
The Desmin Mutation DES-c.735G>C Causes Severe Restrictive Cardiomyopathy by Inducing In-Frame Skipping of Exon-3

 ,
,  , , , ,
, , , , 
Abstract
:1. Introduction
2. Materials and Methods
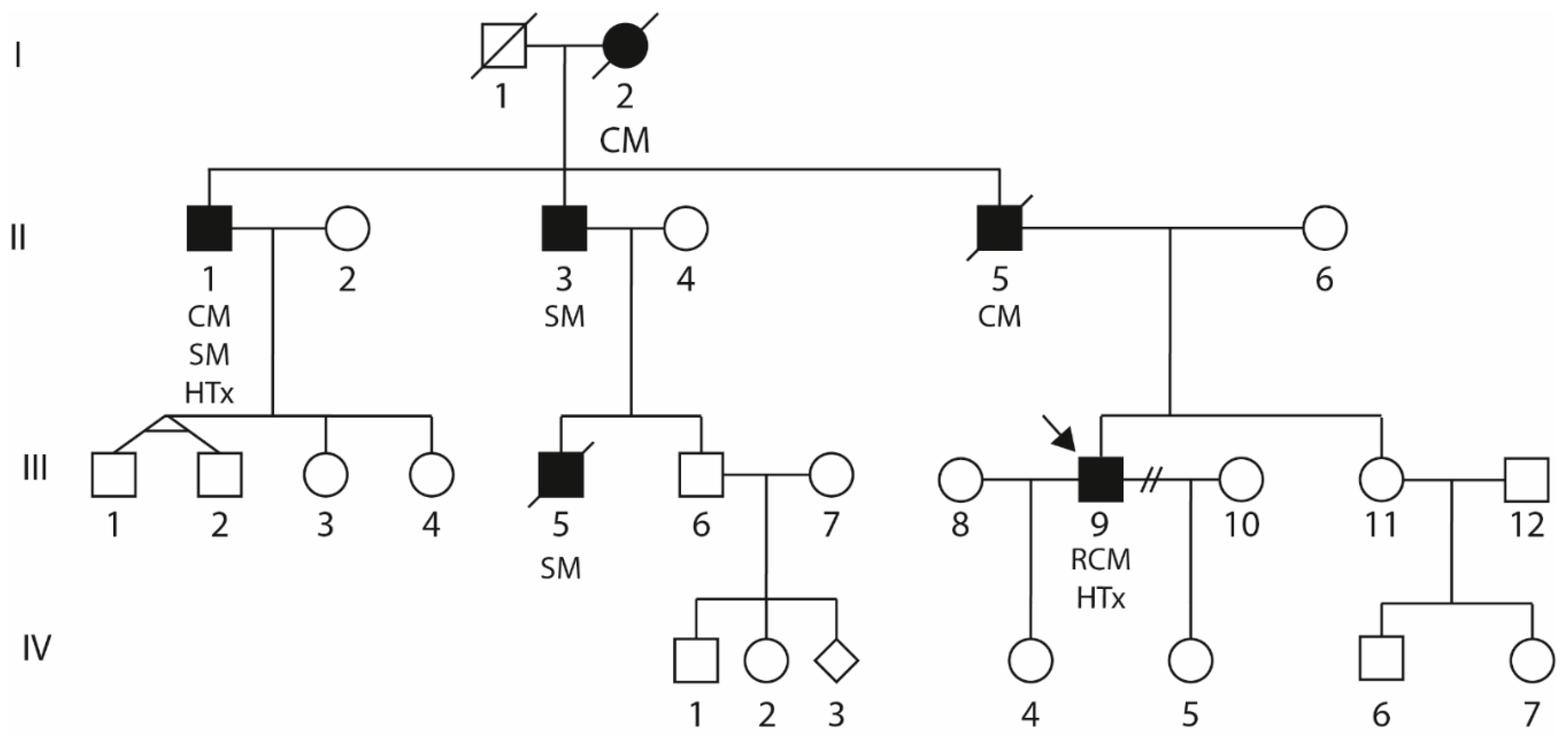
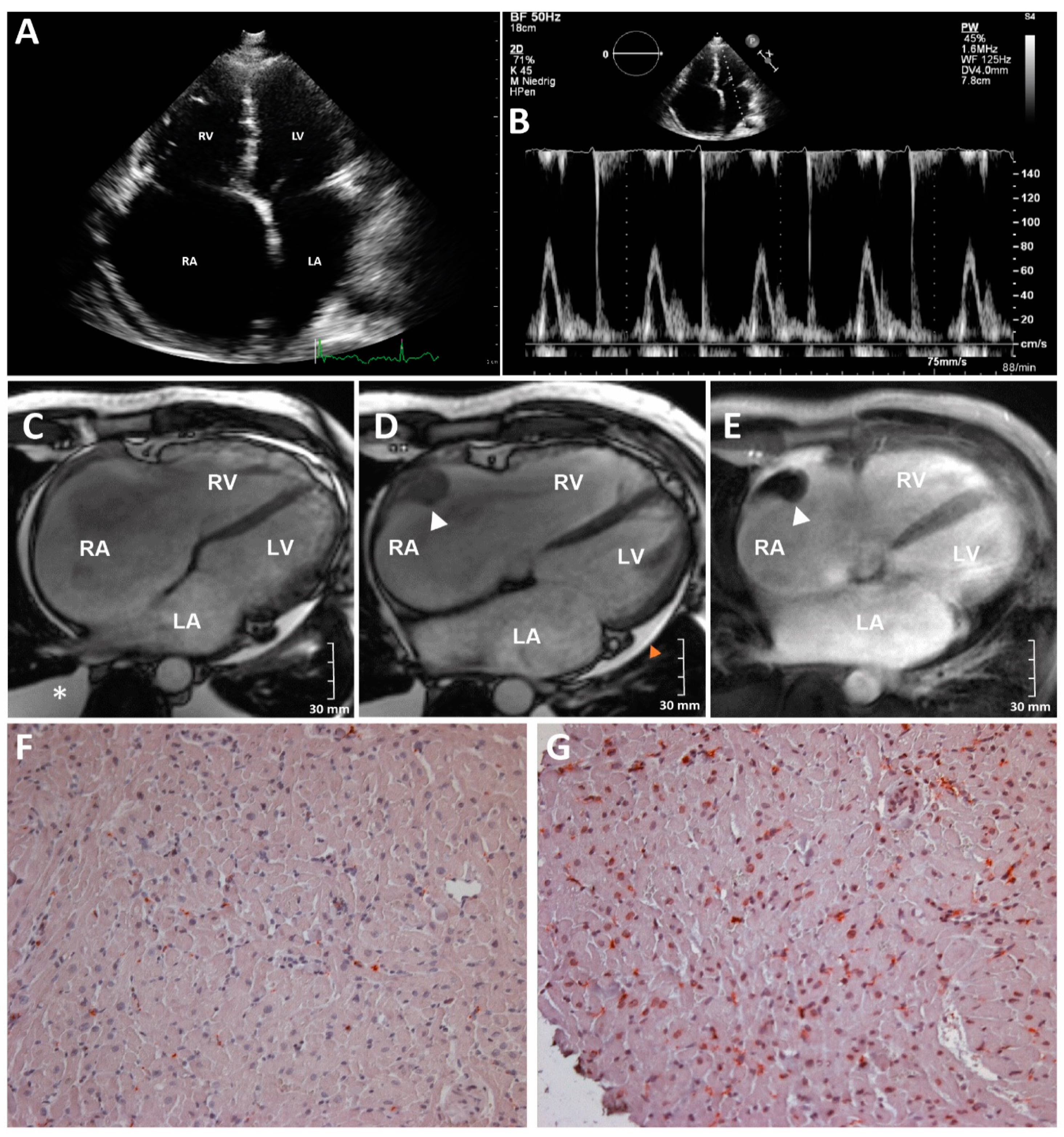
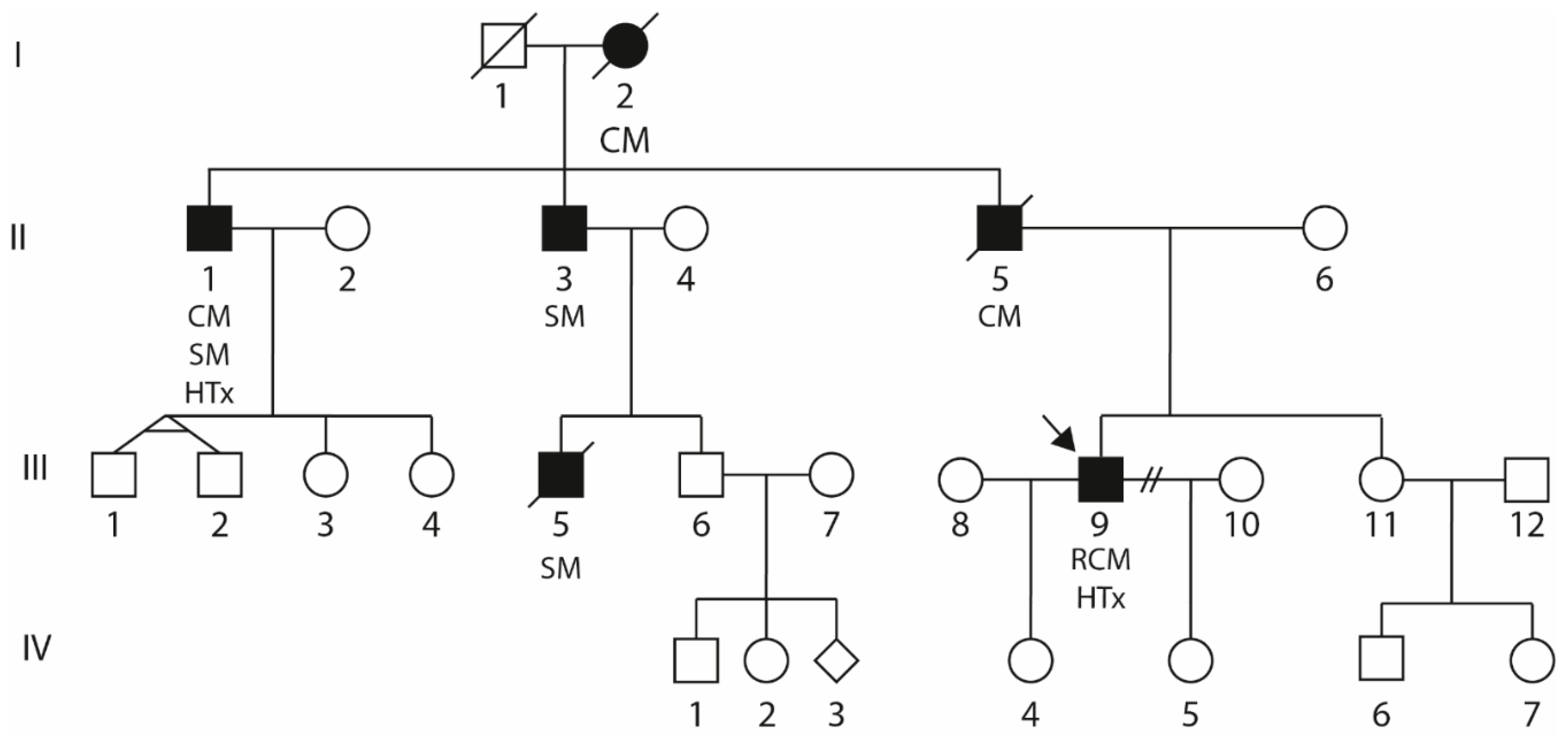
2.1. Clinical Description of the Index Patient (III-9)
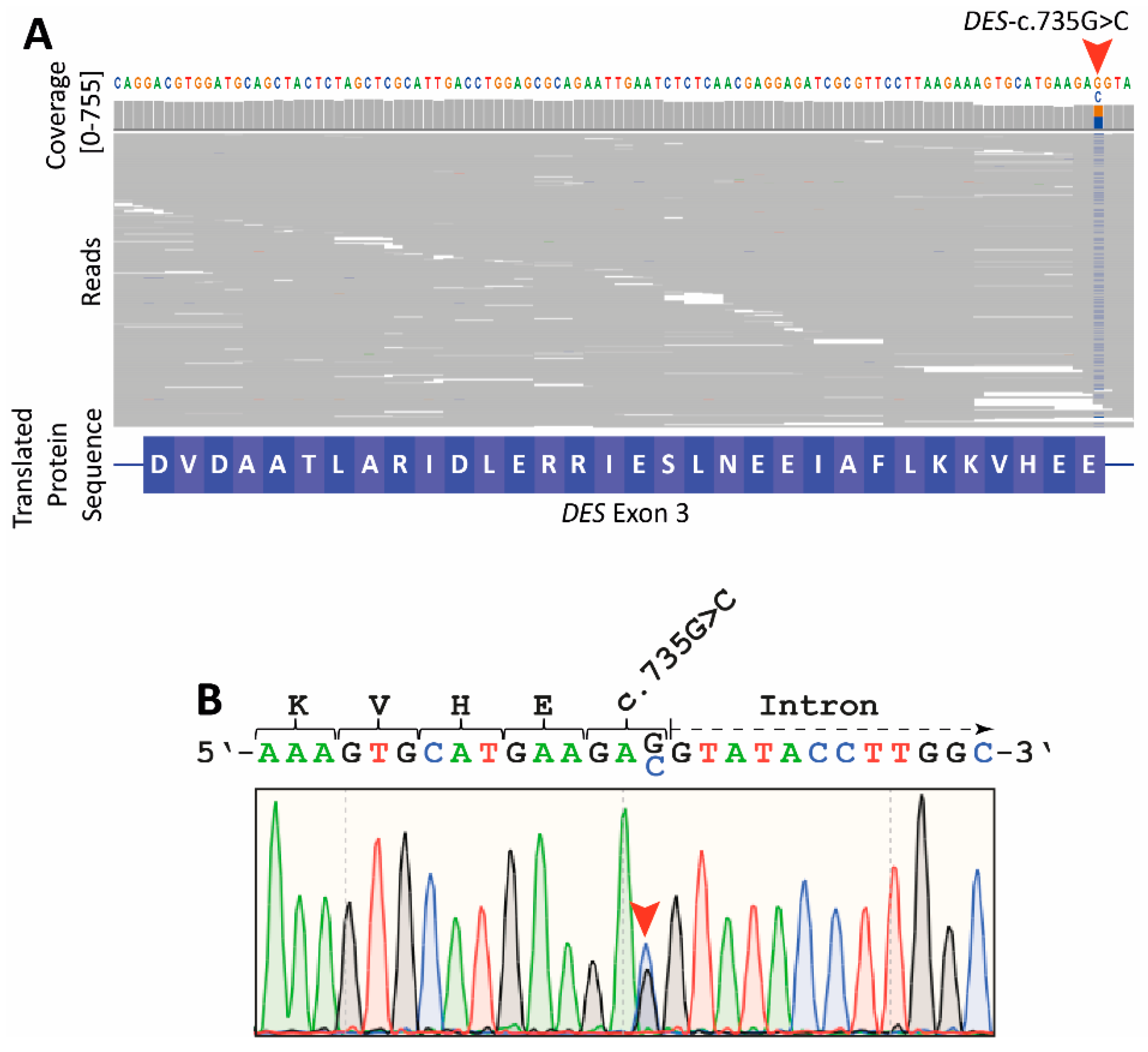
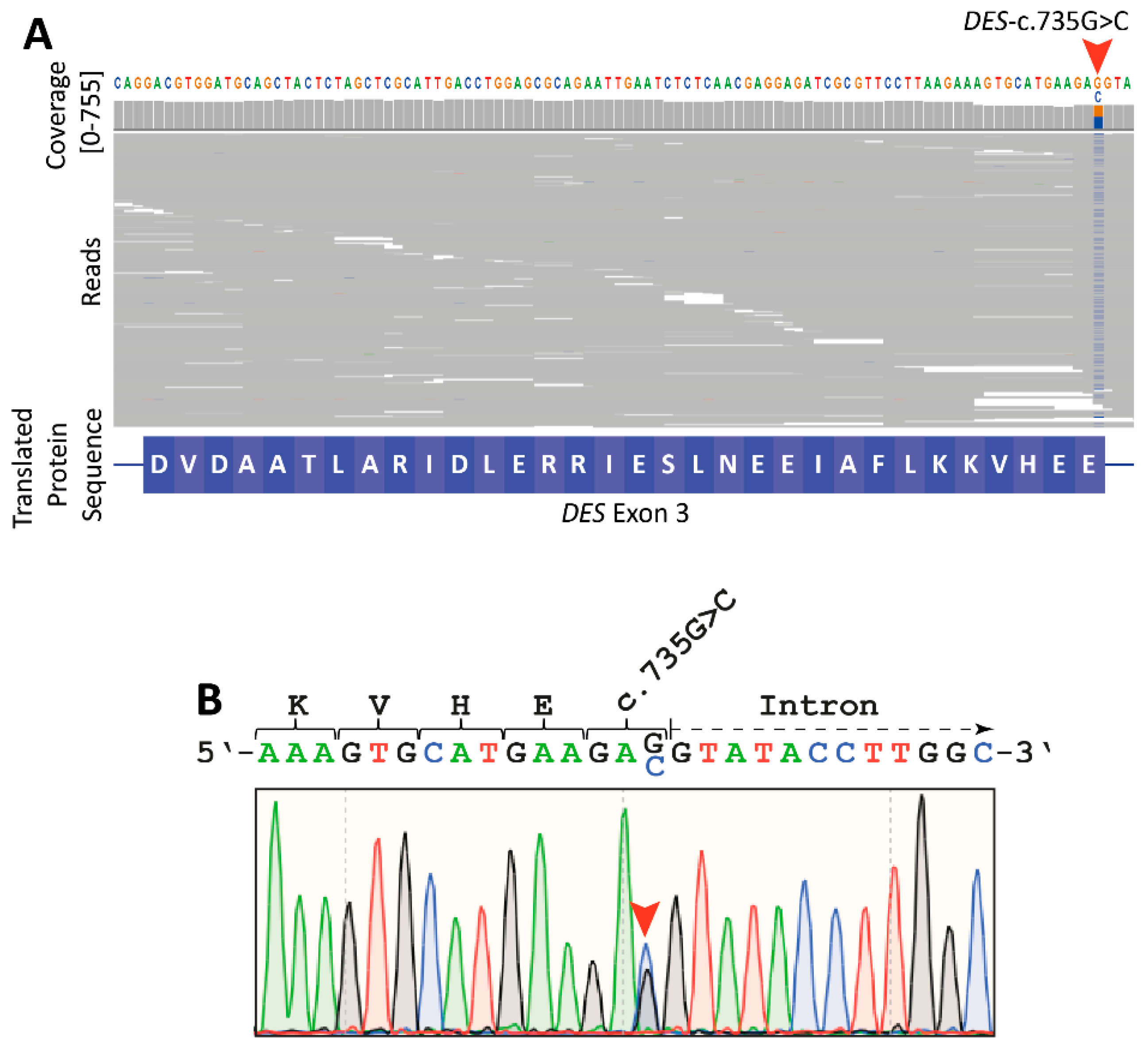
2.2. Genetic Analyses
2.3. Reverse Transcription Polymerase Chain Reaction
2.4. Amplicon Nanopore Sequencing
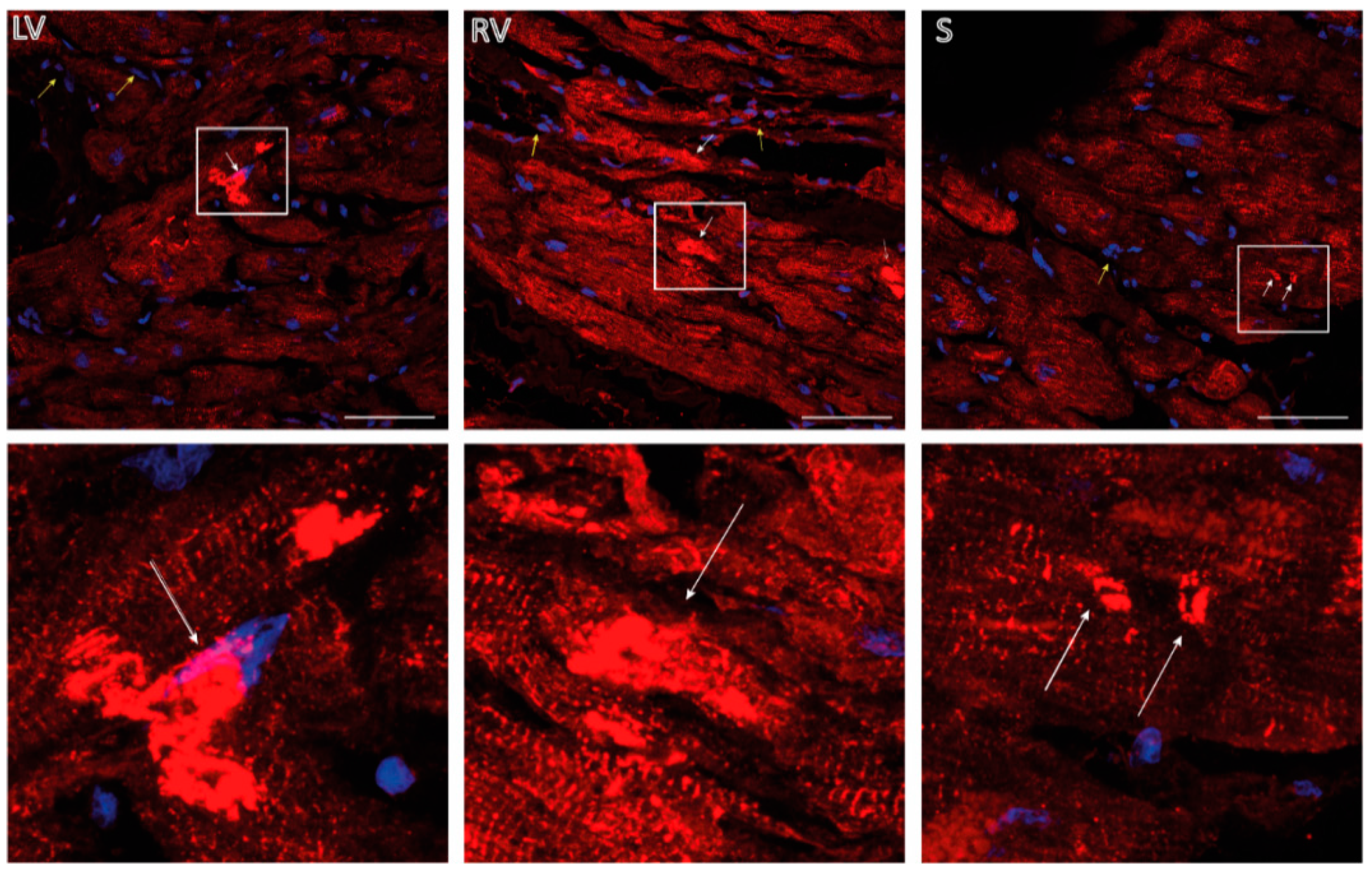
2.5. Immunohistochemistry
2.6. Plasmid Generation
2.7. Cell Culture and Confocal Microscopy
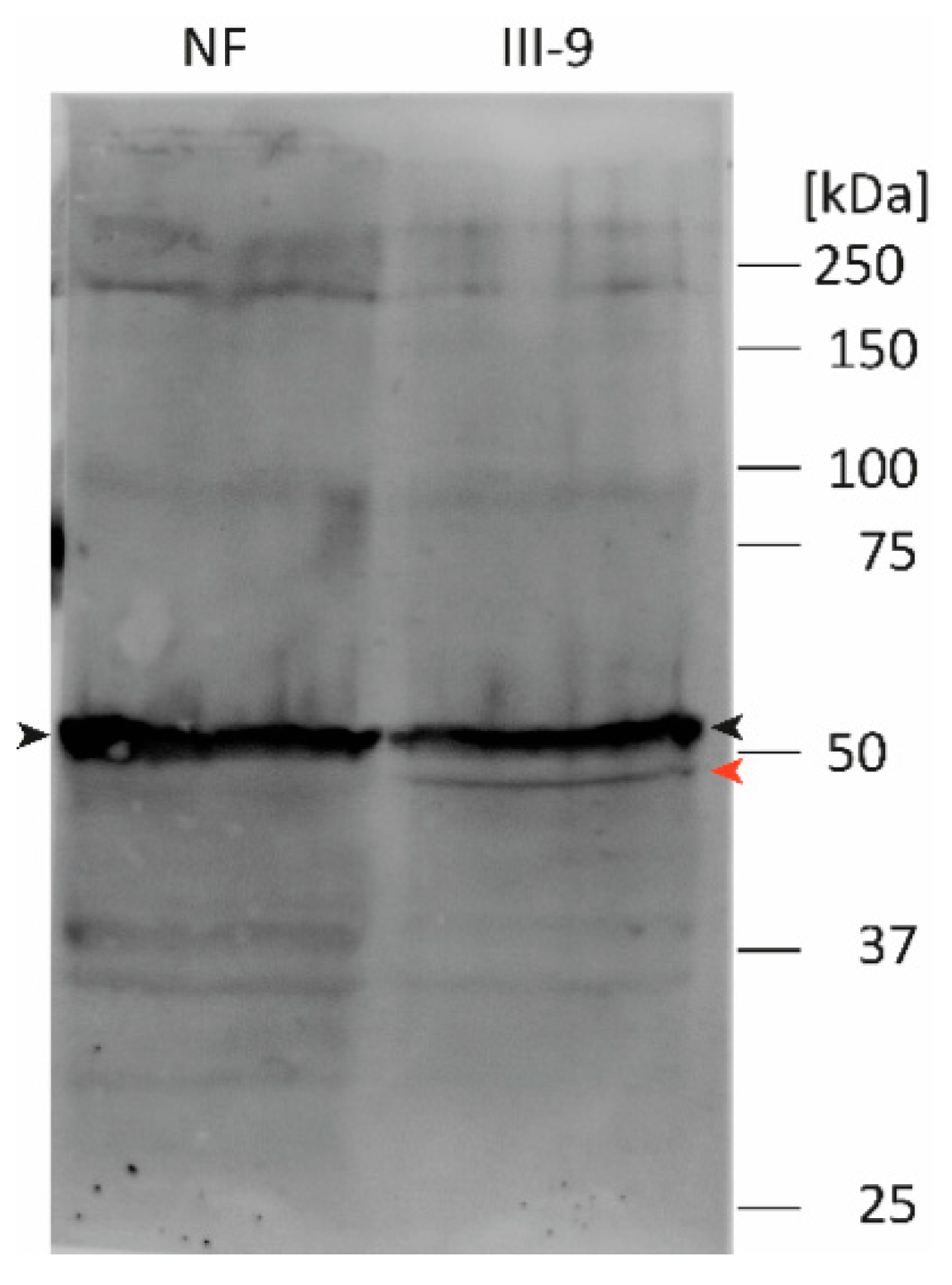
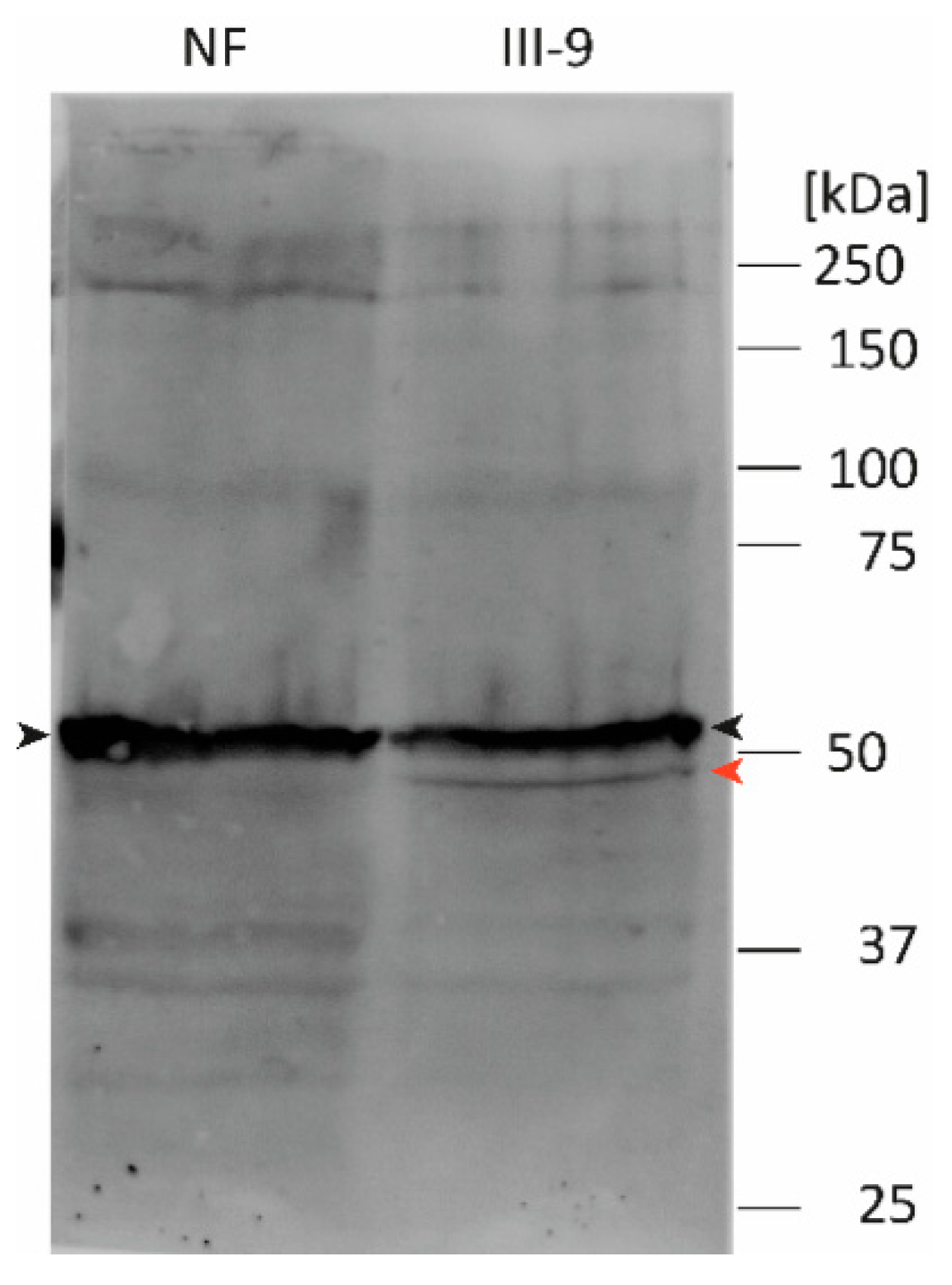
2.8. Western Blot Analysis
2.9. Statistical Analysis
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Brodehl, A.; Pour Hakimi, S.A.; Stanasiuk, C.; Ratnavadivel, S.; Hendig, D.; Gaertner, A.; Gerull, B.; Gummert, J.; Paluszkiewicz, L.; Milting, H. Restrictive Cardiomyopathy is Caused by a Novel Homozygous Desmin (DES) Mutation p.Y122H Leading to a Severe Filament Assembly Defect. Genes 2019, 10, 918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodehl, A.; Dieding, M.; Biere, N.; Unger, A.; Klauke, B.; Walhorn, V.; Gummert, J.; Schulz, U.; Linke, W.A.; Gerull, B.; et al. Functional characterization of the novel DES mutation p.L136P associated with dilated cardiomyopathy reveals a dominant filament assembly defect. J. Mol. Cell. Cardiol. 2016, 91, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, I.; Dieding, M.; Klauke, B.; Brodehl, A.; Gaertner-Rommel, A.; Walhorn, V.; Gummert, J.; Schulz, U.; Paluszkiewicz, L.; Anselmetti, D.; et al. A novel desmin (DES) indel mutation causes severe atypical cardiomyopathy in combination with atrioventricular block and skeletal myopathy. Mol. Genet. Genom. Med. 2018, 6, 288–293. [Google Scholar] [CrossRef] [Green Version]
