
Clinically Translatable Approaches of Inhibiting TGF-β to Target Cancer Stem Cells in TNBC

Abstract
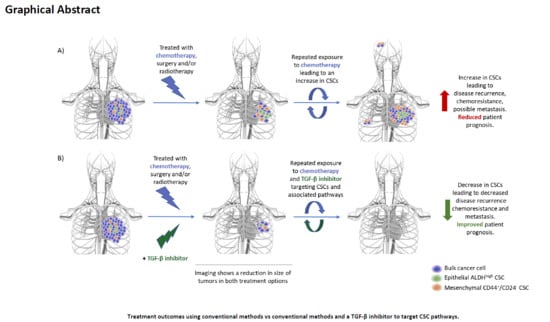
1. Introduction
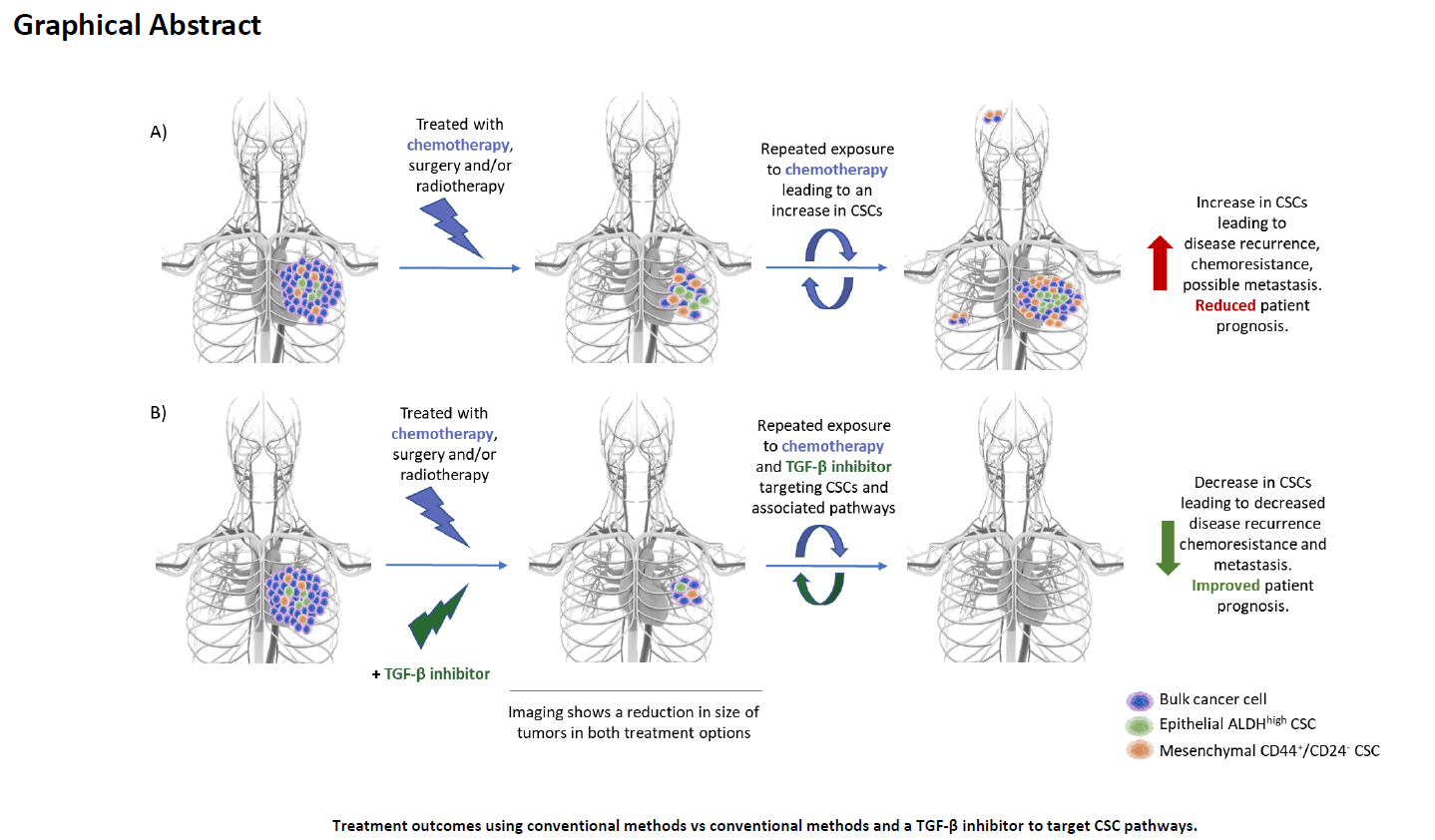
1.1. Overview of TGF-Β Signaling
1.2. The Complicated Regulation of TGF-β in Cancer
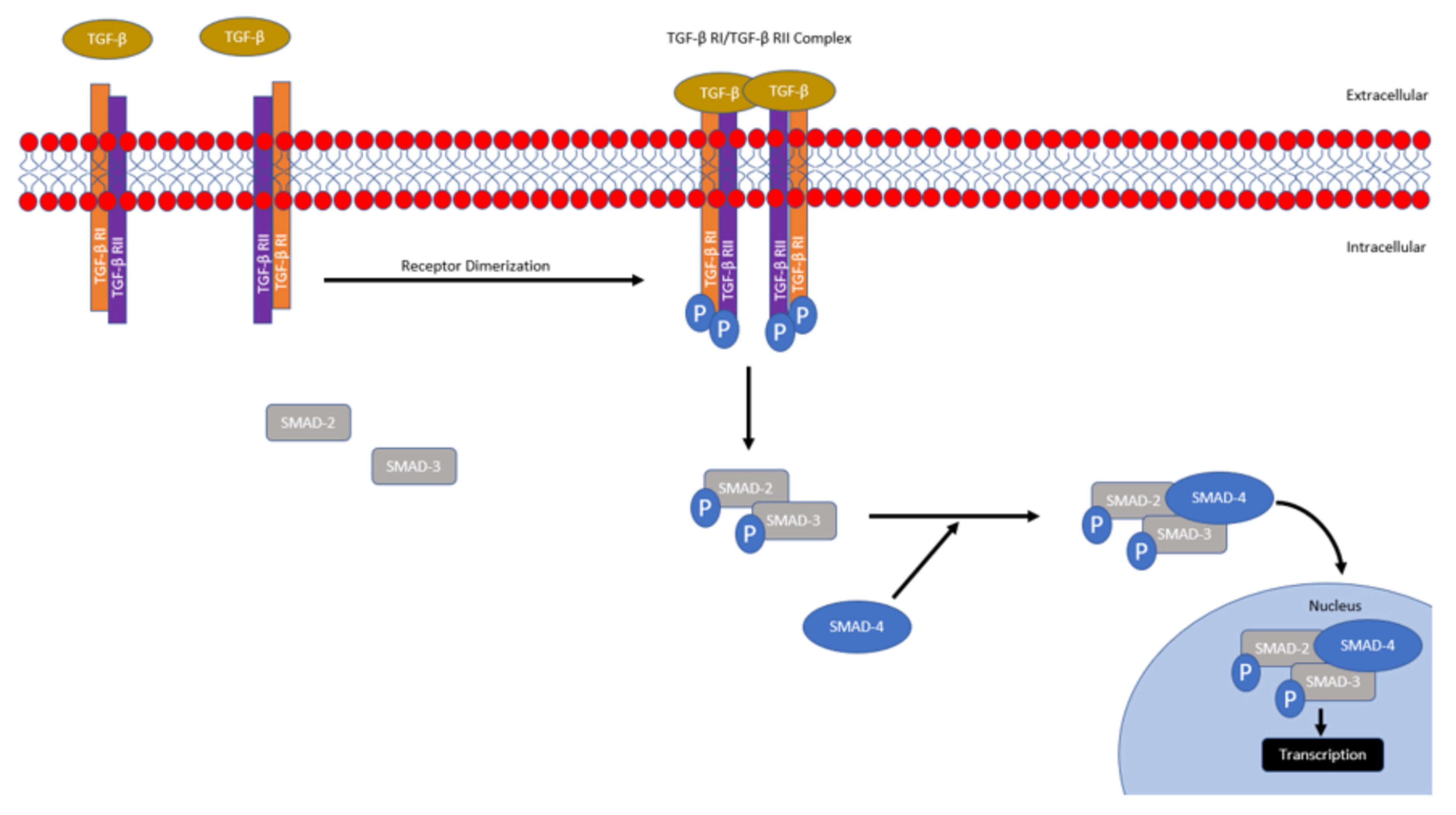
1.3. Clinical Correlates of Dysregulated TGF-β Signaling
1.4. Clinical Importance of CSCs in TNBC
1.5. TGF-β as a Therapeutic Target to Inhibit TNBC and Its CSC Population

{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Clinical Trial Number | Mechanism of Action | Tumor Type | Results |
|---|---|---|---|---|
| Imiquimod [71] | NCT00821964 | Inducer of interferon-gamma, known to inhibit TGF-β | Male breast cancer, recurrent breast cancer, skin metastases, stage IV breast cancer | Treatment using TLR-7 agonist, imiquimod and systemic albumin bound paclitaxel for recurrent chest wall lesion in breast cancer is effective in inducing disease regressing with a response rate of 20–30% [85]. |
| Fenretinide | NCT00001378 | Retinoid inducing dimerization of retinoid acid receptors which has been shown to regulate affect multiple signal transduction pathways, including IGF, TGF-β, and AP-1 [72] | Breast cancer and neoplasms | No results posted. |
| Fresolimumab [52] | NCT01401062 | A human anti-(TGF-β) monoclonal antibody | Metastatic breast cancer | TGF-β blockade using fresolimumab during radiotherapy in metastatic breast cancer was well tolerated. Patients receiving a higher dosage of fresolimumab (10 mg/kg) had a longer median overall survival compared to lower dosage (1 mg/kg). The higher-dosage arm also expressed greater systemic immune response with improved peripheral mononuclear cell count and increased CD8 central memory pool [88]. |
| Bevacizumab | NCT01959490 | Humanized monoclonal antibody that targets VEGF-A and has demonstrated inhibited TGF-β following treatment [73] | Breast cancer stages II–IIIC | 60% of patients treated with bevacizumab and combination chemotherapy (doxorubicin + cyclophosphamide + paclitaxel) achieved pathological complete response (pCR, absence of invasive cancer in breast or lymph nodes after neoadjuvant chemotherapy). Data accessed from clinicaltrials.gov (accessed on 9 September 2021). |
| Galunisertib (LY2157299) | NCT02423343 NCT02178358 NCT02734160 NCT01246986 | A small-molecule inhibitor of the TGF-β receptor I kinase | Solid tumors, NSCLC, HCC recurrent | NCT02423343—galunisertib + nivolumab had a progression-free survival of 5.26 and 5.39 months, with an overall survival of 11.99 and 14.52 months in two cohorts. Data accessed from clinicaltrials.gov (accessed on 9 September 2021). NCT02734160—Co-administration of galunisertib with durvalumab resulted in a 25% disease control rate, with a medial overall survival and progression-free survival of 5.72 months (95% CI: 4.01 to 8.38) and 1.87 months (95% CI: 1.58 to 3.09) [89]. NCT01246986—The overall survival of 160 mg treated vs. 300 mg using galunisertib was 7.3 and 16.8 months, respectively. Median overall survival of TGF-β1 responders vs. non-responders was 11.2 and 5.3 months, respectively [90]. |
| Vactosertib (TEW-7197) | NCT02160106 | Potent TGF-Β receptor ALK4/ALK5 inhibitor | Advanced-stage solid tumors | No results posted. |
