
Inflammation in Duchenne Muscular Dystrophy–Exploring the Role of Neutrophils in Muscle Damage and Regeneration


{kind=link}
{kind=link}
Abstract
:1. Introduction
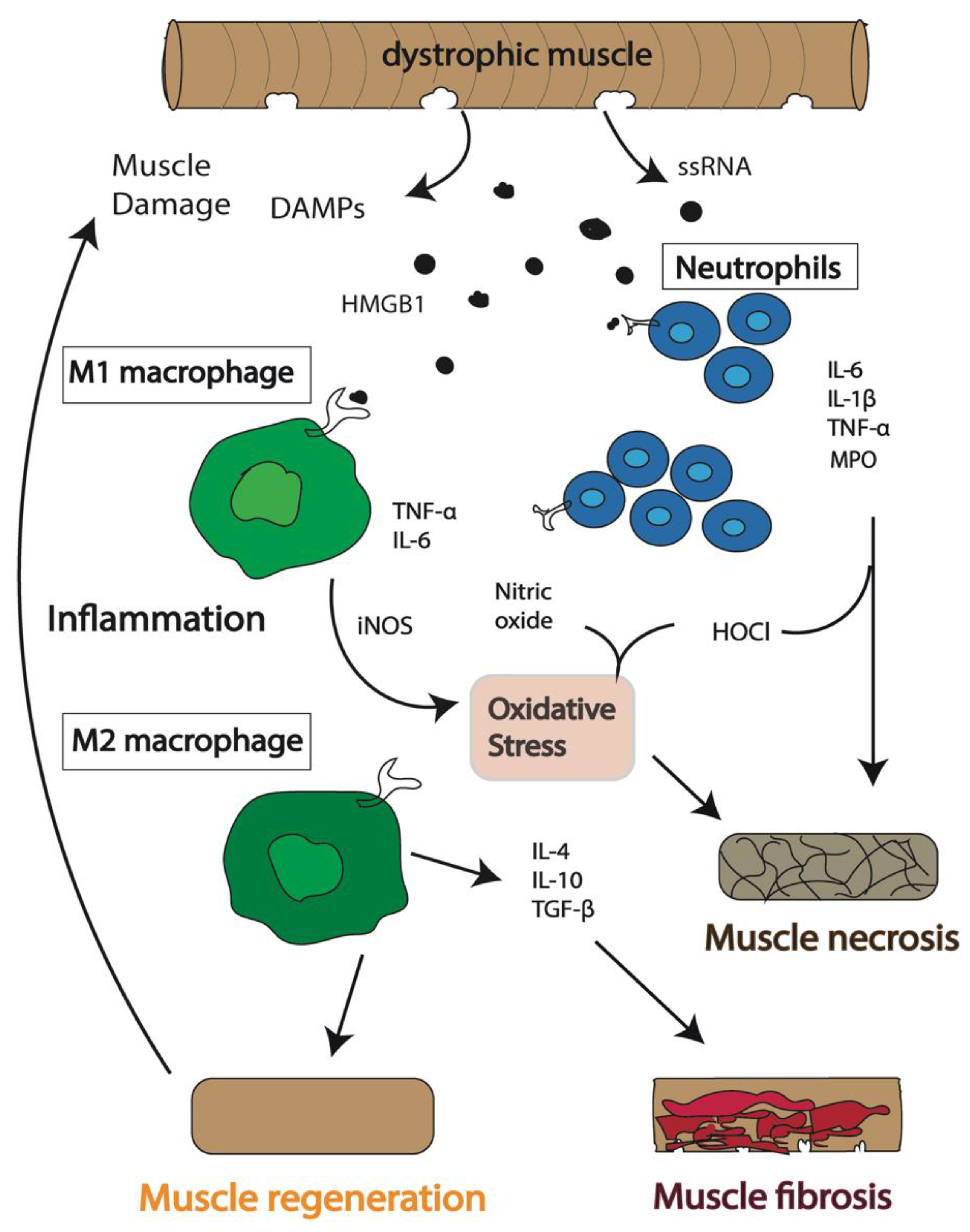
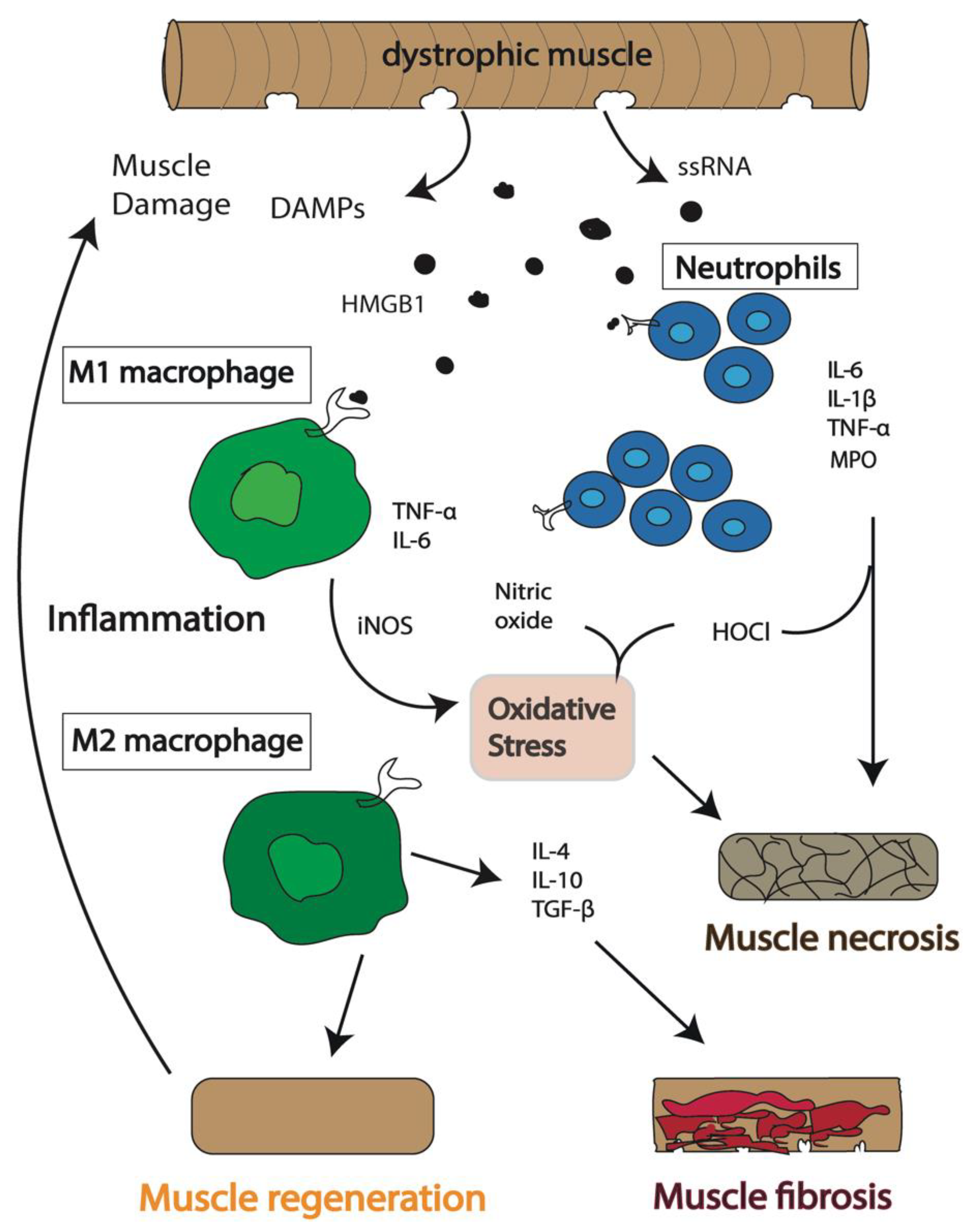
2. Persistent Activation of the Immune System Induces a Chronic Inflammatory State in DMD
3. Which Immune Cells Are the Key Players in DMD Pathogenesis?
3.1. Macrophages
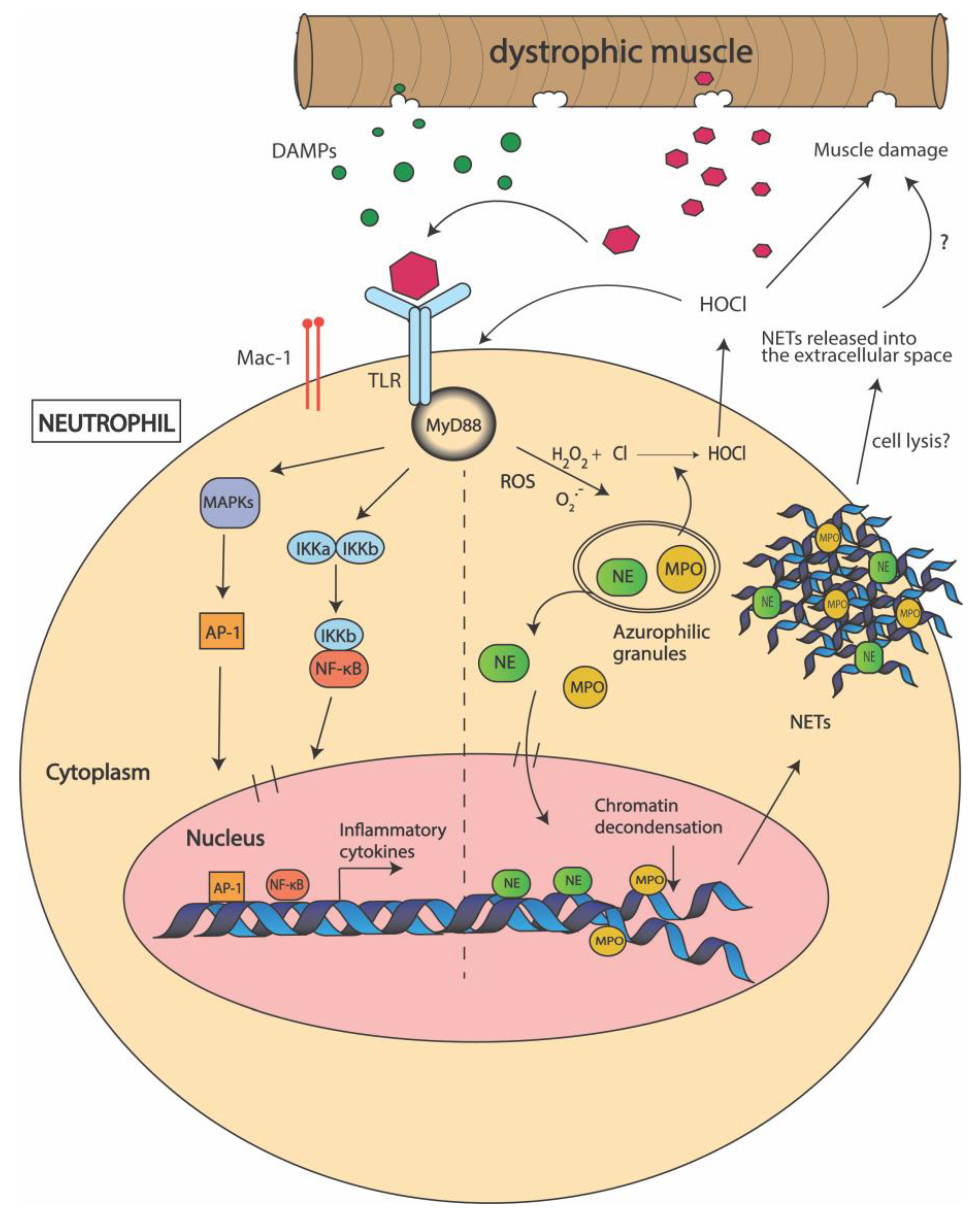
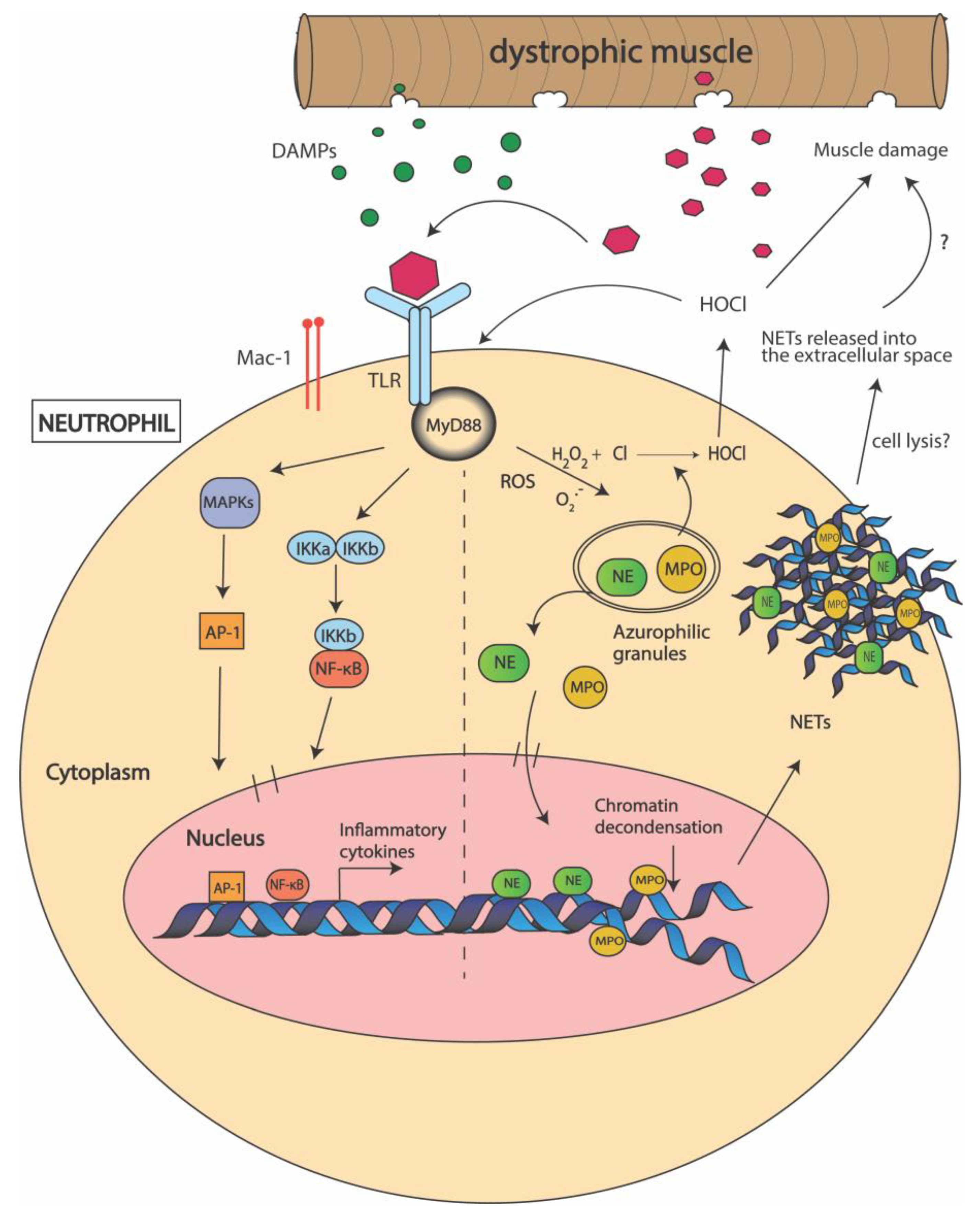
3.2. Neutrophils
4. Does Myeloperoxidase (MPO) Production Contribute to DMD Pathogenesis?
5. Can Neutrophil Elastase (NE) Be Used as a Target to Improve Muscle Regeneration in DMD?
6. Are Neutrophil Extracellular Traps (NETs) Formed in Dystrophic Muscle?
7. Impact of Aged Neutrophil Populations on Chronic Inflammation
8. Additional Factors Affecting Neutrophil Activation
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Blake, D.J.; Weir, A.; Newey, S.E.; Davies, K.E. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol. Rev. 2002, 82, 291–329. [Google Scholar] [CrossRef] [Green Version]
- Omairi, S.; Hau, K.-L.; Collins-Hooper, H.; Scott, C.; Vaiyapuri, S.; Torelli, S.; Montanaro, F.; Matsakas, A.; Patel, K. Regulation of the dystrophin-associated glycoprotein complex composition by the metabolic properties of muscle fibres. Sci. Rep. 2019, 9, 2770. [Google Scholar] [CrossRef] [Green Version]
- Tidball, J.G.; Welc, S.S.; Wehling-Henricks, M. Immunobiology of inherited muscular dystrophies. Compr. Physiol. 2018, 8, 1313–1356. [Google Scholar] [CrossRef]
- Nowak, K.J.; Davies, K.E. Duchenne muscular dystrophy and dystrophin: Pathogenesis and opportunities for treatment. EMBO Rep. 2004, 5, 872–876. [Google Scholar] [CrossRef]
- Grounds, M.D.; Terrill, J.R.; Al-Mshhdani, B.A.; Duong, M.N.; Radley-Crabb, H.G.; Arthur, P.G. Biomarkers for Duchenne muscular dystrophy: Myonecrosis, inflammation and oxidative stress. Dis. Model. Mech. 2020, 13, dmm043638. [Google Scholar] [CrossRef]
