
Dysfunction in the Cystic Fibrosis Transmembrane Regulator in Chronic Obstructive Pulmonary Disease as a Potential Target for Personalised Medicine



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. CFTR: Structure and Function
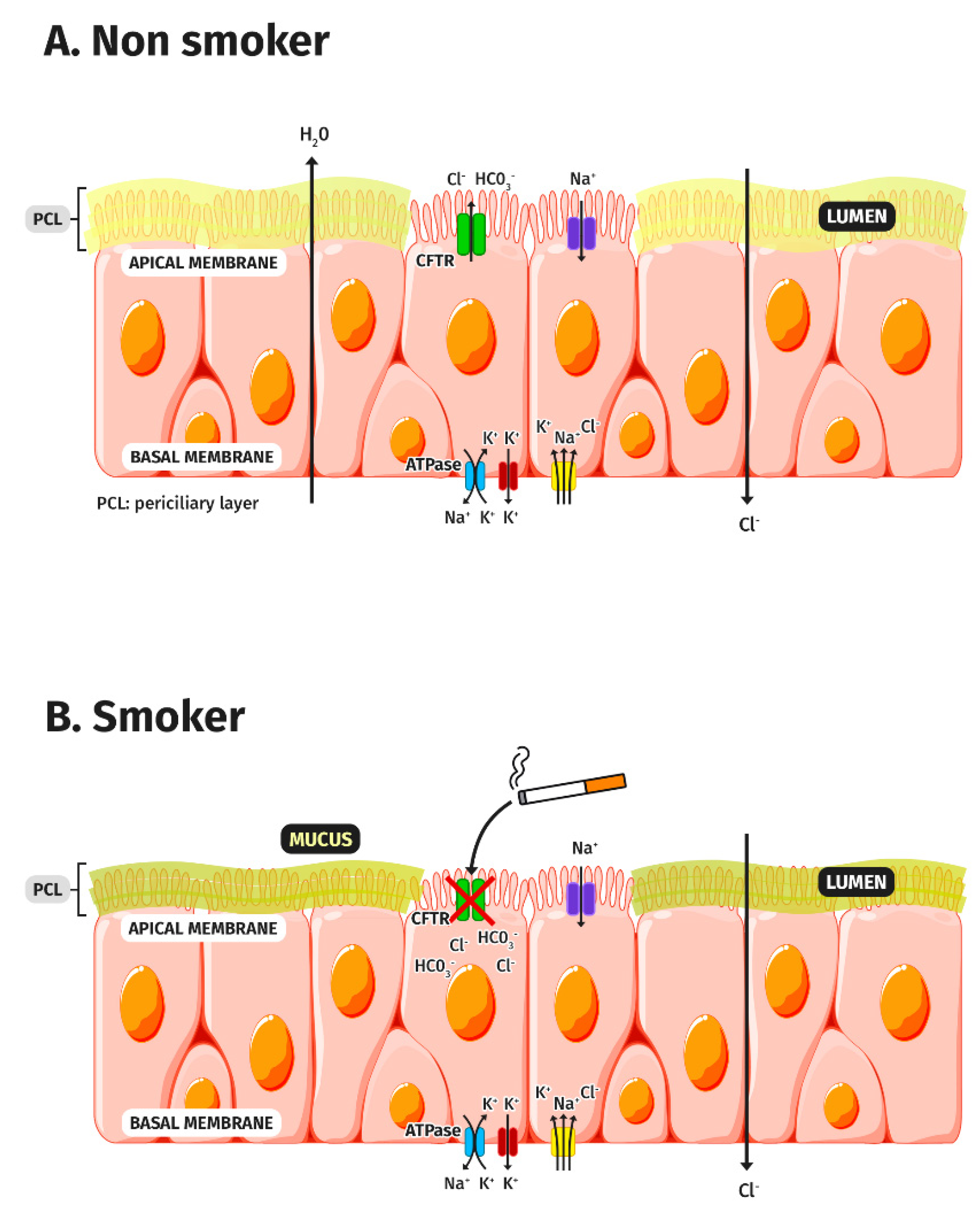
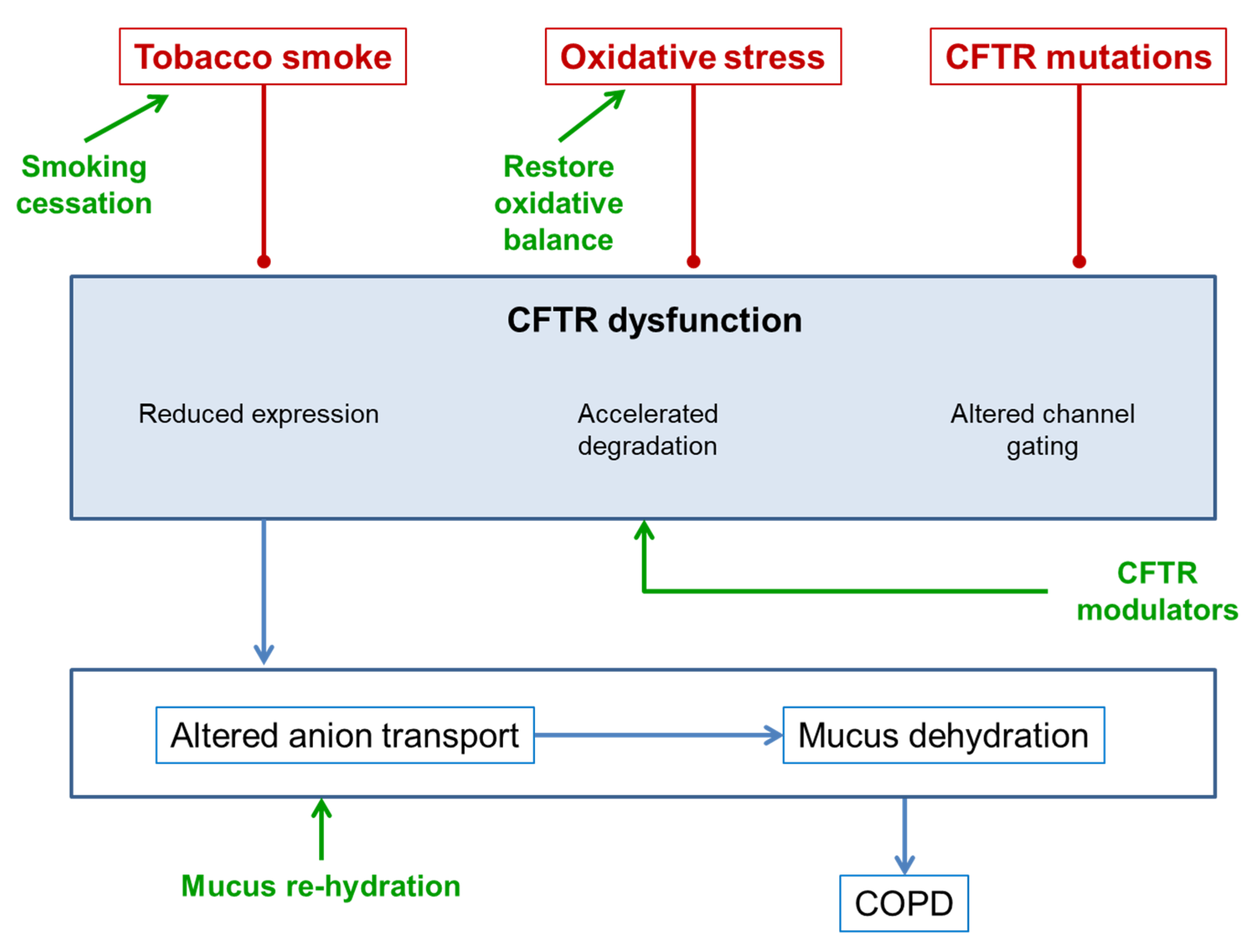
3. CFTR Dysfunction in COPD
3.1. CFTR and Tobacco Smoke
3.2. CFTR and Oxidative Stress
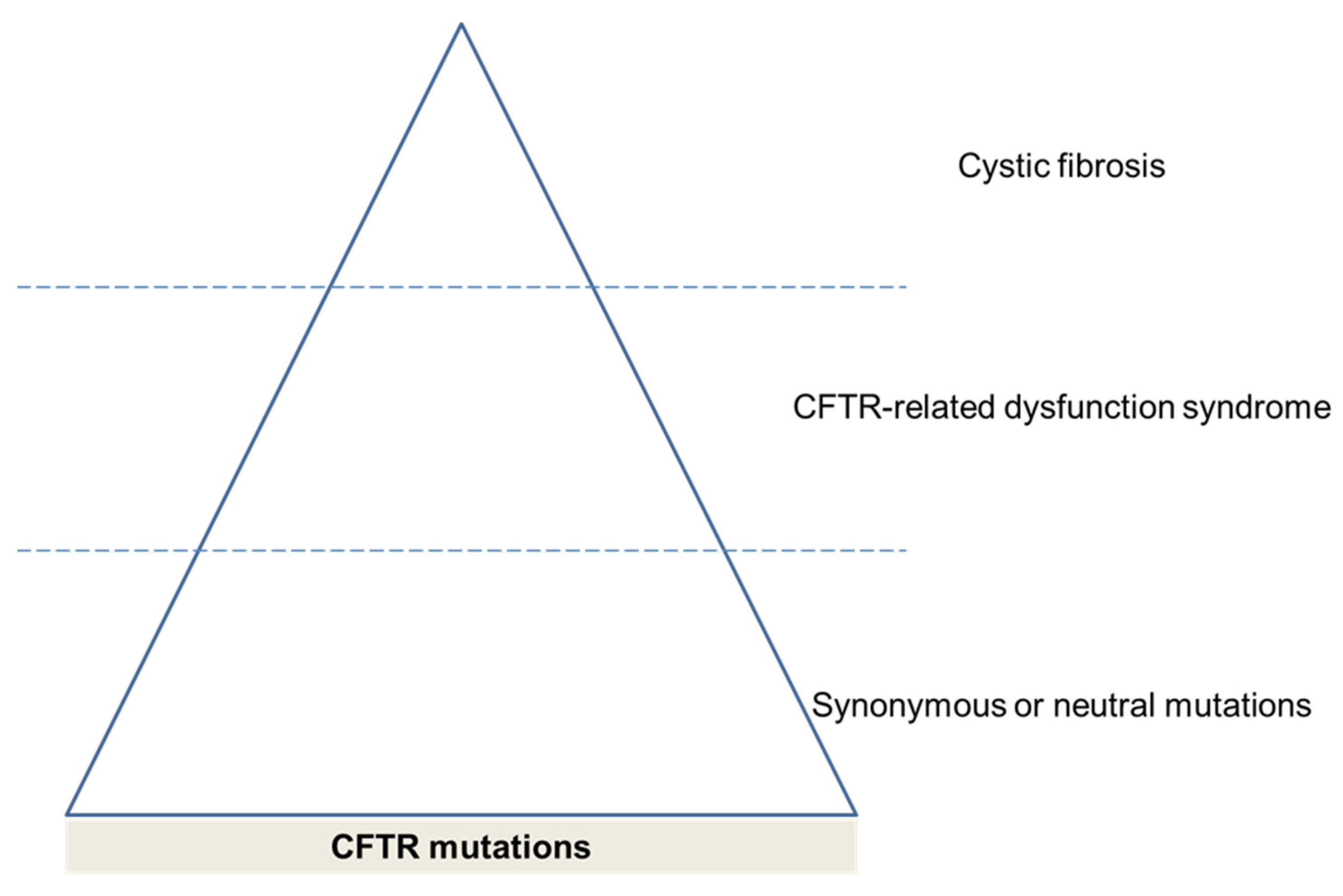
3.3. CFTR Mutations
4. Consequences of CFTR Dysfunction in COPD
5. Treatments for Improving CFTR Function in COPD
5.1. Smoking Cessation
5.2. Rehydration of Mucus
5.3. Antioxidants
5.4. Phosphodiesterase Inhibitors
6. CFTR Modulators
6.1. Ivacaftor and COPD
6.2. Icenticaftor and COPD
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Capistrano, S.J.; van Reyk, D.; Chen, H.; Oliver, B.G. Evidence of biomass smoke exposure as a causative factor for the development of copd. Toxics 2017, 5, 36. [Google Scholar] [CrossRef]
- Kraim-Leleu, M.; Lesage, F.X.; Drame, M.; Lebargy, F.; Deschamps, F. Occupational risk factors for copd: A case-control study. PLoS ONE 2016, 11, e0158719. [Google Scholar] [CrossRef] [PubMed]
- López-Campos, J.L.; Calle Rubio, M.; Izquierdo Alonso, J.L.; Fernández-Villar, A.; Abascal-Bolado, B.; Alcázar, B.; García-Río, F.; Peces-Barba, G.; Serra Batlles, J.; Martínez Garcerán, J.J.; et al. Forum copd working group consensus on the diagnosis, treatment and follow-up of copd. Arch. Bronconeumol. 2021, 57, 596–599. [Google Scholar] [CrossRef] [PubMed]
- Miravitlles, M.; Calle, M.; Molina, J.; Almagro, P.; Gomez, J.T.; Trigueros, J.A.; Cosio, B.G.; Casanova, C.; Lopez-Campos, J.L.; Riesco, J.A.; et al. Spanish copd guidelines (gesepoc) 2021: Updated pharmacological treatment of stable copd. Arch. Bronconeumol. 2021, in press. [Google Scholar] [CrossRef]
- Celli, B.R.; Decramer, M.; Wedzicha, J.A.; Wilson, K.C.; Agusti, A.A.; Criner, G.J.; MacNee, W.; Make, B.J.; Rennard, S.I.; Stockley, R.A.; et al. An official american thoracic society/european respiratory society statement: Research questions in copd. Eur. Respir. Rev. 2015, 24, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Agusti, A.; Faner, R. Chronic obstructive pulmonary disease pathogenesis. Clin. Chest Med. 2020, 41, 307–314. [Google Scholar] [CrossRef]
- Fernandez Fernandez, E.; De Santi, C.; De Rose, V.; Greene, C.M. CFTR dysfunction in cystic fibrosis and chronic obstructive pulmonary disease. Expert Rev. Respir. Med. 2018, 12, 483–492. [Google Scholar] [CrossRef]
- Rowe, S.M.; Jones, I.; Dransfield, M.T.; Haque, N.; Gleason, S.; Hayes, K.A.; Kulmatycki, K.; Yates, D.P.; Danahay, H.; Gosling, M.; et al. Efficacy and safety of the CFTR potentiator icenticaftor (qbw251) in copd: Results from a phase 2 randomized trial. Int. J. Chron. Obstruct. Pulmon. Dis. 2020, 15, 2399–2409. [Google Scholar] [CrossRef]