- Clemen, C.S.; Herrmann, H.; Strelkov, S.V.; Schroder, R. Desminopathies: Pathology and mechanisms. Acta Neuropathol. 2013, 125, 47–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar, H.; Strelkov, S.V.; Sjoberg, G.; Aebi, U.; Herrmann, H. The biology of desmin filaments: How do mutations affect their structure, assembly, and organisation? J. Struct. Biol. 2004, 148, 137–152. [Google Scholar] [CrossRef]
- Maggi, L.; Mavroidis, M.; Psarras, S.; Capetanaki, Y.; Lattanzi, G. Skeletal and Cardiac Muscle Disorders Caused by Mutations in Genes Encoding Intermediate Filament Proteins. Int. J. Mol. Sci. 2021, 22, 4256. [Google Scholar] [CrossRef] [PubMed]
- Marakhonov, A.V.; Brodehl, A.; Myasnikov, R.P.; Sparber, P.A.; Kiseleva, A.V.; Kulikova, O.V.; Meshkov, A.N.; Zharikova, A.A.; Koretsky, S.N.; Kharlap, M.S.; et al. Noncompaction cardiomyopathy is caused by a novel in-frame desmin (DES) deletion mutation within the 1A coiled-coil rod segment leading to a severe filament assembly defect. Hum. Mutat. 2019, 40, 734–741. [Google Scholar] [CrossRef]
- Vernengo, L.; Chourbagi, O.; Panuncio, A.; Lilienbaum, A.; Batonnet-Pichon, S.; Bruston, F.; Rodrigues-Lima, F.; Mesa, R.; Pizzarossa, C.; Demay, L.; et al. Desmin myopathy with severe cardiomyopathy in a Uruguayan family due to a codon deletion in a new location within the desmin 1A rod domain. Neuromuscul. Disord. 2010, 20, 178–187. [Google Scholar] [CrossRef]
- Clemen, C.S.; Stockigt, F.; Strucksberg, K.H.; Chevessier, F.; Winter, L.; Schutz, J.; Bauer, R.; Thorweihe, J.M.; Wenzel, D.; Schlotzer-Schrehardt, U.; et al. The toxic effect of R350P mutant desmin in striated muscle of man and mouse. Acta Neuropathol. 2015, 129, 297–315. [Google Scholar] [CrossRef] [Green Version]