| NIS793 | NCT02947165 | mAb that binds to human TGF-Β and prevents of activation of downstream signaling | Breast, lung, hepatocellula, colorectal, pancreatic and renal cancer | No results posted. |
1.6. Conclusions
2. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Dagenais, G.R.; Leong, D.P.; Rangarajan, S.; Lanas, F.; Lopez-Jaramillo, P.; Gupta, R.; Diaz, R.; Avezum, A.; Oliveira, G.B.; Wielgosz, A. Variations in common diseases, hospital admissions, and deaths in middle-aged adults in 21 countries from five continents (PURE): A prospective cohort study. Lancet 2020, 395, 785–794. [Google Scholar] [CrossRef]
- Anders, C.K.; Carey, L.A. Biology, metastatic patterns, and treatment of patients with triple-negative breast cancer. Clin. Breast Cancer 2009, 9, S73–S81. [Google Scholar] [CrossRef] [PubMed]
- Bauer, K.R.; Brown, M.; Cress, R.D.; Parise, C.A.; Caggiano, V.J.C. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: A population-based study from the California cancer Registry. Cancer 2007, 109, 1721–1728. [Google Scholar] [CrossRef] [PubMed]
- Colak, S.; ten Dijke, P. Targeting TGF-β signaling in cancer. Trends Cancer 2017, 3, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Hering, S.; Isken, F.; Knabbe, C.; Janott, J.; Jost, C.; Pommer, A.; Muhr, G.; Schatz, H.; Pfeiffer, A.F.H. TGFβ1 and TGFβ2 mRNA and protein expression in human bone samples. Exp. Clin. Endocrinol. Diabetes 2001, 109, 217–226. [Google Scholar] [CrossRef]
- Liu, S.; Chen, S.; Zeng, J. TGF-β signaling: A complex role in tumorigenesis. Mol. Med. Rep. 2018, 17, 699–704. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Vo, B.T.; Morton, D., Jr.; Komaragiri, S.; Millena, A.C.; Leath, C.; Khan, S.A. TGF-β effects on prostate cancer cell migration and invasion are mediated by PGE2 through activation of PI3K/AKT/mTOR pathway. Endocrinology 2013, 154, 1768–1779. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad pathways in TGF-β signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef]
- Moustakas, A.; Heldin, C.-H. Non-Smad TGF-β signals. J. Cell Sci. 2005, 118, 3573–3584. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, A.; McGarry, S.; Lam, K.M.; El-Sahli, S.; Chambers, J.; Kaczmarek, S.; Li, L.; Addison, C.; Dimitroulakos, J.; Arnaout, A.; et al. Co-inhibition of mTORC1, HDAC and ESR1α retards the growth of triple-negative breast cancer and suppresses cancer stem cells. Cell Death Dis. 2018, 9, 815. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.-Y.; Jiang, Z.; Ben-David, Y.; Woodgett, J.R.; Zacksenhaus, E. Molecular stratification within triple-negative breast cancer subtypes. Sci. Rep. 2019, 9, 19107. [Google Scholar] [CrossRef]
- Chaudhury, A.; Howe, P.H. The tale of transforming growth factor-beta (TGFβ) signaling: A soigné enigma. IUBMB Life 2009, 61, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Wendt, M.K.; Tian, M.; Schiemann, W.P. Deconstructing the mechanisms and consequences of TGF-β-induced EMT during cancer progression. Cell Tissue Res. 2012, 347, 85–101. [Google Scholar] [CrossRef] [PubMed]
- Principe, D.R.; Doll, J.A.; Bauer, J.; Jung, B.; Munshi, H.G.; Bartholin, L.; Pasche, B.; Lee, C.; Grippo, P.J. TGF-β: Duality of function between tumor prevention and carcinogenesis. JNCI J. Natl. Cancer Inst. 2014, 106, djt369. [Google Scholar] [CrossRef] [PubMed]