- Kunzelmann, K.; Mehta, A. CFTR: A hub for kinases and crosstalk of cAMP and Ca2+. FEBS J. 2013, 280, 4417–4429. [Google Scholar] [CrossRef]
- Cantin, A.M.; Hartl, D.; Konstan, M.W.; Chmiel, J.F. Inflammation in cystic fibrosis lung disease: Pathogenesis and therapy. J. Cyst. Fibros. 2015, 14, 419–430. [Google Scholar] [CrossRef]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.L.; et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef]
- Mingora, C.M.; Flume, P.A. Pulmonary complications in cystic fibrosis: Past, present and future. Chest 2021, 160, 1232–1240. [Google Scholar] [CrossRef] [PubMed]
- Csanády, L.; Vergani, P.; Gadsby, D.C. Structure, gating, and regulation of the CFTR anion channel. Physiol. Rev. 2019, 99, 707–738. [Google Scholar] [CrossRef] [PubMed]
- Stutts, M.J.; Canessa, C.M.; Olsen, J.C.; Hamrick, M.; Cohn, J.A.; Rossier, B.C.; Boucher, R.C. CFTR as a cAMP-dependent regulator of sodium channels. Science 1995, 269, 847–850. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Li, H.; Yuan, C.; Luo, M.; Wei, J.; Liu, X. Cigarette smoke-induced acquired dysfunction of cystic fibrosis transmembrane conductance regulator in the pathogenesis of chronic obstructive pulmonary disease. Oxid Med. Cell. Longev. 2018, 2018, 6567578. [Google Scholar] [CrossRef]
- Welsh, M.J. Cigarette smoke inhibition of ion transport in canine tracheal epithelium. J. Clin. Investig. 1983, 71, 1614–1623. [Google Scholar] [CrossRef] [PubMed]
- Clunes, L.A.; Davies, C.M.; Coakley, R.D.; Aleksandrov, A.A.; Henderson, A.G.; Zeman, K.L.; Worthington, E.N.; Gentzsch, M.; Kreda, S.M.; Cholon, D.; et al. Cigarette smoke exposure induces CFTR internalization and insolubility, leading to airway surface liquid dehydration. FASEB J. 2012, 26, 533–545. [Google Scholar] [CrossRef]
- Marklew, A.J.; Patel, W.; Moore, P.J.; Tan, C.D.; Smith, A.J.; Sassano, M.F.; Gray, M.A.; Tarran, R. Cigarette smoke exposure induces retrograde trafficking of CFTR to the endoplasmic reticulum. Sci. Rep. 2019, 9, 13655. [Google Scholar] [CrossRef]
- Rasmussen, J.E.; Sheridan, J.T.; Polk, W.; Davies, C.M.; Tarran, R. Cigarette smoke-induced Ca2+ release leads to cystic fibrosis transmembrane conductance regulator (CFTR) dysfunction. J. Biol. Chem. 2014, 289, 7671–7681. [Google Scholar] [CrossRef] [PubMed]
- Younger, J.M.; Chen, L.; Ren, H.Y.; Rosser, M.F.; Turnbull, E.L.; Fan, C.Y.; Patterson, C.; Cyr, D.M. Sequential quality-control checkpoints triage misfolded cystic fibrosis transmembrane conductance regulator. Cell 2006, 126, 571–582. [Google Scholar] [CrossRef]