- Mastaglia, F.L.; Papadimitriou, J.M.; Kakulas, B.A. Regeneration of muscle in Duchenne muscular dystrophy: An electron microscope study. J. Neurol. Sci. 1970, 11, 425–444. [Google Scholar] [CrossRef]
- Rosenberg, A.S.; Puig, M.; Nagaraju, K.; Hoffman, E.P.; Villalta, S.A.; Rao, V.A.; Wakefield, L.M.; Woodcock, J. Immune-mediated pathology in Duchenne muscular dystrophy. Sci. Transl. Med. 2015, 7, 299rv4. [Google Scholar] [CrossRef] [Green Version]
- Juban, G.; Saclier, M.; Yacoub-Youssef, H.; Kernou, A.; Arnold, L.; Boisson, C.; Ben Larbi, S.; Magnan, M.; Cuvellier, S.; Théret, M.; et al. AMPK activation regulates LTBP4-dependent TGF-β1 secretion by pro-inflammatory macrophages and controls fibrosis in Duchenne muscular dystrophy. Cell Rep. 2018, 25, 2163–2176.e2166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpenter, S.; Karpati, G. Duchenne muscular dystrophy: Plasma membrane loss initiates muscle cell necrosis unless it is repaired. Brain 1979, 102, 147. [Google Scholar] [CrossRef] [PubMed]
- Chaplin, D.D. Overview of the immune response. J. Allergy Clin. Immunol. 2010, 125, S3–S23. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.M.; Reeves, G.; Billman, G.E.; Sturmberg, J.P. Inflammation–nature’s way to efficiently respond to all types of challenges: Implications for understanding and managing “the epidemic” of chronic diseases. Front. Med. 2018, 5, 316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deconinck, N.; Dan, B. Pathophysiology of Duchenne muscular dystrophy: Current hypotheses. Pediatr. Neurol. 2007, 36, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Dort, J.; Fabre, P.; Molina, T.; Dumont, N.A. Macrophages are key regulators of stem cells during skeletal muscle regeneration and diseases. Stem Cells Int. 2019, 2019, 4761427. [Google Scholar] [CrossRef] [PubMed]
- Ratnayake, D.; Nguyen, P.D.; Rossello, F.J.; Wimmer, V.C.; Tan, J.L.; Galvis, L.A.; Julier, Z.; Wood, A.J.; Boudier, T.; Isiaku, A.I.; et al. Macrophages provide a transient muscle stem cell niche via NAMPT secretion. Nature 2021, 591, 281–287. [Google Scholar] [CrossRef]