- Bar, H.; Mucke, N.; Kostareva, A.; Sjoberg, G.; Aebi, U.; Herrmann, H. Severe muscle disease-causing desmin mutations interfere with in vitro filament assembly at distinct stages. Proc. Natl. Acad. Sci. USA 2005, 102, 15099–15104. [Google Scholar] [CrossRef] [Green Version]
- Santhoshkumar, R.; Preethish-Kumar, V.; Polavarapu, K.; Reghunathan, D.; Chaudhari, S.; Satyamoorthy, K.; Vengalil, S.; Nashi, S.; Faruq, M.; Joshi, A.; et al. A Novel L1 Linker Mutation in DES Resulted in Total Absence of Protein. J. Mol. Neurosci. 2021. [Google Scholar] [CrossRef]
- Riley, L.G.; Waddell, L.B.; Ghaoui, R.; Evesson, F.J.; Cummings, B.B.; Bryen, S.J.; Joshi, H.; Wang, M.X.; Brammah, S.; Kritharides, L.; et al. Recessive DES cardio/myopathy without myofibrillar aggregates: Intronic splice variant silences one allele leaving only missense L190P-desmin. Eur. J. Hum. Genet. 2019, 27, 1267–1273. [Google Scholar] [CrossRef] [PubMed]
- Cimiotti, D.; Budde, H.; Hassoun, R.; Jaquet, K. Genetic Restrictive Cardiomyopathy: Causes and Consequences—An Integrative Approach. Int. J. Mol. Sci. 2021, 22, 558. [Google Scholar] [CrossRef] [PubMed]
- Cimiotti, D.; Fujita-Becker, S.; Mohner, D.; Smolina, N.; Budde, H.; Wies, A.; Morgenstern, L.; Gudkova, A.; Sejersen, T.; Sjoberg, G.; et al. Infantile restrictive cardiomyopathy: cTnI-R170G/W impair the interplay of sarcomeric proteins and the integrity of thin filaments. PLoS ONE 2020, 15, e0229227. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Ferrier, R.A.; Hamilton, S.J.; Greenway, S.C.; Brundler, M.A.; Yu, W.; Gibson, W.T.; McKinnon, M.L.; McGillivray, B.; Alvarez, N.; et al. Mutations in FLNC are Associated with Familial Restrictive Cardiomyopathy. Hum. Mutat. 2016, 37, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Tucker, N.R.; McLellan, M.A.; Hu, D.; Ye, J.; Parsons, V.A.; Mills, R.W.; Clauss, S.; Dolmatova, E.; Shea, M.A.; Milan, D.J.; et al. Novel Mutation in FLNC (Filamin C) Causes Familial Restrictive Cardiomyopathy. Circ. Cardiovasc. Genet. 2017, 10, e001780. [Google Scholar] [CrossRef] [Green Version]