- Dransfield, M.T.; Wilhelm, A.M.; Flanagan, B.; Courville, C.; Tidwell, S.L.; Raju, S.V.; Gaggar, A.; Steele, C.; Tang, L.P.; Liu, B.; et al. Acquired cystic fibrosis transmembrane conductance regulator dysfunction in the lower airways in copd. Chest 2013, 144, 498–506. [Google Scholar] [CrossRef]
- Raju, S.V.; Jackson, P.L.; Courville, C.A.; McNicholas, C.M.; Sloane, P.A.; Sabbatini, G.; Tidwell, S.; Tang, L.P.; Liu, B.; Fortenberry, J.A.; et al. Cigarette smoke induces systemic defects in cystic fibrosis transmembrane conductance regulator function. Am. J. Respir. Crit. Care Med. 2013, 188, 1321–1330. [Google Scholar] [CrossRef]
- Stevens, J.F.; Maier, C.S. Acrolein: Sources, metabolism, and biomolecular interactions relevant to human health and disease. Mol. Nutr. Food Res. 2008, 52, 7–25. [Google Scholar] [CrossRef]
- Alexander, N.S.; Blount, A.; Zhang, S.; Skinner, D.; Hicks, S.B.; Chestnut, M.; Kebbel, F.A.; Sorscher, E.J.; Woodworth, B.A. Cystic fibrosis transmembrane conductance regulator modulation by the tobacco smoke toxin acrolein. Laryngoscope 2012, 122, 1193–1197. [Google Scholar] [CrossRef] [PubMed]
- Hassan, F.; Xu, X.; Nuovo, G.; Killilea, D.W.; Tyrrell, J.; Da Tan, C.; Tarran, R.; Diaz, P.; Jee, J.; Knoell, D.; et al. Accumulation of metals in gold4 copd lungs is associated with decreased CFTR levels. Respir. Res. 2014, 15, 69. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Larrauri, A.; Presa, N.; Dominguez-Herrera, A.; Ouro, A.; Trueba, M.; Gomez-Muñoz, A. Role of bioactive sphingolipids in physiology and pathology. Essays Biochem. 2020, 64, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Bodas, M.; Min, T.; Mazur, S.; Vij, N. Critical modifier role of membrane-cystic fibrosis transmembrane conductance regulator-dependent ceramide signaling in lung injury and emphysema. J. Immunol. 2011, 186, 602–613. [Google Scholar] [CrossRef]
- Newsholme, P.; Cruzat, V.F.; Keane, K.N.; Carlessi, R.; de Bittencourt, P.I., Jr. Molecular mechanisms of ros production and oxidative stress in diabetes. Biochem. J. 2016, 473, 4527–4550. [Google Scholar] [CrossRef]
- Brieger, K.; Schiavone, S.; Miller, F.J., Jr.; Krause, K.H. Reactive oxygen species: From health to disease. Swiss Med. Wkly. 2012, 142, w13659. [Google Scholar] [CrossRef]
- Xu, X.; Huang, H.; Yin, X.; Fang, H.; Shen, X. Effect of lentivirus-mediated CFTR overexpression on oxidative stress injury and inflammatory response in the lung tissue of copd mouse model. Biosci. Rep. 2020, 40, BSR20193667. [Google Scholar] [CrossRef]
- Cantin, A.M.; Bilodeau, G.; Ouellet, C.; Liao, J.; Hanrahan, J.W. Oxidant stress suppresses CFTR expression. Am. J. Physiol. Cell Physiol. 2006, 290, C262–C270. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, Z.; Leir, S.H.; Harris, A. Oxidative stress regulates CFTR gene expression in human airway epithelial cells through a distal antioxidant response element. Am. J. Respir. Cell Mol. Biol. 2015, 52, 387–396. [Google Scholar] [CrossRef]
