro, A.; Martín, C.; Civeira, F.; Sánchez-Hernández, R.M. Predicted pathogenic mutations in STAP1 are not associated with clinically defined familial hypercholesterolemia. Atherosclerosis 2020, 292, 143–151. [Google Scholar] [CrossRef]
- Loaiza, N.; Hartgers, M.L.; Reeskamp, L.F.; Balder, J.W.; Rimbert, A.; Bazioti, V.; Wolters, J.C.; Winkelmeijer, M.; Jansen, H.P.G.; Dallinga-Thie, G.M.; et al. Taking One Step Back in Familial Hypercholesterolemia: STAP1 Does Not Alter Plasma LDL (Low-Density Lipoprotein) Cholesterol in Mice and Humans. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 973–985. [Google Scholar] [CrossRef]
- Stitziel, N.O.; Fouchier, S.W.; Sjouke, B.; Peloso, G.M.; Moscoso, A.M.; Auer, P.L.; Goel, A.; Gigante, B.; Barnes, T.A.; Melander, O.; et al. Exome sequencing and directed clinical phenotyping diagnose cholesterol ester storage disease presenting as autosomal recessive hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2909–2914. [Google Scholar] [CrossRef]
- Sjouke, B.; Defesche, J.C.; de Randamie, J.S.E.; Wiegman, A.; Fouchier, S.W.; Hovingh, G.K. Sequencing for LIPA mutations in patients with a clinical diagnosis of familial hypercholesterolemia. Atherosclerosis 2016, 251, 263–265. [Google Scholar] [CrossRef]
- Vinje, T.; Wierød, L.; Leren, T.P.; Strøm, T.B. Prevalence of cholesteryl ester storage disease among hypercholesterolemic subjects and functional characterization of mutations in the lysosomal acid lipase gene. Mol. Genet. Metab. 2018, 123, 169–176. [Google Scholar] [CrossRef]
- Gilardi, F.; Mitro, N.; Godio, C.; Scotti, E.; Caruso, D.; Crestani, M.; De Fabiani, E. The pharmacological exploitation of cholesterol 7alpha-hydroxylase, the key enzyme in bile acid synthesis: From binding resins to chromatin remodelling to reduce plasma cholesterol. Pharmacol. Ther. 2007, 116, 449–472. [Google Scholar] [CrossRef]
- Pullinger, C.R.; Eng, C.; Salen, G.; Shefer, S.; Batta, A.K.; Erickson, S.K.; Verhagen, A.; Rivera, C.R.; Mulvihill, S.J.; Malloy, M.J.; et al. Human cholesterol 7alpha-hydroxylase (CYP7A1) deficiency has a hypercholesterolemic phenotype. J. Clin. Investig. 2002, 110, 109–117. [Google Scholar] [CrossRef]
- Kajinami, K.; Brousseau, M.E.; Ordovas, J.M.; Schaefer, E.J. A promoter polymorphism in cholesterol 7alpha-hydroxylase interacts with apolipoprotein E genotype in the LDL-lowering response to atorvastatin. Atherosclerosis 2005, 180, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Marduel, M.; Ouguerram, K.; Serre, V.; Bonnefont-Rousselot, D.; Marques-Pinheiro, A.; Erik Berge, K.; Devillers, M.; Luc, G.; Lecerf, J.M.; Tosolini, L.; et al. Description of a large family with autosomal dominant hypercholesterolemia associated with the APOE p.Leu167del mutation. Hum. Mutat. 2013, 34, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Solanas-Barca, M.; de Castro-Orós, I.; Mateo-Gallego, R.; Cofán, M.; Plana, N.; Puzo, J.; Burillo, E.; Martín-Fuentes, P.; Ros, E.; Masana, L.; et al. Apolipoprotein E gene mutations in subjects with mixed hyperlipidemia and a clinical diagnosis of familial combined hyperlipidemia. Atherosclerosis 2012, 222, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Cenarro, A.; Etxebarria, A.; de Castro-Orós, I.; Stef, M.; Bea, A.M.; Palacios, L.; Mateo-Gallego, R.; Benito-Vicente, A.; Ostolaza, H.; Tejedor, T.; et al. The p.Leu167del Mutation in APOE Gene Causes Autosomal Dominant Hypercholesterolemia by Down-regulation of LDL Receptor Expression in Hepatocytes. J. Clin. Endocrinol. Metab. 2016, 101, 2113–2121. [Google Scholar] [CrossRef]
- Kidambi, S.; Patel, S.B. Sitosterolaemia: Pathophysiology, clinical presentation and laboratory diagnosis. J. Clin. Pathol. 2008, 61, 588–594. [Google Scholar] [CrossRef]
- Koeijvoets, K.C.; van der Net, J.B.; Dallinga-Thie, G.M.; Steyerberg, E.W.; Mensink, R.P.; Kastelein, J.J.; Sijbrands, E.J.; Plat, J. ABCG8 gene polymorphisms, plasma cholesterol concentrations, and risk of cardiovascular disease in familial hypercholesterolemia. Atherosclerosis 2009, 204, 453–458. [Google Scholar] [CrossRef]
- Garcia-Rios, A.; Perez-Martinez, P.; Fuentes, F.; Mata, P.; Lopez-Miranda, J.; Alonso, R.; Rodriguez, F.; Garcia-Olid, A.; Ruano, J.; Ordovas, J.M.; et al. Genetic variations at ABCG5/G8 genes modulate plasma lipids concentrations in patients with familial hypercholesterolemia. Atherosclerosis 2010, 210, 486–492. [Google Scholar] [CrossRef]