- Kiselev, A.; Vaz, R.; Knyazeva, A.; Khudiakov, A.; Tarnovskaya, S.; Liu, J.; Sergushichev, A.; Kazakov, S.; Frishman, D.; Smolina, N.; et al. De novo mutations in FLNC leading to early-onset restrictive cardiomyopathy and congenital myopathy. Hum. Mutat. 2018, 39, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Clemen, C.S.; Fischer, D.; Reimann, J.; Eichinger, L.; Muller, C.R.; Muller, H.D.; Goebel, H.H.; Schroder, R. How much mutant protein is needed to cause a protein aggregate myopathy in vivo? Lessons from an exceptional desminopathy. Hum. Mutat. 2009, 30, E490–E499. [Google Scholar] [CrossRef]
- Kulikova, O.; Brodehl, A.; Kiseleva, A.; Myasnikov, R.; Meshkov, A.; Stanasiuk, C.; Gartner, A.; Divashuk, M.; Sotnikova, E.; Koretskiy, S.; et al. The Desmin (DES) Mutation p.A337P Is Associated with Left-Ventricular Non-Compaction Cardiomyopathy. Genes 2021, 12, 121. [Google Scholar] [CrossRef]
- Fischer, B.; Dittmann, S.; Brodehl, A.; Unger, A.; Stallmeyer, B.; Paul, M.; Seebohm, G.; Kayser, A.; Peischard, S.; Linke, W.A.; et al. Functional characterization of novel alpha-helical rod domain desmin (DES) pathogenic variants associated with dilated cardiomyopathy, atrioventricular block and a risk for sudden cardiac death. Int. J. Cardiol. 2021, 329, 167–174. [Google Scholar] [CrossRef]
- Protonotarios, A.; Brodehl, A.; Asimaki, A.; Jager, J.; Quinn, E.; Stanasiuk, C.; Ratnavadivel, S.; Futema, M.; Akhtar, M.M.; Gossios, T.D.; et al. The Novel Desmin Variant p.Leu115Ile Is Associated With a Unique Form of Biventricular Arrhythmogenic Cardiomyopathy. Can. J. Cardiol. 2021, 37, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Klingel, K.; Sauter, M.; Bock, C.T.; Szalay, G.; Schnorr, J.J.; Kandolf, R. Molecular pathology of inflammatory cardiomyopathy. Med. Microbiol. Immunol. 