- Braun, A.P. Cigarette smoke and calcium conspire to impair CFTR function in airway epithelia. Channels 2014, 8, 172–173. [Google Scholar] [CrossRef][Green Version]
- O′Grady, S.M. Oxidative stress, autophagy and airway ion transport. Am. J. Physiol. Cell Physiol. 2019, 316, C16–C32. [Google Scholar] [CrossRef]
- Kunzi, L.; Easter, M.; Hirsch, M.J.; Krick, S. Cystic fibrosis lung disease in the aging population. Front. Pharmacol. 2021, 12, 601438. [Google Scholar] [CrossRef]
- Asensio-Cruz, M.I.; Calero-Acuña, C.; Arellano-Orden, E.; Sánchez-López, V.; Caballero-Eraso, C.; Cejudo, P.; Lopez-Villalobos, J.L.; Lopez-Campos, J.L.; Ortega-Ruiz, F.; Sánchez-Armengol, Á. Differences in overexpression of hypoxia-induced transcription factors and associated biomarkers in three different types of chronic hypoxia. Arch. Bronconeumol. 2020, 57, 555–556. [Google Scholar] [CrossRef]
- Bombieri, C.; Claustres, M.; De Boeck, K.; Derichs, N.; Dodge, J.; Girodon, E.; Sermet, I.; Schwarz, M.; Tzetis, M.; Wilschanski, M.; et al. Recommendations for the classification of diseases as CFTR-related disorders. J. Cyst. Fibros. 2011, 10 (Suppl. 2), S86–S102. [Google Scholar] [CrossRef]
- Pignatti, P.F.; Bombieri, C.; Marigo, C.; Benetazzo, M.; Luisetti, M. Increased incidence of cystic fibrosis gene mutations in adults with disseminated bronchiectasis. Hum. Mol. Genet. 1995, 4, 635–639. [Google Scholar] [CrossRef]
- Tzetis, M.; Efthymiadou, A.; Strofalis, S.; Psychou, P.; Dimakou, A.; Pouliou, E.; Doudounakis, S.; Kanavakis, E. CFTR gene mutations--including three novel nucleotide substitutions--and haplotype background in patients with asthma, disseminated bronchiectasis and chronic obstructive pulmonary disease. Hum. Genet. 2001, 108, 216–221. [Google Scholar] [CrossRef]
- Divac, A.; Nikolic, A.; Mitic-Milikic, M.; Nagorni-Obradovic, L.; Petrovic-Stanojevic, N.; Dopudja-Pantic, V.; Nadaskic, R.; Savic, A.; Radojkovic, D. High frequency of the r75q CFTR variation in patients with chronic obstructive pulmonary disease. J. Cyst. Fibros. 2004, 3, 189–191. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Stankovic, M.; Nikolic, A.; Divac, A.; Tomovic, A.; Petrovic-Stanojevic, N.; Andjelic, M.; Dopudja-Pantic, V.; Surlan, M.; Vujicic, I.; Ponomarev, D.; et al. The CFTR m470v gene variant as a potential modifier of copd severity: Study of serbian population. Genet. Test. 2008, 12, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Cuppens, H.; Lin, W.; Jaspers, M.; Costes, B.; Teng, H.; Vankeerberghen, A.; Jorissen, M.; Droogmans, G.; Reynaert, I.; Goossens, M.; et al. Polyvariant mutant cystic fibrosis transmembrane conductance regulator genes. The polymorphic (tg)m locus explains the partial penetrance of the t5 polymorphism as a disease mutation. J. Clin. Investig. 