- Lamiquiz-Moneo, I.; Baila-Rueda, L.; Bea, A.M.; Mateo-Gallego, R.; Pérez-Calahorra, S.; Marco-Benedí, V.; Martín-Navarro, A.; Ros, E.; Cofán, M.; Rodríguez-Rey, J.C.; et al. ABCG5/G8 gene is associated with hypercholesterolemias without mutation in candidate genes and noncholesterol sterols. J. Clin. Lipidol. 2017, 11, 1432–1440. [Google Scholar] [CrossRef]
- Corral, P.; Geller, A.S.; Polisecki, E.Y.; Lopez, G.I.; Banares, V.G.; Cacciagiu, L.; Berg, G.; Hegele, R.A.; Schaefer, E.J.; Schreier, L.E. Unusual genetic variants associated with hypercholesterolemia in Argentina. Atherosclerosis 2018, 277, 256–261. [Google Scholar] [CrossRef]
- Tada, H.; Okada, H.; Nomura, A.; Yashiro, S.; Nohara, A.; Ishigaki, Y.; Takamura, M.; Kawashiri, M.A. Rare and Deleterious Mutations in ABCG5/ABCG8 Genes Contribute to Mimicking and Worsening of Familial Hypercholesterolemia Phenotype. Circ. J. 2019, 83, 1917–1924. [Google Scholar] [CrossRef]
- Reeskamp, L.F.; Volta, A.; Zuurbier, L.; Defesche, J.C.; Hovingh, G.K.; Grefhorst, A. ABCG5 and ABCG8 genetic variants in familial hypercholesterolemia. J. Clin. Lipidol. 2020, 14, 207–217.e207. [Google Scholar] [CrossRef]
- Wilson, P.A.; Gardner, S.D.; Lambie, N.M.; Commans, S.A.; Crowther, D.J. Characterization of the human patatin-like phospholipase family. J. Lipid Res. 2006, 47, 1940–1949. [Google Scholar] [CrossRef]
- Lange, L.A.; Hu, Y.; Zhang, H.; Xue, C.; Schmidt, E.M.; Tang, Z.Z.; Bizon, C.; Lange, E.M.; Smith, J.D.; Turner, E.H.; et al. Whole-exome sequencing identifies rare and low-frequency coding variants associated with LDL cholesterol. Am. J. Hum. Genet. 2014, 94, 233–245. [Google Scholar] [CrossRef]
- Jang, H.D.; Lee, S.E.; Yang, J.; Lee, H.C.; Shin, D.; Lee, H.; Lee, J.; Jin, S.; Kim, S.; Lee, S.J.; et al. Cyclase-associated protein 1 is a binding partner of proprotein convertase subtilisin/kexin type-9 and is required for the degradation of low-density lipoprotein receptors by proprotein convertase subtilisin/kexin type-9. Eur. Heart J. 2020, 41, 239–252. [Google Scholar] [CrossRef]
- Alonso, R.; Andres, E.; Mata, N.; Fuentes-Jiménez, F.; Badimón, L.; López-Miranda, J.; Padró, T.; Muñiz, O.; Díaz-Díaz, J.L.; Mauri, M.; et al. Lipoprotein(a) Levels in Familial Hypercholesterolemia: An Important Predictor of Cardiovascular Disease Independent of the Type of LDL Receptor Mutation. J. Am. Coll. Cardiol. 2014, 63, 1982–1989. [Google Scholar] [CrossRef]
- Friedewald, W.T.; Levy, R.I.; Fredrickson, D.S. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin. Chem. 1972, 18, 499–502. [Google Scholar] [CrossRef]
- Fatica, E.M.; Meeusen, J.W.; Vasile, V.C.; Jaffe, A.S.; Donato, L.J. Measuring the contribution of Lp(a) cholesterol towards LDL-C interpretation. Clin. Biochem. 2020, 86, 45–51. [Google Scholar] [CrossRef]
- Boerwinkle, E.; Leffert, C.C.; Lin, J.; Lackner, C.; Chiesa, G.; Hobbs, H.H. Apolipoprotein(a) gene accounts for greater than 90% of the variation in plasma lipoprotein(a) concentrations. J. Clin. Investig. 1992, 90, 52–60. [Google Scholar] [CrossRef]
- Welty, F.K. Hypobetalipoproteinemia and abetalipoproteinemia. Curr. Opin. Lipidol. 2014, 25, 161–168. [Google Scholar] [CrossRef]
- Young, S.G.; Northey, S.T.; McCarthy, B.J. Low plasma cholesterol levels caused by a short deletion in the apolipoprotein B gene. Science 1988, 241, 591–593. [Google Scholar] [CrossRef]
- Cohen, J.; Pertsemlidis, A.; Kotowski, I.K.; Graham, R.; Garcia, C.K.; Hobbs, H.H. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat. Genet. 2005, 37, 161–165. [Google Scholar] [CrossRef]
- Musunuru, K.; Pirruccello, J.P.; Do, R.; Peloso, G.M.; Guiducci, C.; Sougnez, C.; Garimella, K.V.; Fisher, S.; Abreu, J.; Barry, A.J.; et al. Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia. N. Engl. J. Med. 2010, 363, 2220–2227. [Google Scholar] [CrossRef]
- Shimizugawa, T.; Ono, M.; Shimamura, M.; Yoshida, K.; Ando, Y.; Koishi, R.; Ueda, K.; Inaba, T.; Minekura, H.; Kohama, T.; et al. ANGPTL3 decreases very low density lipoprotein triglyceride clearance by inhibition of lipoprotein lipase. J. Biol. Chem. 2002, 277, 33742–33748. [Google Scholar] [CrossRef]
- Shoulders, C.C.; Brett, D.J.; Bayliss, J.D.; Narcisi, T.M.; Jarmuz, A.; Grantham, T.T.; Leoni, P.R.; Bhattacharya, S.; Pease, R.J.; Cullen, P.M.; et al. Abetalipoproteinemia is caused by defects of the gene encoding the 97 kDa subunit of a microsomal triglyceride transfer protein. Hum. Mol. Genet. 1993, 2, 2109–2116. [Google Scholar] [CrossRef]
- Hussain, M.M.; Shi, J.; Dreizen, P. Microsomal triglyceride transfer protein and its role in apoB-lipoprotein assembly. J. Lipid Res. 2003, 44, 22–32. [Google Scholar] [CrossRef]