- Wenner, C.E.; Yan, S. Biphasic role of TGF-β1 in signal transduction and crosstalk. J. Cell. Physiol. 2003, 196, 42–50. [Google Scholar] [CrossRef]
- Akhurst, R.J.; Derynck, R. TGF-β signaling in cancer—A double-edged sword. Trends Cell Biol. 2001, 11, S44–S51. [Google Scholar]
- Morrison, C.D.; Parvani, J.G.; Schiemann, W.P. The relevance of the TGF-β Paradox to EMT-MET programs. Cancer Lett. 2013, 341, 30–40. [Google Scholar] [CrossRef]
- Tian, M.; Schiemann, W.P. The TGF-β paradox in human cancer: An update. Future Oncol. 2009, 5, 259–271. [Google Scholar] [CrossRef]
- Yoo, J.; Ghiassi, M.; Jirmanova, L.; Balliet, A.G.; Hoffman, B.; Fornace, A.J.; Liebermann, D.A.; Böttinger, E.P.; Roberts, A.B. Transforming growth factor-β-induced apoptosis is mediated by Smad-dependent expression of GADD45b through p38 activation. J. Biol. Chem. 2003, 278, 43001–43007. [Google Scholar] [CrossRef]
- Huynh, L.K.; Hipolito, C.J.; ten Dijke, P. A Perspective on the Development of TGF-β Inhibitors for Cancer Treatment. Biomolecules 2019, 9, 743. [Google Scholar] [CrossRef]
- Spender, L.; O’Brien, D.; Simpson, D.; Dutt, D.; Gregory, C.; Allday, M.; Clark, L.; Inman, G.J. Differentiation. TGF-β induces apoptosis in human B cells by transcriptional regulation of BIK and BCL-X L. Cell Death Differ. 2009, 16, 593–602. [Google Scholar] [CrossRef]
- Ozaki, I.; Hamajima, H.; Matsuhashi, S.; Mizuta, T. Regulation of TGF-β1-induced pro-apoptotic signaling by growth factor receptors and extracellular matrix receptor integrins in the liver. Front. Physiol. 2011, 2, 78. [Google Scholar] [CrossRef]
- Wiener, Z.; Band, A.M.; Kallio, P.; Högström, J.; Hyvönen, V.; Kaijalainen, S.; Ritvos, O.; Haglund, C.; Kruuna, O.; Robine, S. Oncogenic mutations in intestinal adenomas regulate Bim-mediated apoptosis induced by TGF-β. Proc. Natl. Acad. Sci. USA 2014, 111, E2229–E2236. [Google Scholar] [CrossRef]
- Tang, B.; Yoo, N.; Vu, M.; Mamura, M.; Nam, J.-S.; Ooshima, A.; Du, Z.; Desprez, P.-Y.; Anver, M.R.; Michalowska, A.M. TGF-β can suppress tumorigenesis through effects on the putative cancer stem or early progenitor cell and committed progeny in a breast cancer xenograft model. Cancer Res. 2007, 67, 8643. [Google Scholar] [CrossRef] [PubMed]
- Sikder, H.A.; Devlin, M.K.; Dunlap, S.; Ryu, B.; Alani, R.M. Id proteins in cell growth and tumorigenesis. Cancer Cell 2003, 3, 525–530. [Google Scholar] [CrossRef]
- Bello-DeOcampo, D.; Tindall, D.J. TGF-ß/Smad signaling in prostate cancer. Curr. Drug Targets 2003, 4, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, Y.; Takanashi, S.; Kanehira, Y.; Tsushima, T.; Imai, T.; Okumura, K. Transforming growth factor-β1 level correlates with angiogenesis, tumor progression, and prognosis in patients with nonsmall cell lung carcinoma. Cancer 2001, 91, 964–971. [Google Scholar] [CrossRef]
- Xiong, B.; Gong, L.-L.; Zhang, F.; Hu, M.-B.; Yuan, H.-Y. TGF-β1 expression and angiogenesis in colorectal cancer tissue. World J. Gastroenterol. 2002, 8, 496. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.J.; Blobe, G.C. Dichotomous roles of TGF-β in human cancer. Biochem. Soc. Trans. 2016, 44, 1441–1454. [Google Scholar] [CrossRef]
- Ó hAinmhire, E.; Quartuccio, S.M.; Cheng, W.; Ahmed, R.A.; King, S.M.; Burdette, J.E. Mutation or loss of p53 differentially modifies TGFβ action in ovarian cancer. PLoS ONE 2014, 9, e89553. [Google Scholar] [CrossRef] [PubMed]