1998, 101, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Courville, C.A.; Tidwell, S.; Liu, B.; Accurso, F.J.; Dransfield, M.T.; Rowe, S.M. Acquired defects in CFTR-dependent beta-adrenergic sweat secretion in chronic obstructive pulmonary disease. Respir. Res. 2014, 15, 25. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ni, I.; Ji, C.; Vij, N. Second-hand cigarette smoke impairs bacterial phagocytosis in macrophages by modulating CFTR dependent lipid-rafts. PLoS ONE 2015, 10, e0121200. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Liang, X.; Zhao, M.; Liu, S.L.; Huang, Y.; Idell, S.; Li, X.; Ji, H.L. Correlation of apical fluid-regulating channel proteins with lung function in human copd lungs. PLoS ONE 2014, 9, e109725. [Google Scholar] [CrossRef] [PubMed]
- Åstrand, A.B.; Hemmerling, M.; Root, J.; Wingren, C.; Pesic, J.; Johansson, E.; Garland, A.L.; Ghosh, A.; Tarran, R. Linking increased airway hydration, ciliary beating, and mucociliary clearance through enac inhibition. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, L22–L32. [Google Scholar] [CrossRef]
- Wellmerling, J.H.; Chang, S.W.; Kim, E.; Osman, W.H.; Boyaka, P.N.; Borchers, M.T.; Cormet-Boyaka, E. Reduced expression of the ion channel CFTR contributes to airspace enlargement as a consequence of aging and in response to cigarette smoke in mice. Respir. Res. 2019, 20, 200. [Google Scholar] [CrossRef]
- Li, Q.Y.; Huang, S.G.; Wan, H.Y.; Wu, H.C.; Zhou, T.; Li, M.; Deng, W.W. Effect of smoking cessation on airway inflammation of rats with chronic bronchitis. Chin. Med. J. 2007, 120, 1511–1516. [Google Scholar] [CrossRef]
- Rutgers, S.R.; Postma, D.S.; ten Hacken, N.H.; Kauffman, H.F.; van Der Mark, T.W.; Koeter, G.H.; Timens, W. Ongoing airway inflammation in patients with copd who do not currently smoke. Thorax 2000, 55, 12–18. [Google Scholar] [CrossRef]
- López-Campos, J.L.; Jiménez-Ruiz, C.A.; Meneses Petersen, E.D.; Rabade Castedo, C.; Asensio Sánchez, S.; Vaquero Lozano, P.; Ferrer Espinosa, S.; Pérez Soriano, M.D.P.; de Higes Martínez, E.; García de Llanos, C.; et al. Smoking cessation units as a source of copd diagnoses: Project 1000-200. Arch. Bronconeumol. 2021. [Google Scholar] [CrossRef]
- Cabrera López, C.; Gómez Sáenz, J.T.; Molina París, J.; Trigueros Carrero, J.A.; López-Campos, J.L. Enabling a community approach to respiratory diseases: The hacer copd project. Arch. Bronconeumol. 2021, 57, 442–444. [Google Scholar] [CrossRef]