- Jones, B.; Jones, E.L.; Bonney, S.A.; Patel, H.N.; Mensenkamp, A.R.; Eichenbaum-Voline, S.; Rudling, M.; Myrdal, U.; Annesi, G.; Naik, S.; et al. Mutations in a Sar1 GTPase of COPII vesicles are associated with lipid absorption disorders. Nat. Genet. 2003, 34, 29–31. [Google Scholar] [CrossRef]
- Levy, E.; Poinsot, P.; Spahis, S. Chylomicron retention disease: Genetics, biochemistry, and clinical spectrum. Curr. Opin. Lipidol. 2019, 30, 134–139. [Google Scholar] [CrossRef]
- Zheng, C.; Khoo, C.; Furtado, J.; Sacks, F.M. Apolipoprotein C-III and the metabolic basis for hypertriglyceridemia and the dense low-density lipoprotein phenotype. Circulation 2010, 121, 1722–1734. [Google Scholar] [CrossRef]
- Pollin, T.I.; Damcott, C.M.; Shen, H.; Ott, S.H.; Shelton, J.; Horenstein, R.B.; Post, W.; McLenithan, J.C.; Bielak, L.F.; Peyser, P.A.; et al. A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science 2008, 322, 1702–1705. [Google Scholar] [CrossRef]
- Pokharel, Y.; Sun, W.; Polfus, L.M.; Folsom, A.R.; Heiss, G.; Sharrett, A.R.; Boerwinkle, E.; Ballantyne, C.M.; Hoogeveen, R.C. Lipoprotein associated phospholipase A2 activity, apolipoprotein C3 loss-of-function variants and cardiovascular disease: The Atherosclerosis Risk In Communities Study. Atherosclerosis 2015, 241, 641–648. [Google Scholar] [CrossRef][Green Version]
- Natarajan, P.; Kohli, P.; Baber, U.; Nguyen, K.H.; Sartori, S.; Reilly, D.F.; Mehran, R.; Muntendam, P.; Fuster, V.; Rader, D.J.; et al. Association of APOC3 Loss-of-Function Mutations with Plasma Lipids and Subclinical Atherosclerosis: The Multi-Ethnic BioImage Study. J. Am. Coll. Cardiol. 2015, 66, 2053–2055. [Google Scholar] [CrossRef] [PubMed]
- Wulff, A.B.; Nordestgaard, B.G.; Tybjærg-Hansen, A. APOC3 Loss-of-Function Mutations, Remnant Cholesterol, Low-Density Lipoprotein Cholesterol, and Cardiovascular Risk: Mediation- and Meta-Analyses of 137 895 Individuals. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 660–668. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, V.; Fouchier, S.W.; Motazacker, M.M.; Nelson, J.K.; Defesche, J.C.; Dallinga-Thie, G.M.; Kastelein, J.J.; Kees Hovingh, G.; Zelcer, N. Identification of a loss-of-function inducible degrader of the low-density lipoprotein receptor variant in individuals with low circulating low-density lipoprotein. Eur. Heart J. 2013, 34, 1292–1297. [Google Scholar] [CrossRef] [PubMed]
- Adi, D.; Abuzhalihan, J.; Wang, Y.H.; Baituola, G.; Wu, Y.; Xie, X.; Fu, Z.Y.; Yang, Y.N.; Ma, X.; Li, X.M.; et al. IDOL gene variant is associated with hyperlipidemia in Han population in Xinjiang, China. Sci. Rep. 2020, 10, 14280. [Google Scholar] [CrossRef]
- Cohen, J.C.; Pertsemlidis, A.; Fahmi, S.; Esmail, S.; Vega, G.L.; Grundy, S.M.; Hobbs, H.H. Multiple rare variants in NPC1L1 associated with reduced sterol absorption and plasma low-density lipoprotein levels. Proc. Natl. Acad. Sci. USA 2006, 103, 1810–1815. [Google Scholar] [CrossRef]
- Stitziel, N.O.; Won, H.H.; Morrison, A.C.; Peloso, G.M.; Do, R.; Lange, L.A.; Fontanillas, P.; Gupta, N.; Duga, S.; Goel, A.; et al. Inactivating mutations in NPC1L1 and protection from coronary heart disease. N. Engl. J. Med. 2014, 371, 2072–2082. [Google Scholar] [CrossRef]
- Kotzka, J.; Knebel, B.; Janssen, O.E.; Schaefer, J.R.; Soufi, M.; Jacob, S.; Nitzgen, U.; Muller-Wieland, D. Identification of a gene variant in the master regulator of lipid metabolism SREBP-1 in a family with a novel form of severe combined hypolipidemia. Atherosclerosis 2011, 218, 134–143. [Google Scholar] [CrossRef]
- Björkegren, J.L.M.; Kovacic, J.C.; Dudley, J.T.; Schadt, E.E. Genome-wide significant loci: How important are they? Systems genetics to understand heritability of coronary artery disease and other common complex disorders. J. Am. Coll. Cardiol. 2015, 65, 830–845. [Google Scholar] [CrossRef]
- Winkler, T.W.; Day, F.R.; Croteau-Chonka, D.C.; Wood, A.R.; Locke, A.E.; Mägi, R.; Ferreira, T.; Fall, T.; Graff, M.; Justice, A.E.; et al. Quality control and conduct of genome-wide association meta-analyses. Nat. Protoc. 2014, 9, 1192–1212. [Google Scholar] [CrossRef]
- Xu, C.; Tachmazidou, I.; Walter, K.; Ciampi, A.; Zeggini, E.; Greenwood, C.M. Estimating genome-wide significance for whole-genome sequencing studies. Genet. Epidemiol. 2014, 38, 281–290. [Google Scholar] [CrossRef]
- Naj, A.C. Genotype Imputation in Genome-Wide Association Studies. Curr. Protoc. Hum. Genet. 2019, 102, e84. [Google Scholar] [CrossRef]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef]