- Chang, N.C.; Chevalier, F.P.; Rudnicki, M.A. Satellite cells in muscular dystrophy—Lost in polarity. Trends Mol. Med. 2016, 22, 479–496. [Google Scholar] [CrossRef] [Green Version]
- Land, W.G. The Role of Damage-Associated Molecular Patterns in human diseases: Part I—Promoting inflammation and immunity. Sultan Qaboos Univ. Med. J. 2015, 15, e9–e21. [Google Scholar]
- Petrof, B. The role of innate immunity in dystrophic diaphragm pathology. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Kharraz, Y.; Guerra, J.; Pessina, P.; Serrano, A.L.; Muñoz-Cánoves, P. Understanding the process of fibrosis in Duchenne muscular dystrophy. BioMed. Res. Int. 2014, 2014, 965631. [Google Scholar] [CrossRef]
- Petrillo, S.; Pelosi, L.; Piemonte, F.; Travaglini, L.; Forcina, L.; Catteruccia, M.; Petrini, S.; Verardo, M.; D’Amico, A.; Musarò, A.; et al. Oxidative stress in Duchenne muscular dystrophy: Focus on the NRF2 redox pathway. Hum. Mol. Genet. 2017, 26, 2781–2790. [Google Scholar] [CrossRef]
- Miyatake, S.; Shimizu-Motohashi, Y.; Takeda, S.i.; Aoki, Y. Anti-inflammatory drugs for Duchenne muscular dystrophy: Focus on skeletal muscle-releasing factors. Drug Des. Dev. Ther. 2016, 10, 2745–2758. [Google Scholar] [CrossRef] [Green Version]
- Henriques-Pons, A.; Yu, Q.; Rayavarapu, S.; Cohen, T.V.; Ampong, B.; Cha, H.J.; Jahnke, V.; Van der Meulen, J.; Wang, D.; Jiang, W.; et al. Role of Toll-like receptors in the pathogenesis of dystrophin-deficient skeletal and heart muscle. Hum. Mol. Genet. 2014, 23, 2604–2617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Careccia, G.; Saclier, M.; Tirone, M.; Ruggieri, E.; Principi, E.; Raffaghello, L.; Torchio, S.; Recchia, D.; Canepari, M.; Gorzanelli, A.; et al. Rebalancing expression of HMGB1 redox isoforms to counteract muscular dystrophy. Sci. Transl. Med. 2021, 13, eaay8416. [Google Scholar] [CrossRef] [PubMed]
- Herbelet, S.; Rodenbach, A.; Paepe, B.D.; De Bleecker, J.L. Anti-Inflammatory and general glucocorticoid physiology in skeletal muscles affected by Duchenne muscular dystrophy: Exploration of steroid-sparing agents. Int. J. Mol. Sci. 2020, 21, 4596. [Google Scholar] [CrossRef] [PubMed]