2004, 193, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Tang, A.D.; Soulette, C.M.; van Baren, M.J.; Hart, K.; Hrabeta-Robinson, E.; Wu, C.J.; Brooks, A.N. Full-length transcript characterization of SF3B1 mutation in chronic lymphocytic leukemia reveals downregulation of retained introns. Nat. Commun. 2020, 11, 1438. [Google Scholar] [CrossRef] [Green Version]
- Brodehl, A.; Belke, D.D.; Garnett, L.; Martens, K.; Abdelfatah, N.; Rodriguez, M.; Diao, C.; Chen, Y.X.; Gordon, P.M.; Nygren, A.; et al. Transgenic mice overexpressing desmocollin-2 (DSC2) develop cardiomyopathy associated with myocardial inflammation and fibrotic remodeling. PLoS ONE 2017, 12, e0174019. [Google Scholar] [CrossRef]
- Brodehl, A.; Gaertner-Rommel, A.; Klauke, B.; Grewe, S.A.; Schirmer, I.; Peterschroder, A.; Faber, L.; Vorgerd, M.; Gummert, J.; Anselmetti, D.; et al. The novel alphaB-crystallin (CRYAB) mutation p.D109G causes restrictive cardiomyopathy. Hum. Mutat. 2017, 38, 947–952. [Google Scholar] [CrossRef]
- Brodehl, A.; Hedde, P.N.; Dieding, M.; Fatima, A.; Walhorn, V.; Gayda, S.; Saric, T.; Klauke, B.; Gummert, J.; Anselmetti, D.; et al. Dual color photoactivation localization microscopy of cardiomyopathy-associated desmin mutants. J. Biol. Chem. 2012, 287, 16047–16057. [Google Scholar] [CrossRef] [Green Version]
- Hedberg, K.K.; Chen, L.B. Absence of intermediate filaments in a human adrenal cortex carcinoma-derived cell line. Exp. Cell Res. 1986, 163, 509–517. [Google Scholar] [CrossRef]
- Kubanek, M.; Schimerova, T.; Piherova, L.; Brodehl, A.; Krebsova, A.; Ratnavadivel, S.; Stanasiuk, C.; Hansikova, H.; Zeman, J.; Palecek, T.; et al. Desminopathy: Novel Desmin Variants, a New Cardiac Phenotype, and Further Evidence for Secondary Mitochondrial Dysfunction. J. Clin. Med. 2020, 9, 937. [Google Scholar] [CrossRef] [Green Version]
- CSH Press. Recipe RIPA Lysis Buffer. Cold Spring Harb. Protoc. 2017. [CrossRef]