- Beck, T.; Shorter, T.; Brookes, A.J. GWAS Central: A comprehensive resource for the discovery and comparison of genotype and phenotype data from genome-wide association studies. Nucleic Acids Res. 2020, 48, D933–D940. [Google Scholar] [CrossRef]
- Klimentidis, Y.C.; Arora, A.; Newell, M.; Zhou, J.; Ordovas, J.M.; Renquist, B.J.; Wood, A.C. Phenotypic and Genetic Characterization of Lower LDL Cholesterol and Increased Type 2 Diabetes Risk in the UK Biobank. Diabetes 2020, 69, 2194–2205. [Google Scholar] [CrossRef]
- Martín-Campos, J.M.; Chang, B.; Blanco-Vaca, F.; Chan, L.; Julve, J. Apolipoprotein gene structure and function. In eLS; John Wiley & Sons, Ltd.: Chichester, UK, 2017. [Google Scholar]
- Melendez, Q.M.; Wooten, C.J.; Krishnaji, S.T.; Knagge, K.; Kirchner, D.; Lopez, D. Identification of Novel Proteins Interacting with Proprotein Convertase Subtilisin/Kexin 9. Int. J. Biomed. Investig. 2020, 3, 123. [Google Scholar] [CrossRef]
- Kounnas, M.Z.; Argraves, W.S.; Strickland, D.K. The 39-kDa receptor-associated protein interacts with two members of the low density lipoprotein receptor family, alpha 2-macroglobulin receptor and glycoprotein 330. J. Biol. Chem. 1992, 267, 21162–21166. [Google Scholar] [CrossRef]
- Brooks-Wilson, A.; Marcil, M.; Clee, S.M.; Zhang, L.H.; Roomp, K.; van Dam, M.; Yu, L.; Brewer, C.; Collins, J.A.; Molhuizen, H.O.; et al. Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nat. Genet. 1999, 22, 336–345. [Google Scholar] [CrossRef]
- Herman-Edelstein, M.; Scherzer, P.; Tobar, A.; Levi, M.; Gafter, U. Altered renal lipid metabolism and renal lipid accumulation in human diabetic nephropathy. J. Lipid Res. 2014, 55, 561–572. [Google Scholar] [CrossRef]
- Gai, J.; Ji, M.; Shi, C.; Li, W.; Chen, S.; Wang, Y.; Li, H. FoxO regulates expression of ABCA6, an intracellular ATP-binding-cassette transporter responsive to cholesterol. Int. J. Biochem. Cell Biol. 2013, 45, 2651–2659. [Google Scholar] [CrossRef]
- He, B.; Kang, S.; Chen, Z.; Liu, X.; Wang, J.; Li, X.; Liu, X.; Zheng, L.; Luo, M.; Wang, Y. Hypercholesterolemia risk associated Abca6 does not regulate lipoprotein metabolism in mice or hamster. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 159006. [Google Scholar] [CrossRef]
- Lee, J.H.; Kang, H.S.; Park, H.Y.; Moon, Y.A.; Kang, Y.N.; Oh, B.C.; Song, D.K.; Bae, J.H.; Im, S.S. PPARα-dependent Insig2a overexpression inhibits SREBP-1c processing during fasting. Sci. Rep. 2017, 7, 9958. [Google Scholar] [CrossRef]
- Armendariz, A.D.; Krauss, R.M. Hepatic nuclear factor 1-alpha: Inflammation, genetics, and atherosclerosis. Curr. Opin. Lipidol. 2009, 20, 106–111. [Google Scholar] [CrossRef]
- Li, H.; Dong, B.; Park, S.W.; Lee, H.S.; Chen, W.; Liu, J. Hepatocyte nuclear factor 1alpha plays a critical role in PCSK9 gene transcription and regulation by the natural hypocholesterolemic compound berberine. J. Biol. Chem. 2009, 284, 28885–28895. [Google Scholar] [CrossRef]
- Tavares-Sanchez, O.L.; Rodriguez, C.; Gortares-Moroyoqui, P.; Estrada, M.I. Hepatocyte nuclear factor-4α, a multifunctional nuclear receptor associated with cardiovascular disease and cholesterol catabolism. Int. J. Environ. Health Res. 2015, 25, 126–139. [Google Scholar] [CrossRef]
- Johnston, J.M.; Angyal, A.; Bauer, R.C.; Hamby, S.; Suvarna, S.K.; Baidžajevas, K.; Hegedus, Z.; Dear, T.N.; Turner, M.; Wilson, H.L.; et al. Myeloid Tribbles 1 induces early atherosclerosis via enhanced foam cell expansion. Sci. Adv. 2019, 5, eaax9183. [Google Scholar] [CrossRef]
- Berthier, A.; Johanns, M.; Zummo, F.P.; Lefebvre, P.; Staels, B. PPARs in liver physiology. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166097. [Google Scholar] [CrossRef]
- Li, B.; Cai, S.Y.; Boyer, J.L. The role of the retinoid receptor, RAR/RXR heterodimer, in liver physiology. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166085. [Google Scholar] [CrossRef]
- Kim, H. The transcription factor MafB promotes anti-inflammatory M2 polarization and cholesterol efflux in macrophages. Sci. Rep. 2017, 7, 7591. [Google Scholar] [CrossRef]
- Susan-Resiga, D.; Girard, E.; Essalmani, R.; Roubtsova, A.; Marcinkiewicz, J.; Derbali, R.M.; Evagelidis, A.; Byun, J.H.; Lebeau, P.F.; Austin, R.C.; et al. Asialoglycoprotein receptor 1 is a novel PCSK9-independent ligand of liver LDLR cleaved by furin. J. Biol. Chem. 2021, 297, 101177. [Google Scholar] [CrossRef]
- Conlon, D.M. Role of sortilin in lipid metabolism. Curr. Opin. Lipidol. 2019, 30, 198–204. [Google Scholar] [CrossRef]