- Moresi, V.; Adamo, S.; Berghella, L. The JAK/STAT Pathway in skeletal muscle pathophysiology. Front. Physiol. 2019, 10, 500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelosi, L.; Berardinelli, M.G.; De Pasquale, L.; Nicoletti, C.; D’Amico, A.; Carvello, F.; Moneta, G.M.; Catizone, A.; Bertini, E.; De Benedetti, F.; et al. Functional and morphological improvement of dystrophic muscle by interleukin 6 receptor blockade. EBioMedicine 2015, 2, 285–293. [Google Scholar] [CrossRef] [Green Version]
- Vita, G.L.; Sframeli, M.; Licata, N.; Bitto, A.; Romeo, S.; Frisone, F.; Ciranni, A.; Pallio, G.; Mannino, F.; Aguennouz, M.H.; et al. A Phase 1/2 study of Flavocoxid, an oral NF-κB inhibitor, in Duchenne muscular dystrophy. Brain Sci. 2021, 11, 115. [Google Scholar] [CrossRef]
- Finkel, R.S.; Finanger, E.; Vandenborne, K.; Sweeney, H.L.; Tennekoon, G.; Shieh, P.B.; Willcocks, R.; Walter, G.; Rooney, W.D.; Forbes, S.C.; et al. Disease-modifying effects of edasalonexent, an NF-κB inhibitor, in young boys with Duchenne muscular dystrophy: Results of the MoveDMD phase 2 and open label extension trial. Neuromuscul. Disord. 2021, 31, 385–396. [Google Scholar] [CrossRef]
- Tierney, M.T.; Aydogdu, T.; Sala, D.; Malecova, B.; Gatto, S.; Puri, P.L.; Latella, L.; Sacco, A. STAT3 signaling controls satellite cell expansion and skeletal muscle repair. Nat. Med. 2014, 20, 1182–1186. [Google Scholar] [CrossRef] [Green Version]
- Vu Hong, A.; Sanson, M.; Richard, I.; Israeli, D. A revised model for mitochondrial dysfunction in Duchenne muscular dystrophy. Eur. J. Transl. Myol. 2021. [Google Scholar] [CrossRef]