- Brodehl, A.; Gaertner-Rommel, A.; Milting, H. Molecular insights into cardiomyopathies associated with desmin (DES) mutations. Biophys. Rev. 2018, 10, 983–1006. [Google Scholar] [CrossRef]
- Herrmann, H.; Cabet, E.; Chevalier, N.R.; Moosmann, J.; Schultheis, D.; Haas, J.; Schowalter, M.; Berwanger, C.; Weyerer, V.; Agaimy, A.; et al. Dual Functional States of R406W-Desmin Assembly Complexes Cause Cardiomyopathy With Severe Intercalated Disc Derangement in Humans and in Knock-In Mice. Circulation 2020, 142, 2155–2171. [Google Scholar] [CrossRef]
- Arbustini, E.; Pasotti, M.; Pilotto, A.; Pellegrini, C.; Grasso, M.; Previtali, S.; Repetto, A.; Bellini, O.; Azan, G.; Scaffino, M.; et al. Desmin accumulation restrictive cardiomyopathy and atrioventricular block associated with desmin gene defects. Eur. J. Heart Fail. 2006, 8, 477–483. [Google Scholar] [CrossRef]
- Pinol-Ripoll, G.; Shatunov, A.; Cabello, A.; Larrode, P.; de la Puerta, I.; Pelegrin, J.; Ramos, F.J.; Olive, M.; Goldfarb, L.G. Severe infantile-onset cardiomyopathy associated with a homozygous deletion in desmin. Neuromuscul. Disord. 2009, 19, 418–422. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Vrabie, A.; Goldfarb, L.G.; Shatunov, A.; Nagele, A.; Fritz, P.; Kaczmarek, I.; Goebel, H.H. The enlarging spectrum of desminopathies: New morphological findings, eastward geographic spread, novel exon 3 desmin mutation. Acta Neuropathol. 2005, 109, 411–417. [Google Scholar] [CrossRef]
- Strach, K.; Sommer, T.; Grohe, C.; Meyer, C.; Fischer, D.; Walter, M.C.; Vorgerd, M.; Reilich, P.; Bar, H.; Reimann, J.; et al. Clinical, genetic, and cardiac magnetic resonance imaging findings in primary desminopathies. Neuromuscul. Disord. 2008, 18, 475–482. [Google Scholar] [CrossRef]