- Tan, J.; Che, Y.; Liu, Y.; Hu, J.; Wang, W.; Hu, L.; Zhou, Q.; Wang, H.; Li, J. CELSR2 deficiency suppresses lipid accumulation in hepatocyte by impairing the UPR and elevating ROS level. Faseb J. 2021, 35, e21908. [Google Scholar] [CrossRef] [PubMed]
- Marais, A.D. Apolipoprotein E in lipoprotein metabolism, health and cardiovascular disease. Pathology 2019, 51, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Mahdessian, H.; Taxiarchis, A.; Popov, S.; Silveira, A.; Franco-Cereceda, A.; Hamsten, A.; Eriksson, P.; van’t Hooft, F. TM6SF2 is a regulator of liver fat metabolism influencing triglyceride secretion and hepatic lipid droplet content. Proc. Natl. Acad. Sci. USA 2014, 111, 8913–8918. [Google Scholar] [CrossRef] [PubMed]
- Li, T.T.; Li, T.H.; Peng, J.; He, B.; Liu, L.S.; Wei, D.H.; Jiang, Z.S.; Zheng, X.L.; Tang, Z.H. TM6SF2: A novel target for plasma lipid regulation. Atherosclerosis 2018, 268, 170–176. [Google Scholar] [CrossRef]
- Wright, M.B.; Varona Santos, J.; Kemmer, C.; Maugeais, C.; Carralot, J.P.; Roever, S.; Molina, J.; Ducasa, G.M.; Mitrofanova, A.; Sloan, A.; et al. Compounds targeting OSBPL7 increase ABCA1-dependent cholesterol efflux preserving kidney function in two models of kidney disease. Nat. Commun. 2021, 12, 4662. [Google Scholar] [CrossRef]
- Saborowski, M.; Kullak-Ublick, G.A.; Eloranta, J.J. The human organic cation transporter-1 gene is transactivated by hepatocyte nuclear factor-4alpha. J. Pharmacol. Exp. Ther. 2006, 317, 778–785. [Google Scholar] [CrossRef]
- Baillie, J.K.; Bretherick, A.; Haley, C.S.; Clohisey, S.; Gray, A.; Neyton, L.P.A.; Barrett, J.; Stahl, E.A.; Tenesa, A.; Andersson, R.; et al. Shared activity patterns arising at genetic susceptibility loci reveal underlying genomic and cellular architecture of human disease. PLoS Comput. Biol. 2018, 14, e1005934. [Google Scholar] [CrossRef]
- Parhofer, K.G. Interaction between Glucose and Lipid Metabolism: More than Diabetic Dyslipidemia. Diabetes Metab. J. 2015, 39, 353–362. [Google Scholar] [CrossRef]
- Lin, Q.Z.; Yin, R.X.; Guo, T.; Wu, J.; Sun, J.Q.; Shen, S.W.; Shi, G.Y.; Wu, J.Z.; Liu, C.W.; Pan, S.L. Association of the ST3GAL4 rs11220462 polymorphism and serum lipid levels in the Mulao and Han populations. Lipids Health Dis. 2014, 13, 123. [Google Scholar] [CrossRef]
- Arguinano, A.A.; Ndiaye, N.C.; Masson, C.; Visvikis-Siest, S. Pleiotropy of ABO gene: Correlation of rs644234 with E-selectin and lipid levels. Clin. Chem. Lab. Med. 2018, 56, 748–754. [Google Scholar] [CrossRef]
- Paquette, M.; Dufour, R.; Baass, A. ABO blood group is a cardiovascular risk factor in patients with familial hypercholesterolemia. J. Clin. Lipidol. 2018, 12, 383–389. [Google Scholar] [CrossRef]
- Hegele, R.A.; Brunt, J.H.; Connelly, P.W. Multiple genetic determinants of variation of plasma lipoproteins in Alberta Hutterites. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 861–871. [Google Scholar] [CrossRef]
- Aulchenko, Y.S.; Ripatti, S.; Lindqvist, I.; Boomsma, D.; Heid, I.M.; Pramstaller, P.P.; Penninx, B.W.J.H.; Janssens, A.C.J.W.; Wilson, J.F.; Spector, T.; et al. Loci influencing lipid levels and coronary heart disease risk in 16 European population cohorts. Nat. Genet. 2008, 41, 47. [Google Scholar] [CrossRef]
- Lutsey, P.L.; Rasmussen-Torvik, L.J.; Pankow, J.S.; Alonso, A.; Smolenski, D.J.; Tang, W.; Coresh, J.; Volcik, K.A.; Ballantyne, C.M.; Boerwinkle, E.; et al. Relation of Lipid Gene Scores to Longitudinal Trends in Lipid Levels and Incidence of Abnormal Lipid Levels Among Individuals of European Ancestry. Circ. Cardiovasc. Genet. 2012, 5, 73–80. [Google Scholar] [CrossRef][Green Version]
- Nomura, A.; Sato, T.; Tada, H.; Kannon, T.; Hosomichi, K.; Tsujiguchi, H.; Nakamura, H.; Takamura, M.; Tajima, A.; Kawashiri, M.A. Polygenic risk scores for low-density lipoprotein cholesterol and familial hypercholesterolemia. J. Hum. Genet. 2021, 66, 1079–1087. [Google Scholar] [CrossRef]
- Kuchenbaecker, K.; Telkar, N.; Reiker, T.; Walters, R.G.; Lin, K.; Eriksson, A.; Gurdasani, D.; Gilly, A.; Southam, L.; Tsafantakis, E.; et al. The transferability of lipid loci across African, Asian and European cohorts. Nat. Commun. 2019, 10, 4330. [Google Scholar] [CrossRef]
- Talmud, P.J.; Shah, S.; Whittall, R.; Futema, M.; Howard, P.; Cooper, J.A.; Harrison, S.C.; Li, K.; Drenos, F.; Karpe, F.; et al. Use of low-density lipoprotein cholesterol gene score to distinguish patients with polygenic and monogenic familial hypercholesterolaemia: A case-control study. Lancet 2013, 381, 1293–1301. [Google Scholar] [CrossRef]