- Torraca, V.; Masud, S.; Spaink, H.P.; Meijer, A.H. Macrophage-pathogen interactions in infectious diseases: New therapeutic insights from the zebrafish host model. Dis. Model. Mech. 2014, 7, 785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villalta, S.A.; Deng, B.; Rinaldi, C.; Wehling-Henricks, M.; Tidball, J.G. IFN-γ promotes muscle damage in the mdx mouse model of Duchenne muscular dystrophy by suppressing M2 macrophage activation and inhibiting muscle cell proliferation. J. Immunol. 2011, 187, 5419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acharyya, S.; Villalta, S.A.; Bakkar, N.; Bupha-Intr, T.; Janssen, P.M.L.; Carathers, M.; Li, Z.-W.; Beg, A.A.; Ghosh, S.; Sahenk, Z.; et al. Interplay of IKK/NF-kappaB signaling in macrophages and myofibers promotes muscle degeneration in Duchenne muscular dystrophy. J. Clin. Investig. 2007, 117, 889–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kharraz, Y.; Guerra, J.; Mann, C.J.; Serrano, A.L.; Muñoz-Cánoves, P. Macrophage plasticity and the role of inflammation in skeletal muscle repair. Mediat. Inflamm. 2013, 2013, 491497. [Google Scholar] [CrossRef] [Green Version]
- Villalta, S.A.; Nguyen, H.X.; Deng, B.; Gotoh, T.; Tidball, J.G. Shifts in macrophage phenotypes and macrophage competition for arginine metabolism affect the severity of muscle pathology in muscular dystrophy. Hum. Mol. Genet. 2009, 18, 482–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro, A.F.; Souza, L.S.; Almeida, C.F.; Ishiba, R.; Fernandes, S.A.; Guerrieri, D.A.; Santos, A.L.F.; Onofre-Oliveira, P.C.G.; Vainzof, M. Muscle satellite cells and impaired late stage regeneration in different murine models for muscular dystrophies. Sci. Rep. 2019, 9, 11842. [Google Scholar] [CrossRef]
- Filippelli, R.L.; Chang, N.C. Empowering muscle stem cells for the treatment of Duchenne muscular dystrophy. Cells Tissues Organs 2021. [Google Scholar] [CrossRef]
- Vaughan, D.; Kretz, O.; Alqallaf, A.; Mitchell, R.; von der Heide, J.L.; Vaiyapuri, S.; Matsakas, A.; Pasternack, A.; Collins-Hooper, H.; Ritvos, O.; et al. Diminution in sperm quantity and quality in mouse models of Duchenne muscular dystrophy induced by a myostatin-based muscle growth-promoting intervention. Eur. J. Transl. Myol. 2020, 30, 276–285. [Google Scholar] [CrossRef]