- Conery, A.R.; Cao, Y.; Thompson, E.A.; Townsend, C.M.; Ko, T.C.; Luo, K. Akt interacts directly with Smad3 to regulate the sensitivity to TGF-β-induced apoptosis. Nat. Cell Biol. 2004, 6, 366–372. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Huang, Y.-H.; Chen, M.; Su, J.; Zou, Y.; Bardeesy, N.; Iacobuzio-Donahue, C.A.; Massagué, J. TGF-β tumor suppression through a lethal EMT. Cell 2016, 164, 1015–1030. [Google Scholar] [CrossRef]
- Ji, L.; Xu, J.; Liu, J.; Amjad, A.; Zhang, K.; Liu, Q.; Zhou, L.; Xiao, J.; Li, X. Mutant p53 promotes tumor cell malignancy by both positive and negative regulation of the transforming growth factor β (TGF-β) pathway. J. Biol. Chem. 2015, 290, 11729–11740. [Google Scholar] [CrossRef] [PubMed]
- Massihnia, D.; Galvano, A.; Fanale, D.; Perez, A.; Castiglia, M.; Incorvaia, L.; Listì, A.; Rizzo, S.; Cicero, G.; Bazan, V. Triple negative breast cancer: Shedding light onto the role of pi3k/akt/mtor pathway. Oncotarget 2016, 7, 60712. [Google Scholar] [CrossRef] [PubMed]
- Giltnane, J.M.; Balko, J.M. Rationale for targeting the Ras/MAPK pathway in triple-negative breast cancer. Discov. Med. 2014, 17, 275–283. [Google Scholar]
- Xu, J.; Prosperi, J.R.; Choudhury, N.; Olopade, O.I.; Goss, K.H. β-Catenin is required for the tumorigenic behavior of triple-negative breast cancer cells. PLoS ONE 2015, 10, e0117097. [Google Scholar] [CrossRef]
- Jiang, H.-L.; Sun, H.-F.; Gao, S.-P.; Li, L.-D.; Hu, X.; Wu, J.; Jin, W. Loss of RAB1B promotes triple-negative breast cancer metastasis by activating TGF-β/SMAD signaling. Oncotarget 2015, 6, 16352. [Google Scholar] [CrossRef][Green Version]
- Ding, M.-J.; Su, K.; Cui, G.Z.; Yang, W.H.; Chen, L.; Yang, M.; Liu, Y.-Q.; Dai, D.-L. Association between transforming growth factor-β1 expression and the clinical features of triple negative breast cancer. Oncol. Lett. 2016, 11, 4040–4044. [Google Scholar] [CrossRef]
- Zhang, M.; Wu, J.; Mao, K.; Deng, H.; Yang, Y.; Zhou, E.; Liu, J. Role of transforming growth factor-β1 in triple negative breast cancer patients. Int. J. Surg. 2017, 45, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Wu, Z.; Qi, Y.; Wu, Q.; Yang, F. Expression of TGF-beta1 and the mechanism of invasiveness and metastasis induced by TGF-beta1 in breast cancer. Chin. J. Oncol. 2009, 31, 679–682. [Google Scholar]
- Berger, A.C.; Korkut, A.; Kanchi, R.S.; Hegde, A.M.; Lenoir, W.; Liu, W.; Liu, Y.; Fan, H.; Shen, H.; Ravikumar, V. A comprehensive pan-cancer molecular study of gynecologic and breast cancers. Cancer Cell 2018, 33, 690–705.e9. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304.e6. [Google Scholar] [CrossRef] [PubMed]
- Hachim, M.Y.; Hachim, I.Y.; Dai, M.; Ali, S.; Lebrun, J.-J. Differential expression of TGFβ isoforms in breast cancer highlights different roles during breast cancer progression. Tumor Biol. 2018, 40, 1010428317748254. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.-T.; Ryu, C.J. Cancer stem cell surface markers on normal stem cells. BMB Rep. 2017, 50, 285. [Google Scholar] [CrossRef]
- Gasch, C.; Ffrench, B.; O’Leary, J.J.; Gallagher, M.F. Catching moving targets: Cancer stem cell hierarchies, therapy-resistance & considerations for clinical intervention. Mol. Cancer 2017, 16, 43. [Google Scholar] [CrossRef]