- Teslovich, T.M.; Musunuru, K.; Smith, A.V.; Edmondson, A.C.; Stylianou, I.M.; Koseki, M.; Pirruccello, J.P.; Ripatti, S.; Chasman, D.I.; Willer, C.J.; et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010, 466, 707–713. [Google Scholar] [CrossRef]
- Wang, J.; Dron, J.S.; Ban, M.R.; Robinson, J.F.; McIntyre, A.D.; Alazzam, M.; Zhao, P.J.; Dilliott, A.A.; Cao, H.; Huff, M.W.; et al. Polygenic versus monogenic causes of hypercholesterolemia ascertained clinically. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2439–2445. [Google Scholar] [CrossRef]
- Kathiresan, S.; Willer, C.J.; Peloso, G.M.; Demissie, S.; Musunuru, K.; Schadt, E.E.; Kaplan, L.; Bennett, D.; Li, Y.; Tanaka, T.; et al. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat. Genet. 2009, 41, 56–65. [Google Scholar] [CrossRef]
- Futema, M.; Shah, S.; Cooper, J.A.; Li, K.; Whittall, R.A.; Sharifi, M.; Goldberg, O.; Drogari, E.; Mollaki, V.; Wiegman, A.; et al. Refinement of variant selection for the LDL cholesterol genetic risk score in the diagnosis of the polygenic form of clinical familial hypercholesterolemia and replication in samples from 6 countries. Clin. Chem. 2015, 61, 231–238. [Google Scholar] [CrossRef]
- Mariano, C.; Futema, M.; Humphries, S.E.; Bourbon, M. Applicability of the low-density lipoprotein cholesterol gene score in a south european population. Atherosclerosis 2017, 263, e99–e100. [Google Scholar] [CrossRef]
- Durst, R.; Ibe, U.K.; Shpitzen, S.; Schurr, D.; Eliav, O.; Futema, M.; Whittall, R.; Szalat, A.; Meiner, V.; Knobler, H.; et al. Molecular genetics of familial hypercholesterolemia in Israel-revisited. Atherosclerosis 2017, 257, 55–63. [Google Scholar] [CrossRef]
- Rieck, L.; Bardey, F.; Grenkowitz, T.; Bertram, L.; Helmuth, J.; Mischung, C.; Spranger, J.; Steinhagen-Thiessen, E.; Bobbert, T.; Kassner, U.; et al. Mutation spectrum and polygenic score in German patients with familial hypercholesterolemia. Clin. Genet. 2020, 98, 457–467. [Google Scholar] [CrossRef]
- Martín-Campos, J.M.; Ruiz-Nogales, S.; Ibarretxe, D.; Ortega, E.; Sánchez-Pujol, E.; Royuela-Juncadella, M.; Vila, À.; Guerrero, C.; Zamora, A.; Soler, I.F.C.; et al. Polygenic Markers in Patients Diagnosed of Autosomal Dominant Hypercholesterolemia in Catalonia: Distribution of Weighted LDL-c-Raising SNP Scores and Refinement of Variant Selection. Biomedicines 2020, 8, 353. [Google Scholar] [CrossRef]
- Khera, A.V.; Chaffin, M.; Aragam, K.G.; Haas, M.E.; Roselli, C.; Choi, S.H.; Natarajan, P.; Lander, E.S.; Lubitz, S.A.; Ellinor, P.T.; et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 2018, 50, 1219–1224. [Google Scholar] [CrossRef]
- Cupido, A.J.; Tromp, T.R.; Hovingh, G.K. The clinical applicability of polygenic risk scores for LDL-cholesterol: Considerations, current evidence and future perspectives. Curr. Opin. Lipidol. 2021, 32, 112–116. [Google Scholar] [CrossRef]
- Forsberg, L.A.; Gisselsson, D.; Dumanski, J.P. Mosaicism in health and disease—Clones picking up speed. Nat. Rev. Genet. 2016, 18, 128–142. [Google Scholar] [CrossRef]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef]
- Rodríguez-Nóvoa, S.; Rodríguez-Jiménez, C.; Alonso, C.; Rodriguez-Laguna, L.; Gordo, G.; Martinez-Glez, V.; García Polo, I. Familial hypercholesterolemia: A single-nucleotide variant (SNV) in mosaic at the low density lipoprotein receptor (LDLR). Atherosclerosis 2020, 311, 37–43. [Google Scholar] [CrossRef]
- Knopp, R.H.; Bonet, B.; Zhu, X. Lipid Metabolism in Pregnancy. In Principles of Perinatal—Neonatal Metabolism; Springer: New York, NY, USA, 1998; pp. 221–258. [Google Scholar]
- Brizzi, P.; Tonolo, G.; Esposito, F.; Puddu, L.; Dessole, S.; Maioli, M.; Milia, S. Lipoprotein metabolism during normal pregnancy. Am. J. Obstet. Gynecol. 1999, 181, 430–434. [Google Scholar] [CrossRef]
- Edison, R.J.; Berg, K.; Remaley, A.; Kelley, R.; Rotimi, C.; Stevenson, R.E.; Muenke, M. Adverse birth outcome among mothers with low serum cholesterol. Pediatrics 2007, 120, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Hoy, W.E.; Nicol, J.L. The Barker hypothesis confirmed: Association of low birth weight with all-cause natural deaths in young adult life in a remote Australian Aboriginal community. J. Dev. Orig. Health Dis. 2019, 10, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, R.; Yang, W.; Xu, H.; Song, R.; Qi, X.; Xu, W. Association of low birth weight with cardiometabolic diseases in Swedish twins: A population-based cohort study. BMJ Open 2021, 11, e048030. [Google Scholar] [CrossRef]