- Németh, T.; Sperandio, M.; Mócsai, A. Neutrophils as emerging therapeutic targets. Nat. Rev. Drug Discov. 2020, 19, 253–275. [Google Scholar] [CrossRef]
- Rosales, C. Neutrophil: A cell with many roles in inflammation or several cell types? Front. Physiol. 2018, 9, 113. [Google Scholar] [CrossRef]
- Jones, H.R.; Robb, C.T.; Perretti, M.; Rossi, A.G. The role of neutrophils in inflammation resolution. Semin. Immunol. 2016, 28, 137–145. [Google Scholar] [CrossRef]
- Häger, M.; Cowland, J.B.; Borregaard, N. Neutrophil granules in health and disease. J. Intern. Med. 2010, 268, 25–34. [Google Scholar] [CrossRef]
- Sheshachalam, A.; Srivastava, N.; Mitchell, T.; Lacy, P.; Eitzen, G. Granule protein processing and regulated secretion in neutrophils. Front. Immunol. 2014, 5, 448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terrill, J.R.; Duong, M.N.; Turner, R.; Le Guiner, C.; Boyatzis, A.; Kettle, A.J.; Grounds, M.D.; Arthur, P.G. Levels of inflammation and oxidative stress, and a role for taurine in dystropathology of the Golden Retriever Muscular Dystrophy dog model for Duchenne muscular dystrophy. Redox Biol. 2016, 9, 276–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aratani, Y. Myeloperoxidase: Its role for host defense, inflammation, and neutrophil function. Arch. Biochem. Biophys. 2018, 640, 47–52. [Google Scholar] [CrossRef]
- Hodgetts, S.; Radley, H.; Davies, M.; Grounds, M.D. Reduced necrosis of dystrophic muscle by depletion of host neutrophils, or blocking TNFα function with Etanercept in mdx mice. Neuromuscul. Disord. 2006, 16, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Terrill, J.R.P.; Grounds, M.D.; Arthur, P.G. Increased taurine in pre-weaned juvenile mdx mice greatly reduces the acute onset of myofibre necrosis and dystropathology and prevents inflammation. PLoS Curr. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Lau, D.; Mollnau, H.; Eiserich, J.P.; Freeman, B.A.; Daiber, A.; Gehling, U.M.; Brümmer, J.; Rudolph, V.; Münzel, T.; Heitzer, T.; et al. Myeloperoxidase mediates neutrophil activation by association with CD11b/CD18 integrins. Proc. Natl. Acad. Sci. USA 2005, 102, 431–436. [Google Scholar] [CrossRef] [Green Version]