- Ayob, A.Z.; Ramasamy, T.S. Cancer stem cells as key drivers of tumour progression. J. Biomed. Sci. 2018, 25, 20. [Google Scholar] [CrossRef] [PubMed]
- Agliano, A.; Calvo, A.; Box, C. The challenge of targeting cancer stem cells to halt metastasis. Semin. Cancer Biol. 2017, 44, 25–42. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cong, Y.; Wang, D.; Sun, Y.; Deng, L.; Liu, Y.; Martin-Trevino, R.; Shang, L.; McDermott, S.P.; Landis, M.D. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. 2014, 2, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, A.; McGarry, S.; Han, X.; Liu, S.; Wang, L. CSCs in Breast Cancer—One Size Does Not Fit All: Therapeutic Advances in Targeting Heterogeneous Epithelial and Mesenchymal CSCs. Cancers 2019, 11, 1128. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Sulaiman, A.; Sulaiman, B.; Khouri, L.; McGarry, S.; Nessim, C.; Arnaout, A.; Li, X.; Addison, C.; Dimitroulakos, J.; Wang, L. Both bulk and cancer stem cell subpopulations in triple-negative breast cancer are susceptible to Wnt, HDAC, and ER α coinhibition. FEBS Lett. 2016, 590, 4606–4616. [Google Scholar] [CrossRef]
- Sulaiman, A.; McGarry, S.; Li, L.; Jia, D.; Ooi, S.; Addison, C.; Dimitroulakos, J.; Arnaout, A.; Nessim, C.; Yao, Z. Dual inhibition of Wnt and Yes-associated protein signaling retards the growth of triple-negative breast cancer in both mesenchymal and epithelial states. Mol. Oncol. 2018, 12, 423–440. [Google Scholar] [CrossRef]
- Formenti, S.C.; Hawtin, R.E.; Dixit, N.; Evensen, E.; Lee, P.; Goldberg, J.D.; Li, X.; Vanpouille-Box, C.; Schaue, D.; McBride, W.H.; et al. Baseline T cell dysfunction by single cell network profiling in metastatic breast cancer patients. J. Immunother. Cancer 2019, 7, 177. [Google Scholar] [CrossRef] [PubMed]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef]
- Sarmiento-Castro, A.; Caamaño-Gutiérrez, E.; Sims, A.H.; Hull, N.J.; James, M.I.; Santiago-Gómez, A.; Eyre, R.; Clark, C.; Brown, M.E.; Brooks, M.D. Increased expression of interleukin-1 receptor characterizes anti-estrogen-resistant ALDH+ breast cancer stem cells. Stem Cell Rep. 2020, 15, 307–316. [Google Scholar] [CrossRef]
- Wu, Y.; Tran, T.; Dwabe, S.; Sarkissyan, M.; Kim, J.; Nava, M.; Clayton, S.; Pietras, R.; Farias-Eisner, R.; Vadgama, J.V.; et al. A83-01 inhibits TGF-β-induced upregulation of Wnt3 and epithelial to mesenchymal transition in HER2-overexpressing breast cancer cells. Breast Cancer Res. Treat. 2017, 163, 449–460. [Google Scholar] [CrossRef]
- Sulaiman, A.; Yao, Z.; Wang, L. Re-evaluating the role of epithelial-mesenchymal-transition in cancer progression. Cancers 2018, 32, 81. [Google Scholar]
- Sulaiman, A.; McGarry, S.; El-Sahli, S.; Li, L.; Chambers, J.; Phan, A.; Cote, M.; Cron, G.O.; Alain, T.; Le, Y. Co-targeting bulk tumor and cscs in clinically translatable TNBC patient-derived xenografts via combination nanotherapy. Mol. Cancer Ther. 2019, 18, 1755–1764. [Google Scholar] [CrossRef]
- Jia, D.; Tan, Y.; Liu, H.; Ooi, S.; Li, L.; Wright, K.; Bennett, S.; Addison, C.L.; Wang, L. Cardamonin reduces chemotherapy-enriched breast cancer stem-like cells in vitro and in vivo. Oncotarget 2016, 7, 771. [Google Scholar] [CrossRef]