- Mall, M.A. Unplugging mucus in cystic fibrosis and chronic obstructive pulmonary disease. Ann. Am. Thorac Soc. 2016, 13 (Suppl. 2), S177–S185. [Google Scholar] [CrossRef]
- Daviskas, E.; Robinson, M.; Anderson, S.D.; Bye, P.T. Osmotic stimuli increase clearance of mucus in patients with mucociliary dysfunction. J. Aerosol Med. 2002, 15, 331–341. [Google Scholar] [CrossRef]
- Mall, M.A.; Hartl, D. CFTR: Cystic fibrosis and beyond. Eur. Respir. J. 2014, 44, 1042–1054. [Google Scholar] [CrossRef]
- Donaldson, S.H.; Bennett, W.D.; Zeman, K.L.; Knowles, M.R.; Tarran, R.; Boucher, R.C. Mucus clearance and lung function in cystic fibrosis with hypertonic saline. N. Engl. J. Med. 2006, 354, 241–250. [Google Scholar] [CrossRef]
- Decramer, M.; Rutten-van Molken, M.; Dekhuijzen, P.N.; Troosters, T.; van Herwaarden, C.; Pellegrino, R.; van Schayck, C.P.; Olivieri, D.; Del Donno, M.; De Backer, W.; et al. Effects of n-acetylcysteine on outcomes in chronic obstructive pulmonary disease (bronchitis randomized on nac cost-utility study, broncus): A randomised placebo-controlled trial. Lancet 2005, 365, 1552–1560. [Google Scholar] [CrossRef]
- Zheng, J.P.; Kang, J.; Huang, S.G.; Chen, P.; Yao, W.Z.; Yang, L.; Bai, C.X.; Wang, C.Z.; Wang, C.; Chen, B.Y.; et al. Effect of carbocisteine on acute exacerbation of chronic obstructive pulmonary disease (peace study): A randomised placebo-controlled study. Lancet 2008, 371, 2013–2018. [Google Scholar] [CrossRef]
- Mroz, M.S.; Harvey, B.J. Ursodeoxycholic acid inhibits enac and na/k pump activity to restore airway surface liquid height in cystic fibrosis bronchial epithelial cells. Steroids 2019, 151, 108461. [Google Scholar] [CrossRef]
- Vij, N. Nano-based rescue of dysfunctional autophagy in chronic obstructive lung diseases. Expert Opin. Drug Deliv. 2017, 14, 483–489. [Google Scholar] [CrossRef]
- Sun, X.; Qiu, J.; Strong, S.A.; Green, L.S.; Wasley, J.W.; Blonder, J.P.; Colagiovanni, D.B.; Mutka, S.C.; Stout, A.M.; Richards, J.P.; et al. Discovery of potent and novel s-nitrosoglutathione reductase inhibitors devoid of cytochrome p450 activities. Bioorg. Med. Chem. Lett. 2011, 21, 5849–5853. [Google Scholar] [CrossRef]
- Bodas, M.; Silverberg, D.; Walworth, K.; Brucia, K.; Vij, N. Augmentation of s-nitrosoglutathione controls cigarette smoke-induced inflammatory-oxidative stress and chronic obstructive pulmonary disease-emphysema pathogenesis by restoring cystic fibrosis transmembrane conductance regulator function. Antioxid. Redox Signal. 2017, 27, 433–451. [Google Scholar] [CrossRef]
- Bodas, M.; Pehote, G.; Silverberg, D.; Gulbins, E.; Vij, N. Autophagy augmentation alleviates cigarette smoke-induced CFTR-dysfunction, ceramide-accumulation and copd-emphysema pathogenesis. Free Radic. Biol. Med. 2019, 131, 81–97. [Google Scholar] [CrossRef]