- Martinez, L.; Ermolova, N.V.; Ishikawa, T.-O.; Stout, D.B.; Herschman, H.R.; Spencer, M.J. A reporter mouse for optical imaging of inflammation in mdx muscles. Skelet. Muscle 2015, 5, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azcutia, V.; Kelm, M.; Luissint, A.-C.; Boerner, K.; Flemming, S.; Quiros, M.; Newton, G.; Nusrat, A.; Luscinskas, F.W.; Parkos, C.A. Neutrophil expressed CD47 regulates CD11b/CD18-dependent neutrophil transepithelial migration in the intestine in vivo. Mucosal Immunol. 2021, 14, 331–341. [Google Scholar] [CrossRef]
- Mojumdar, K.; Liang, F.; Giordano, C.; Lemaire, C.; Danialou, G.; Okazaki, T.; Bourdon, J.; Rafei, M.; Galipeau, J.; Divangahi, M.; et al. Inflammatory monocytes promote progression of Duchenne muscular dystrophy and can be therapeutically targeted via CCR2. EMBO Mol. Med. 2014, 6, 1476–1492. [Google Scholar] [CrossRef]
- Domon, H.; Nagai, K.; Maekawa, T.; Oda, M.; Yonezawa, D.; Takeda, W.; Hiyoshi, T.; Tamura, H.; Yamaguchi, M.; Kawabata, S.; et al. Neutrophil elastase subverts the immune response by cleaving toll-like receptors and cytokines in Pneumococcal Pneumonia. Front. Immunol. 2018, 9, 732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gramegna, A.; Amati, F.; Terranova, L.; Sotgiu, G.; Tarsia, P.; Miglietta, D.; Calderazzo, M.A.; Aliberti, S.; Blasi, F. Neutrophil elastase in bronchiectasis. Respir. Res. 2017, 18, 211. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-H.; Chung, J.H.; Son, B.S.; Kim, Y.J.; Lee, S.G. Effect of a neutrophil elastase inhibitor on ventilator-induced lung injury in rats. J. Thorac. Dis. 2014, 6, 1681–1689. [Google Scholar]
- Yao, W.; Han, X.; Guan, Y.; Guan, J.; Wu, S.; Chen, C.; Li, H.; Hei, Z. Neutrophil elastase inhibitors suppress oxidative stress in lung during liver transplantation. Oxid. Med. Cell Longev. 2019, 2019, 7323986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arecco, N.; Clarke, C.J.; Jones, F.K.; Simpson, D.M.; Mason, D.; Beynon, R.J.; Pisconti, A. Elastase levels and activity are increased in dystrophic muscle and impair myoblast cell survival, proliferation and differentiation. Sci. Rep. 2016, 6, 24708. [Google Scholar] [CrossRef] [Green Version]