- Sulaiman, A.; McGarry, S.; Chambers, J.; Al-Kadi, E.; Phan, A.; Li, L.; Mediratta, K.; Dimitroulakos, J.; Addison, C.; Li, X. Targeting hypoxia sensitizes TNBC to cisplatin and promotes inhibition of both bulk and cancer stem cells. Int. J. Mol. Sci. 2020, 21, 5788. [Google Scholar] [CrossRef] [PubMed]
- Creighton, C.J.; Li, X.; Landis, M.; Dixon, J.M.; Neumeister, V.M.; Sjolund, A.; Rimm, D.L.; Wong, H.; Rodriguez, A.; Herschkowitz, J.I. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc. Natl. Acad. Sci. USA 2009, 106, 13820–13825. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.D.; Fan, F.; Camp, E.R.; van Buren, G.; Liu, W.; Somcio, R.; Gray, M.J.; Cheng, H.; Hoff, P.M.; Ellis, L.M. Chronic oxaliplatin resistance induces epithelial-to-mesenchymal transition in colorectal cancer cell lines. Clin. Cancer Res. 2006, 12, 4147–4153. [Google Scholar] [CrossRef]
- Kajiyama, H.; Hosono, S.; Terauchi, M.; Shibata, K.; Ino, K.; Yamamoto, E.; Nomura, S.; Nawa, A.; Kikkawa, F. Twist expression predicts poor clinical outcome of patients with clear cell carcinoma of the ovary. Oncology 2006, 71, 394–401. [Google Scholar] [CrossRef]
- Kajiyama, H.; Shibata, K.; Terauchi, M.; Yamashita, M.; Ino, K.; Nawa, A.; Kikkawa, F. Chemoresistance to paclitaxel induces epithelial-mesenchymal transition and enhances metastatic potential for epithelial ovarian carcinoma cells. Int. J. Oncol. 2007, 31, 277–283. [Google Scholar] [CrossRef]
- Sulaiman, A.; McGarry, S.; El-Sahli, S.; Li, L.; Chambers, J.; Phan, A.; Al-Kadi, E.; Kahiel, Z.; Farah, E.; Ji, G. Nanoparticles Loaded with Wnt and YAP/Mevalonate Inhibitors in Combination with Paclitaxel Stop the Growth of TNBC Patient-Derived Xenografts and Diminish Tumorigenesis. Adv. Ther. 2020, 3, 2000123. [Google Scholar] [CrossRef]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef]
- Papadaki, M.A.; Stoupis, G.; Theodoropoulos, P.A.; Mavroudis, D.; Georgoulias, V.; Agelaki, S. Circulating tumor cells with stemness and epithelial-to-mesenchymal transition features are chemoresistant and predictive of poor outcome in metastatic breast cancer. Mol. Cancer Ther. 2019, 18, 437–447. [Google Scholar] [CrossRef]
- Jia, D.; Li, L.; Andrew, S.; Allan, D.; Li, X.; Lee, J.; Ji, G.; Yao, Z.; Gadde, S.; Figeys, D.; et al. An autocrine inflammatory forward-feedback loop after chemotherapy withdrawal facilitates the repopulation of drug-resistant breast cancer cells. Cell Death Dis. 2017, 8, e2932. [Google Scholar] [CrossRef] [PubMed]
- Bhola, N.E.; Balko, J.M.; Dugger, T.C.; Kuba, M.G.; Sánchez, V.; Sanders, M.; Stanford, J.; Cook, R.S.; Arteaga, C.L. TGF-β inhibition enhances chemotherapy action against triple-negative breast cancer. J. Clin. Investig. 2013, 123, 1348–1358. [Google Scholar] [CrossRef]
- Yadav, P.; Shankar, B.S. Radio resistance in breast cancer cells is mediated through TGF-β signalling, hybrid epithelial-mesenchymal phenotype and cancer stem cells. Biomed. Pharmacother. 2019, 111, 119–130. [Google Scholar] [CrossRef]
- Huang, M.; Zhang, F.; Xu, Y.; Wang, H.; Lin, S.; Zhang, Y. The comparison of epirubicin-treated MCF-7 mammosphere cells to the treated monolayer cells. Clin. Oncol. 2009, 27, e13542. [Google Scholar]