- Wahbi, K.; Behin, A.; Charron, P.; Dunand, M.; Richard, P.; Meune, C.; Vicart, P.; Laforet, P.; Stojkovic, T.; Becane, H.M.; et al. High cardiovascular morbidity and mortality in myofibrillar myopathies due to DES gene mutations: A 10-year longitudinal study. Neuromuscul. Disord. 2012, 22, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodehl, A.; Dieding, M.; Klauke, B.; Dec, E.; Madaan, S.; Huang, T.; Gargus, J.; Fatima, A.; Saric, T.; Cakar, H.; et al. The novel desmin mutant p.A120D impairs filament formation, prevents intercalated disk localization, and causes sudden cardiac death. Circ. Cardiovasc. Genet. 2013, 6, 615–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ojrzynska, N.; Bilinska, Z.T.; Franaszczyk, M.; Ploski, R.; Grzybowski, J. Restrictive cardiomyopathy due to novel desmin gene mutation. Kardiol. Pol. 2017, 75, 723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khudiakov, A.; Kostina, D.; Zlotina, A.; Nikulina, T.; Sergushichev, A.; Gudkova, A.; Tomilin, A.; Malashicheva, A.; Kostareva, A. Generation of iPSC line from desmin-related cardiomyopathy patient carrying splice site mutation of DES gene. Stem Cell Res. 2017, 24, 77–80. [Google Scholar] [CrossRef]
- Park, K.Y.; Dalakas, M.C.; Goebel, H.H.; Ferrans, V.J.; Semino-Mora, C.; Litvak, S.; Takeda, K.; Goldfarb, L.G. Desmin splice variants causing cardiac and skeletal myopathy. J. Med. Genet. 2000, 37, 851–857. [Google Scholar] [CrossRef]
- Gudkova, A.; Kostareva, A.; Sjoberg, G.; Smolina, N.; Turalchuk, M.; Kuznetsova, I.; Rybakova, M.; Edstrom, L.; Shlyakhto, E.; Sejersen, T. Diagnostic challenge in desmin cardiomyopathy with transformation of clinical phenotypes. Pediatr. Cardiol. 2013, 34, 467–470. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
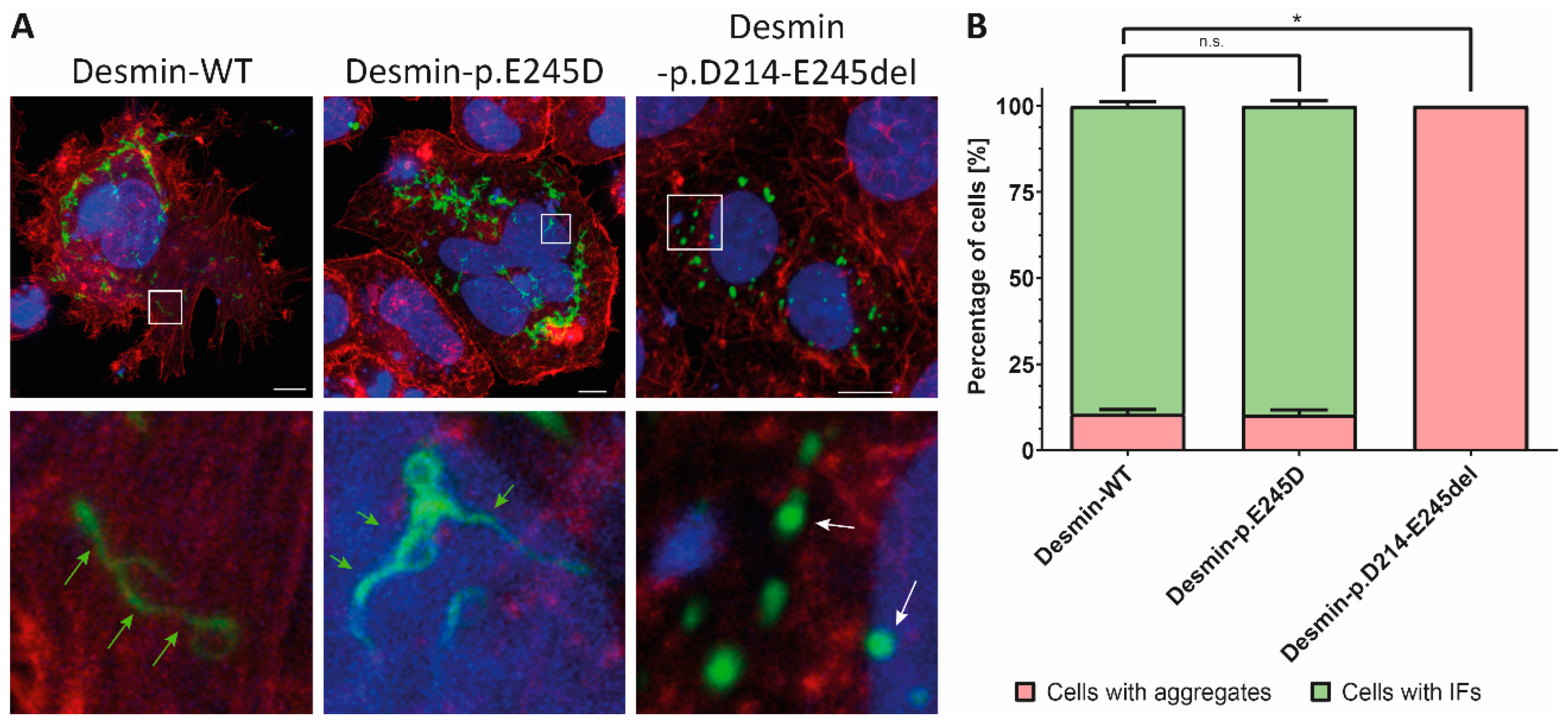{kind=link}
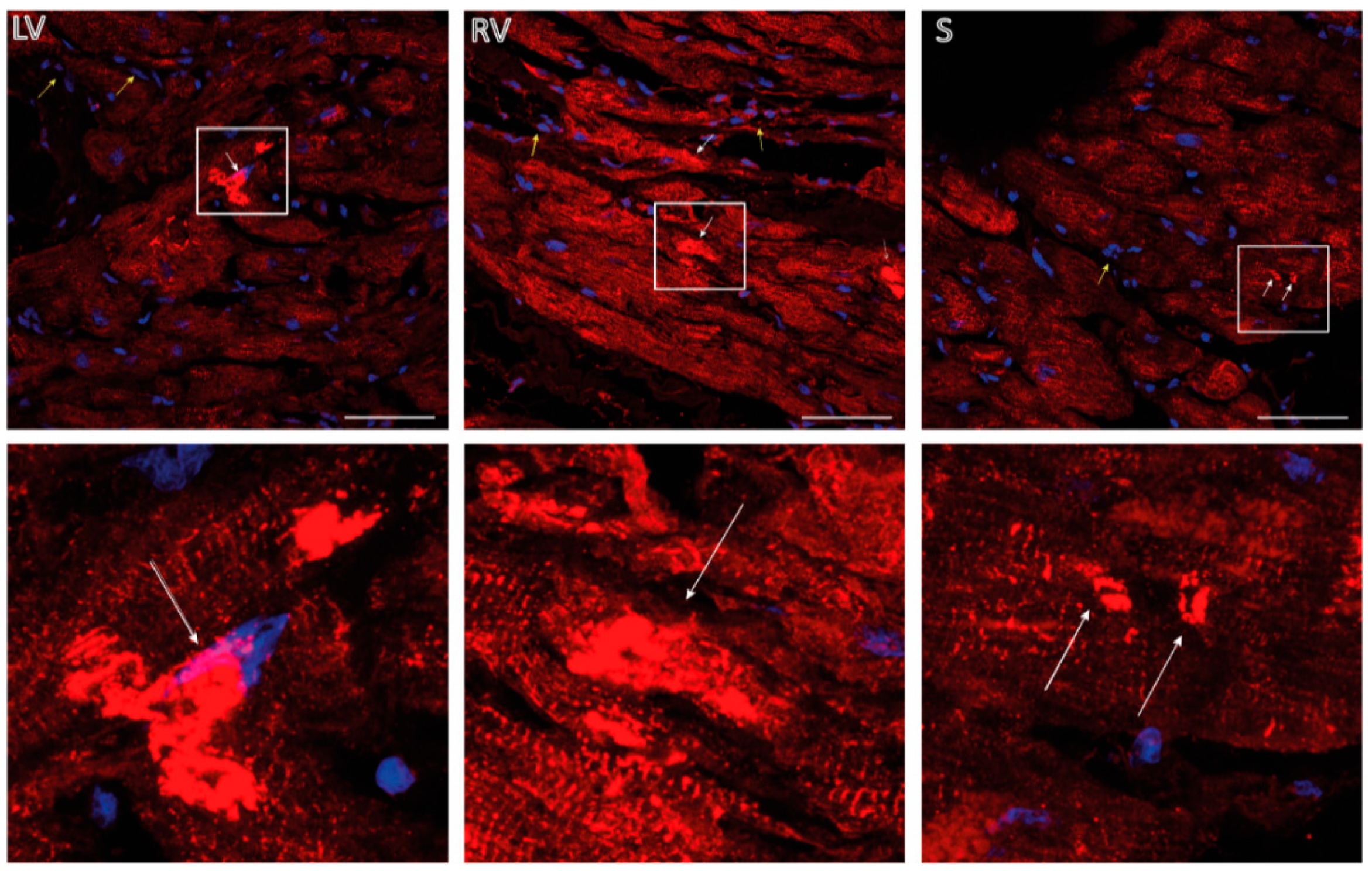{kind=link}
| Name | Sequence (5′-3′) | Application |
|---|---|---|
| DES_3F | GGAAGAAGCAGAGAACAATTTGGC | Sanger sequencing |
| DES_3R | ACCTGGACCTGCTGTTCCTG | Sanger sequencing |