- Catov, J.M.; Ness, R.B.; Wellons, M.F.; Jacobs, D.R.; Roberts, J.M.; Gunderson, E.P. Prepregnancy lipids related to preterm birth risk: The coronary artery risk development in young adults study. J. Clin. Endocrinol. Metab. 2010, 95, 3711–3718. [Google Scholar] [CrossRef]
- Napoli, C.; D’Armiento, F.P.; Mancini, F.P.; Postiglione, A.; Witztum, J.L.; Palumbo, G.; Palinski, W. Fatty streak formation occurs in human fetal aortas and is greatly enhanced by maternal hypercholesterolemia. Intimal accumulation of low density lipoprotein and its oxidation precede monocyte recruitment into early atherosclerotic lesions. J. Clin. Investig. 1997, 100, 2680–2690. [Google Scholar] [CrossRef]
- Versmissen, J.; Botden, I.P.; Huijgen, R.; Oosterveer, D.M.; Defesche, J.C.; Heil, T.C.; Muntz, A.; Langendonk, J.G.; Schinkel, A.F.; Kastelein, J.J.; et al. Maternal inheritance of familial hypercholesterolemia caused by the V408M low-density lipoprotein receptor mutation increases mortality. Atherosclerosis 2011, 219, 690–693. [Google Scholar] [CrossRef]
- Paquette, M.; Fantino, M.; Bernard, S.; Baass, A. Paternal inheritance predicts earlier cardiovascular event onset in patients with familial hypercholesterolemia. Atherosclerosis 2021, 329, 9–13. [Google Scholar] [CrossRef]
- Marco-Benedí, V.; Laclaustra, M.; Bea, A.M.; Suarez-Tembra, M.; Plana, N.; Pinto, X.; Brea, A.; Sanchez-Hernandez, R.M.; Civeira, F. Maternally inherited hypercholesterolemia does not modify the cardiovascular phenotype in familial hypercholesterolemia. Atherosclerosis 2021, 320, 47–52. [Google Scholar] [CrossRef]
- Mendelson, M.M.; Lyass, A.; O’Donnell, C.J.; D’Agostino, R.B., Sr.; Levy, D. Association of Maternal Prepregnancy Dyslipidemia with Adult Offspring Dyslipidemia in Excess of Anthropometric, Lifestyle, and Genetic Factors in the Framingham Heart Study. JAMA Cardiol. 2016, 1, 26–35. [Google Scholar] [CrossRef]
- Narverud, I.; van Lennep, J.R.; Christensen, J.J.; Versmissen, J.; Gran, J.M.; Iversen, P.O.; Aukrust, P.; Halvorsen, B.; Ueland, T.; Ulven, S.M.; et al. Maternal inheritance does not predict cholesterol levels in children with familial hypercholesterolemia. Atherosclerosis 2015, 243, 155–160. [Google Scholar] [CrossRef]
- Mathew, J.; Huang, S.C.; Dumolt, J.H.; Patel, M.S.; Rideout, T.C. Maternal hypercholesterolemia programs dyslipidemia in adult male mouse progeny. Reproduction 2020, 160, 1–10. [Google Scholar] [CrossRef]
- Braun, K.V.E.; Dhana, K.; de Vries, P.S.; Voortman, T.; van Meurs, J.B.J.; Uitterlinden, A.G.; Hofman, A.; Hu, F.B.; Franco, O.H.; Dehghan, A. Epigenome-wide association study (EWAS) on lipids: The Rotterdam Study. Clin. Epigenetics 2017, 9, 15. [Google Scholar] [CrossRef]
- Hedman, Å.K.; Mendelson, M.M.; Marioni, R.E.; Gustafsson, S.; Joehanes, R.; Irvin, M.R.; Zhi, D.; Sandling, J.K.; Yao, C.; Liu, C.; et al. Epigenetic Patterns in Blood Associated with Lipid Traits Predict Incident Coronary Heart Disease Events and Are Enriched for Results from Genome-Wide Association Studies. Circ. Cardiovasc. Genet. 2017, 10, e001487. [Google Scholar] [CrossRef]
- Mittelstraß, K.; Waldenberger, M. DNA methylation in human lipid metabolism and related diseases. Curr. Opin. Lipidol. 2018, 29, 116–124. [Google Scholar] [CrossRef]
- Dekkers, K.F.; Slagboom, P.E.; Jukema, J.W.; Heijmans, B.T. The multifaceted interplay between lipids and epigenetics. Curr. Opin. Lipidol. 2016, 27, 288–294. [Google Scholar] [CrossRef]
- Bekkering, S.; Quintin, J.; Joosten, L.A.; van der Meer, J.W.; Netea, M.G.; Riksen, N.P. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1731–1738. [Google Scholar] [CrossRef]
- Waszak, S.M.; Delaneau, O.; Gschwind, A.R.; Kilpinen, H.; Raghav, S.K.; Witwicki, R.M.; Orioli, A.; Wiederkehr, M.; Panousis, N.I.; Yurovsky, A.; et al. Population Variation and Genetic Control of Modular Chromatin Architecture in Humans. Cell 2015, 162, 1039–1050. [Google Scholar] [CrossRef]
- Yu, C.-E.; Cudaback, E.; Foraker, J.; Thomson, Z.; Leong, L.; Lutz, F.; Gill, J.A.; Saxton, A.; Kraemer, B.; Navas, P.; et al. Epigenetic signature and enhancer activity of the human APOE gene. Hum. Mol. Genet. 2013, 22, 5036–5047. [Google Scholar] [CrossRef]
- Malhotra, P.; Soni, V.; Kumar, A.; Anbazhagan, A.N.; Dudeja, A.; Saksena, S.; Gill, R.K.; Dudeja, P.K.; Alrefai, W.A. Epigenetic Modulation of Intestinal Cholesterol Transporter Niemann-Pick C1-like 1 (NPC1L1) Gene Expression by DNA Methylation*. J. Biol. Chem. 2014, 289, 23132–23140. [Google Scholar] [CrossRef]
- Gomez-Alonso, M.D.C.; Kretschmer, A.; Wilson, R.; Pfeiffer, L.; Karhunen, V.; Seppälä, I.; Zhang, W.; Mittelstraß, K.; Wahl, S.; Matias-Garcia, P.R.; et al. DNA methylation and lipid metabolism: An EWAS of 226 metabolic measures. Clin. Epigenetics 2021, 13, 7. [Google Scholar] [CrossRef]