- Xu, X.; Zhang, L.; He, X.; Zhang, P.; Sun, C.; Xu, X.; Lu, Y.; Li, F. TGF-β plays a vital role in triple-negative breast cancer (TNBC) drug-resistance through regulating stemness, EMT and apoptosis. Biochem. Biophys. Res. Commun. 2018, 502, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Wang, K.; Chen, Y. Ophiopogonin D suppresses TGF-β1-mediated metastatic behavior of MDA-MB-231 breast carcinoma cells via regulating ITGB1/FAK/Src/AKT/β-catenin/MMP-9 signaling axis. Toxicol. In Vitro 2020, 69, 104973. [Google Scholar] [CrossRef]
- Sun, X.; He, Z.; Guo, L.; Wang, C.; Lin, C.; Ye, L.; Wang, X.; Li, Y.; Yang, M.; Liu, S.; et al. ALG3 contributes to stemness and radioresistance through regulating glycosylation of TGF-β receptor II in breast cancer. J. Exp. Clin. Cancer Res. 2021, 40, 149. [Google Scholar] [CrossRef]
- Partridge, E.A.; Le Roy, C.; Di Guglielmo, G.M.; Pawling, J.; Cheung, P.; Granovsky, M.; Nabi, I.R.; Wrana, J.L.; Dennis, J.W. Regulation of cytokine receptors by Golgi N-glycan processing and endocytosis. Science 2004, 306, 120–124. [Google Scholar] [CrossRef]
- Schech, A.; Kazi, A.; Yu, S.; Shah, P.; Sabnis, G. Histone Deacetylase Inhibitor Entinostat Inhibits Tumor-Initiating Cells in Triple-Negative Breast Cancer Cells. Mol. Cancer Ther. 2015, 14, 1848–1857. [Google Scholar] [CrossRef]
- Liu, N.; He, S.; Ma, L.; Ponnusamy, M.; Tang, J.; Tolbert, E.; Bayliss, G.; Zhao, T.C.; Yan, H.; Zhuang, S. Blocking the class I histone deacetylase ameliorates renal fibrosis and inhibits renal fibroblast activation via modulating TGF-beta and EGFR signaling. PLoS ONE 2013, 8, e54001. [Google Scholar] [CrossRef]
- Wahdan-Alaswad, R.; Harrell, J.C.; Fan, Z.; Edgerton, S.M.; Liu, B.; Thor, A.D. Metformin attenuates transforming growth factor beta (TGF-β) mediated oncogenesis in mesenchymal stem-like/claudin-low triple negative breast cancer. Cell Cycle 2016, 15, 1046–1059. [Google Scholar] [CrossRef]
- Holmgaard, R.B.; Schaer, D.A.; Li, Y.; Castaneda, S.P.; Murphy, M.Y.; Xu, X.; Inigo, I.; Dobkin, J.; Manro, J.R.; Iversen, P.W. Targeting the TGFβ pathway with galunisertib, a TGFβRI small molecule inhibitor, promotes anti-tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint blockade. J. Immunother. Cancer 2018, 6, 47. [Google Scholar] [CrossRef] [PubMed]
- Salazar, L.G.; Lu, H.; Reichow, J.L.; Childs, J.S.; Coveler, A.L.; Higgins, D.M.; Waisman, J.; Allison, K.H.; Dang, Y.; Disis, M.L. Topical Imiquimod Plus Nab-paclitaxel for Breast Cancer Cutaneous Metastases: A Phase 2 Clinical Trial. JAMA Oncol. 2017, 3, 969–973. [Google Scholar] [CrossRef]
- Gucev, Z.S.; Oh, Y.; Kelley, K.M.; Rosenfeld, R.G. Insulin-like growth factor binding protein 3 mediates retinoic acid-and transforming growth factor β2-induced growth inhibition in human breast cancer cells. Cancer Res. 1996, 56, 1545–1550. [Google Scholar] [PubMed]
- Varadan, V.; Kamalakaran, S.; Gilmore, H.; Banerjee, N.; Janevski, A.; Miskimen, K.L.; Williams, N.; Basavanhalli, A.; Madabhushi, A.; Lezon-Geyda, K. Brief-exposure to preoperative bevacizumab reveals a TGF-β signature predictive of response in HER2-negative breast cancers. Int. J. Cancer 2016, 138, 747–757. [Google Scholar] [CrossRef]
- Formenti, S.C.; Lee, P.; Adams, S.; Goldberg, J.D.; Li, X.; Xie, M.W.; Ratikan, J.A.; Felix, C.; Hwang, L.; Faull, K.F. Focal irradiation and systemic TGFβ blockade in metastatic breast cancer. Clin. Cancer Res. 2018, 24, 2493–2504. [Google Scholar] [CrossRef] [PubMed]