| oligo(dT)18 | TTTTTTTTTTTTTTTTTT | Reverse transcription |
| DES_for | ATGAGCCAGGCCTACTCGTC | RT-PCR |
| DES_rev | GAGCACTTCATGCTGCTGCTG | RT-PCR |
| DES_E245D_for | TAAGAAAGTGCATGAAGACGAGATCCGTGAGTTGCAG | SDM |
| DES_E245D_rev | CTGCAACTCACGGATCTCGTCTTCATGCACTTTCTTA | SDM |
| DES_E3_Del_for | GCTGCCTTCCGAGCGGAGATCCGTGAGTTG | SDM |
| DES_E3_Del_rev | CAACTCACGGATCTCCGCTCGGAAGGCAGC | SDM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brodehl, A.; Hain, C.; Flottmann, F.; Ratnavadivel, S.; Gaertner, A.; Klauke, B.; Kalinowski, J.; Körperich, H.; Gummert, J.; Paluszkiewicz, L.; et al. The Desmin Mutation DES-c.735G>C Causes Severe Restrictive Cardiomyopathy by Inducing In-Frame Skipping of Exon-3. Biomedicines 2021, 9, 1400. https://doi.org/10.3390/biomedicines9101400
Brodehl A, Hain C, Flottmann F, Ratnavadivel S, Gaertner A, Klauke B, Kalinowski J, Körperich H, Gummert J, Paluszkiewicz L, et al. The Desmin Mutation DES-c.735G>C Causes Severe Restrictive Cardiomyopathy by Inducing In-Frame Skipping of Exon-3. Biomedicines. 2021; 9(10):1400. https://doi.org/10.3390/biomedicines9101400
Chicago/Turabian StyleBrodehl, Andreas, Carsten Hain, Franziska Flottmann, Sandra Ratnavadivel, Anna Gaertner, Bärbel Klauke, Jörn Kalinowski, Hermann Körperich, Jan Gummert, Lech Paluszkiewicz, and et al. 2021. "The Desmin Mutation DES-c.735G>C Causes Severe Restrictive Cardiomyopathy by Inducing In-Frame Skipping of Exon-3" Biomedicines 9, no. 10: 1400. https://doi.org/10.3390/biomedicines9101400
APA StyleBrodehl, A., Hain, C., Flottmann, F., Ratnavadivel, S., Gaertner, A., Klauke, B., Kalinowski, J., Körperich, H., Gummert, J., Paluszkiewicz, L., Deutsch, M.-A., & Milting, H. (2021). The Desmin Mutation DES-c.735G>C Causes Severe Restrictive Cardiomyopathy by Inducing In-Frame Skipping of Exon-3. Biomedicines, 9(10), 1400. https://doi.org/10.3390/biomedicines9101400