- Zaman, K.; Sawczak, V.; Zaidi, A.; Butler, M.; Bennett, D.; Getsy, P.; Zeinomar, M.; Greenberg, Z.; Forbes, M.; Rehman, S.; et al. Augmentation of CFTR maturation by s-nitrosoglutathione reductase. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 310, L263–L270. [Google Scholar] [CrossRef]
- Nguyen, J.P.; Bianca, M.; Huff, R.D.; Tiessen, N.; Inman, M.D.; Hirota, J.A. Modulation of cAMP metabolism for CFTR potentiation in human airway epithelial cells. Sci. Rep. 2021, 11, 904. [Google Scholar] [CrossRef]
- Padda, I.S.; Tripp, J. Phosphodiesterase inhibitors. In Statpearls; StatPearls Publishing LLC: Treasure Island, FL, USA, 2021. [Google Scholar]
- Lam, J.; Tonnu-Mihara, I.; Kenyon, N.J.; Kuhn, B.T. Comparative effectiveness of roflumilast and azithromycin for the treatment of chronic obstructive pulmonary disease. Chronic Obstr. Pulm. Dis. 2021. [Google Scholar] [CrossRef]
- Martin, C.; Burgel, P.R.; Roche, N. Inhaled dual phosphodiesterase 3/4 inhibitors for the treatment of patients with copd: A short review. Int. J. Chron. Obstruct. Pulmon. Dis. 2021, 16, 2363–2373. [Google Scholar] [CrossRef]
- Lambert, J.A.; Raju, S.V.; Tang, L.P.; McNicholas, C.M.; Li, Y.; Courville, C.A.; Farris, R.F.; Coricor, G.E.; Smoot, L.H.; Mazur, M.M.; et al. Cystic fibrosis transmembrane conductance regulator activation by roflumilast contributes to therapeutic benefit in chronic bronchitis. Am. J. Respir. Cell Mol. Biol. 2014, 50, 549–558. [Google Scholar] [CrossRef]
- Schmid, A.; Baumlin, N.; Ivonnet, P.; Dennis, J.S.; Campos, M.; Krick, S.; Salathe, M. Roflumilast partially reverses smoke-induced mucociliary dysfunction. Respir. Res. 2015, 16, 135. [Google Scholar] [CrossRef] [PubMed]
- Raju, S.V.; Rasmussen, L.; Sloane, P.A.; Tang, L.P.; Libby, E.F.; Rowe, S.M. Roflumilast reverses CFTR-mediated ion transport dysfunction in cigarette smoke-exposed mice. Respir. Res. 2017, 18, 173. [Google Scholar] [CrossRef] [PubMed]
- Tyrrell, J.; Qian, X.; Freire, J.; Tarran, R. Roflumilast combined with adenosine increases mucosal hydration in human airway epithelial cultures after cigarette smoke exposure. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, L1068–L1077. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Patel, S.D.; Bono, T.R.; Rowe, S.M.; Solomon, G.M. CFTR targeted therapies: Recent advances in cystic fibrosis and possibilities in other diseases of the airways. Eur. Respir. Rev. 2020, 29, 190068. [Google Scholar] [CrossRef]
- Spanò, V.; Venturini, A.; Genovese, M.; Barreca, M.; Raimondi, M.V.; Montalbano, A.; Galietta, L.J.V.; Barraja, P. Current development of CFTR potentiators in the last decade. Eur. J. Med. Chem. 2020, 204, 112631. [Google Scholar] [CrossRef]