- Melisi, D.; Oh, D.-Y.; Hollebecque, A.; Calvo, E.; Varghese, A.; Borazanci, E.; Macarulla, T.; Merz, V.; Zecchetto, C.; Zhao, Y. Safety and activity of the TGFβ receptor I kinase inhibitor galunisertib plus the anti-PD-L1 antibody durvalumab in metastatic pancreatic cancer. J. Immunother. Cancer 2021, 9, e002068. [Google Scholar] [CrossRef] [PubMed]
- Giannelli, G.; Santoro, A.; Kelley, R.K.; Gane, E.; Paradis, V.; Cleverly, A.; Smith, C.; Estrem, S.T.; Man, M.; Wang, S. Biomarkers and overall survival in patients with advanced hepatocellular carcinoma treated with TGF-βRI inhibitor galunisertib. PLoS ONE 2020, 15, e0222259. [Google Scholar] [CrossRef]


| Inhibitor | Clinical Trial Number | Mechanism of Action | Tumor Type |
|---|---|---|---|
| AVID200 | NCT03834662 | A receptor ectodomain trap that inhibits TGF-β1 and TGF-β3 | Malignant solid tumors |
| Bintrafusp alfa (M7824) | NCT04246489 NCT04551950 NCT03833661 | Bifunctional fusion protein with a ectodomain of TGF-ΒRII fused to human IgG1 blocking PD-L1 | Uterine cervical neoplasms, biliary tract cancer, cholangiocarcinoma, gallbladder cancer |
| Fresolimumab | NCT02581787 | A recombinant anti-TGB growth factor antibody against TGF-Β1,2,3 | Stage IA-B NSCLC |
| Vactosertib (TEW-7197) | NCT03732274 NCT04103645 | Potent TGF-Β receptor ALK4/ALK5 inhibitor | Metastatic NSCLC Myeloproliferative neoplasm |
| GT90001 | NCT03893695 | Fully human anti-ALK1 monoclonal antibody that inhibits TGF-Β and ALK-1 | Metastatic HCC |
| Nimotuzumab | NCT00957086 | Recombinant humanized murine immune antibody that blocks TGF and EGF | Squamous-cell carcinoma of the head and neck |
| LY3200882 | NCT02937272 | An ATP competitive inhibitor of the serine-threonine kinase domain of TGF-βRI | Solid tumors |
| Galunisertib (LY2157299) | NCT02423343 NCT02906397 NCT02178358 NCT03206177 | A small-molecule inhibitor of the TGF-β receptor I kinase | Solid tumors, NSCLC, HCC recurrent, lung neoplasms, esophageal neoplasms, stomach neoplasms, hepatocellular carcinoma |
| Losartan | NCT01821729 | Angiotensin II receptor antagonist | Pancreatic cancer |
| M7824 | NCT03833661 | A fusion protein inhibitor comprised of human TGF-βRII fused to the extracellular domain of human linked to the C-terminus of human anti-PD-L1 heavy chain | Biliary tract cancer, cholangiocarcinoma, gallbladder cancer |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sulaiman, A.; McGarry, S.; Chilumula, S.C.; Kandunuri, R.; Vinod, V. Clinically Translatable Approaches of Inhibiting TGF-β to Target Cancer Stem Cells in TNBC. Biomedicines 2021, 9, 1386. https://doi.org/10.3390/biomedicines9101386
Sulaiman A, McGarry S, Chilumula SC, Kandunuri R, Vinod V. Clinically Translatable Approaches of Inhibiting TGF-β to Target Cancer Stem Cells in TNBC. Biomedicines. 2021; 9(10):1386. https://doi.org/10.3390/biomedicines9101386
Chicago/Turabian StyleSulaiman, Andrew, Sarah McGarry, Sai Charan Chilumula, Rohith Kandunuri, and Vishak Vinod. 2021. "Clinically Translatable Approaches of Inhibiting TGF-β to Target Cancer Stem Cells in TNBC" Biomedicines 9, no. 10: 1386. https://doi.org/10.3390/biomedicines9101386
APA StyleSulaiman, A., McGarry, S., Chilumula, S. C., Kandunuri, R., & Vinod, V. (2021). Clinically Translatable Approaches of Inhibiting TGF-β to Target Cancer Stem Cells in TNBC. Biomedicines, 9(10), 1386. https://doi.org/10.3390/biomedicines9101386