- Reeskamp, L.F.; Venema, A.; Pereira, J.P.B.; Levin, E.; Nieuwdorp, M.; Groen, A.K.; Defesche, J.C.; Grefhorst, A.; Henneman, P.; Hovingh, G.K. Differential DNA methylation in familial hypercholesterolemia. EBioMedicine 2020, 61, 103079. [Google Scholar] [CrossRef]
- Griffin, B.A.; Walker, C.G.; Jebb, S.A.; Moore, C.; Frost, G.S.; Goff, L.; Sanders, T.A.B.; Lewis, F.; Griffin, M.; Gitau, R.; et al. APOE4 Genotype Exerts Greater Benefit in Lowering Plasma Cholesterol and Apolipoprotein B than Wild Type (E3/E3), after Replacement of Dietary Saturated Fats with Low Glycaemic Index Carbohydrates. Nutrients 2018, 10, 1524. [Google Scholar] [CrossRef]
- Hellstrand, S.; Ericson, U.; Schulz, C.A.; Drake, I.; Gullberg, B.; Hedblad, B.; Engström, G.; Orho-Melander, M.; Sonestedt, E. Genetic susceptibility to dyslipidemia and incidence of cardiovascular disease depending on a diet quality index in the Malmö Diet and Cancer cohort. Genes Nutr. 2016, 11, 20. [Google Scholar] [CrossRef] [PubMed]
- Cranley, J.; MacRae, C.A. A New Approach to an Old Problem: One Brave Idea. Circ. Res. 2018, 122, 1172–1175. [Google Scholar] [CrossRef] [PubMed]
- Benton, M.L.; Abraham, A.; LaBella, A.L.; Abbot, P.; Rokas, A.; Capra, J.A. The influence of evolutionary history on human health and disease. Nat. Rev. Genet. 2021, 22, 269–283. [Google Scholar] [CrossRef] [PubMed]

| Gene | ID | Position 1 | Type | Change 2 | MA 3 | Risk Allele | Effect 4 |
|---|---|---|---|---|---|---|---|
| PRS from Talmud et al., 2013 [129] | |||||||
| PCSK9 | rs2479409 | 1:55,038,977 | 5’-UTR | c.-861G>A | G | G | 0.052 |
| CELSR2 | rs629301 | 1:109,275,684 | 3-’UTR | c.*1635G>T | G | T | 0.146 |
| APOB | rs1367117 | 2:21,041,028 | missense | c.293C>T (p.Thr98Ile) | T | T | 0.105 |
| ABCG8 | rs4299376 | 2:43,845,437 | intronic | c.166-718G>T | G | G | 0.071 |
| HFE | rs1800562 | 6:26,092,913 | missense | c.845G>A (p.Cys282Tyr) | A | G | 0.057 |
| MYLIP | rs3757354 | 6:16,127,176 | upstream | c.-2147C>T | T | C | 0.037 |
| SLC22A1 | rs1564348 | 6:160,157,828 | intronic | c.1599-688T>C | C | T | 0.050 |
| ST3GAL4 | rs11220462 | 11:126,374,057 | intronic | c.-61+18215G>A | A | A | 0.050 |
| NYNRIN | rs8017377 | 14:24,414,681 | missense | c.2932G>A (p.Ala978Thr) | A | A | 0.029 |
| LDLR | rs6511720 | 19:11,091,630 | intronic | c.67+2015G>T | T | G | 0.181 |
| APOE | rs429358 | 19:44,908,684 | missense | c.388T>C (p.Cys130Arg) | C | ||
| APOE | rs7412 | 19:44,908,822 | missense | c.526C>T (p.Arg176Cys) | T | ||
| APOE genotype | E2/E2 | −0.9 | |||||
| E2/E3 | −0.4 | ||||||
| E2/E4 | −0.2 | ||||||
| E3/E3 | 0.0 | ||||||
| E3/E4 | 0.1 | ||||||
| E4/E4 | 0.2 | ||||||
| PRS from Wang et al., 2016 [131] | |||||||
| PCSK9 | rs11206510 | 1:55,030,366 | intergenic | T>C | C | T | 0.090 |
| CELSR2 | rs12740374 | 1:109,274,968 | 3-’UTR | c.*919G>T | T | G | 0.230 |
| APOB | rs515135 | 2:21,063,185 | intergenic | T>C | T | C | 0.160 |
| ABCG8 | rs6544713 | 2:43,846,742 | intronic | c.322+431T>C | T | T | 0.150 |
| HMGCR | rs3846663 | 5:75,359,901 | intronic | c.2458-84C>T | T | T | 0.070 |
| TIMD4 | rs1501908 | 5:156,971,158 | intergenic | C>G | G | C | 0.070 |
| HNF1A | rs2650000 | 12:120,951,159 | intergenic | C>A | A | A | 0.070 |
| LDLR | rs6511720 | 19:11,091,630 | intronic | c.67+2015G>T | T | G | 0.260 |
| SUGP1 (NCAN) | rs10401969 | 19:19,296,909 | intronic | c.1243+80T>C | C | T | 0.050 |
| MAFB | rs6102059 | 20:40,600,144 | intergenic | C>T | T | C | 0.060 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martín-Campos, J.M. Genetic Determinants of Plasma Low-Density Lipoprotein Cholesterol Levels: Monogenicity, Polygenicity, and “Missing” Heritability. Biomedicines 2021, 9, 1728. https://doi.org/10.3390/biomedicines9111728
Martín-Campos JM. Genetic Determinants of Plasma Low-Density Lipoprotein Cholesterol Levels: Monogenicity, Polygenicity, and “Missing” Heritability. Biomedicines. 2021; 9(11):1728. https://doi.org/10.3390/biomedicines9111728
Chicago/Turabian StyleMartín-Campos, Jesús Maria. 2021. "Genetic Determinants of Plasma Low-Density Lipoprotein Cholesterol Levels: Monogenicity, Polygenicity, and “Missing” Heritability" Biomedicines 9, no. 11: 1728. https://doi.org/10.3390/biomedicines9111728
APA StyleMartín-Campos, J. M. (2021). Genetic Determinants of Plasma Low-Density Lipoprotein Cholesterol Levels: Monogenicity, Polygenicity, and “Missing” Heritability. Biomedicines, 9(11), 1728. https://doi.org/10.3390/biomedicines9111728