- Sloane, P.A.; Shastry, S.; Wilhelm, A.; Courville, C.; Tang, L.P.; Backer, K.; Levin, E.; Raju, S.V.; Li, Y.; Mazur, M.; et al. A pharmacologic approach to acquired cystic fibrosis transmembrane conductance regulator dysfunction in smoking related lung disease. PLoS ONE 2012, 7, e39809. [Google Scholar] [CrossRef] [PubMed]
- Raju, S.V.; Lin, V.Y.; Liu, L.; McNicholas, C.M.; Karki, S.; Sloane, P.A.; Tang, L.; Jackson, P.L.; Wang, W.; Wilson, L.; et al. The cystic fibrosis transmembrane conductance regulator potentiator ivacaftor augments mucociliary clearance abrogating cystic fibrosis transmembrane conductance regulator inhibition by cigarette smoke. Am. J. Respir. Cell Mol. Biol. 2017, 56, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Solomon, G.M.; Hathorne, H.; Liu, B.; Raju, S.V.; Reeves, G.; Acosta, E.P.; Dransfield, M.T.; Rowe, S.M. Pilot evaluation of ivacaftor for chronic bronchitis. Lancet Respir. Med. 2016, 4, e32–e33. [Google Scholar] [CrossRef]
- Grand, D.L.; Gosling, M.; Baettig, U.; Bahra, P.; Bala, K.; Brocklehurst, C.; Budd, E.; Butler, R.; Cheung, A.K.; Choudhury, H.; et al. Discovery of icenticaftor (qbw251), a cystic fibrosis transmembrane conductance regulator potentiator with clinical efficacy in cystic fibrosis and chronic obstructive pulmonary disease. J. Med. Chem. 2021, 64, 7241–7260. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carrasco-Hernández, L.; Quintana-Gallego, E.; Calero, C.; Reinoso-Arija, R.; Ruiz-Duque, B.; López-Campos, J.L. Dysfunction in the Cystic Fibrosis Transmembrane Regulator in Chronic Obstructive Pulmonary Disease as a Potential Target for Personalised Medicine. Biomedicines 2021, 9, 1437. https://doi.org/10.3390/biomedicines9101437
Carrasco-Hernández L, Quintana-Gallego E, Calero C, Reinoso-Arija R, Ruiz-Duque B, López-Campos JL. Dysfunction in the Cystic Fibrosis Transmembrane Regulator in Chronic Obstructive Pulmonary Disease as a Potential Target for Personalised Medicine. Biomedicines. 2021; 9(10):1437. https://doi.org/10.3390/biomedicines9101437
Chicago/Turabian StyleCarrasco-Hernández, Laura, Esther Quintana-Gallego, Carmen Calero, Rocío Reinoso-Arija, Borja Ruiz-Duque, and José Luis López-Campos. 2021. "Dysfunction in the Cystic Fibrosis Transmembrane Regulator in Chronic Obstructive Pulmonary Disease as a Potential Target for Personalised Medicine" Biomedicines 9, no. 10: 1437. https://doi.org/10.3390/biomedicines9101437
APA StyleCarrasco-Hernández, L., Quintana-Gallego, E., Calero, C., Reinoso-Arija, R., Ruiz-Duque, B., & López-Campos, J. L. (2021). Dysfunction in the Cystic Fibrosis Transmembrane Regulator in Chronic Obstructive Pulmonary Disease as a Potential Target for Personalised Medicine. Biomedicines, 9(10), 1437. https://doi.org/10.3390/biomedicines9101437