- Iba, T.; Murai, M.; Nagaoka, I.; Tabe, Y. Neutrophil extracellular traps, damage-associated molecular patterns, and cell death during sepsis. Acute Med. Surg. 2013, 1, 2–9. [Google Scholar] [CrossRef]
- Lee, K.H.; Kronbichler, A.; Park, D.D.; Park, Y.; Moon, H.; Kim, H.; Choi, J.H.; Choi, Y.; Shim, S.; Lyu, I.S.; et al. Neutrophil extracellular traps (NETs) in autoimmune diseases: A comprehensive review. Autoimmun. Rev. 2017, 16, 1160–1173. [Google Scholar] [CrossRef]
- Hu, Z.; Murakami, T.; Tamura, H.; Reich, J.; Kuwahara-Arai, K.; Iba, T.; Tabe, Y.; Nagaoka, I. Neutrophil extracellular traps induce IL-1β production by macrophages in combination with lipopolysaccharide. Int. J. Mol. Med. 2017, 39, 549–558. [Google Scholar] [CrossRef] [Green Version]
- Edwards, N.J.; Hwang, C.; Marini, S.; Pagani, C.A.; Spreadborough, P.J.; Rowe, C.J.; Yu, P.; Mei, A.; Visser, N.; Li, S.; et al. The role of neutrophil extracellular traps and TLR signaling in skeletal muscle ischemia reperfusion injury. FASEB J. 2020, 34, 15753–15770. [Google Scholar] [CrossRef]
- Wang, J. Neutrophils in tissue injury and repair. Cell Tissue Res. 2018, 371, 531–539. [Google Scholar] [CrossRef] [Green Version]
- Colom, B.; Bodkin, J.V.; Beyrau, M.; Woodfin, A.; Ody, C.; Rourke, C.; Chavakis, T.; Brohi, K.; Imhof, B.A.; Nourshargh, S. Leukotriene B4-neutrophil elastase axis drives neutrophil reverse transendothelial cell migration in vivo. Immunity 2015, 42, 1075–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adrover, J.M.; Nicolás-Ávila, J.A.; Hidalgo, A. Aging: A temporal dimension for neutrophils. Trends Immunol. 2016, 37, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Uhl, B.; Vadlau, Y.; Zuchtriegel, G.; Nekolla, K.; Sharaf, K.; Gaertner, F.; Massberg, S.; Krombach, F.; Reichel, C.A. Aged neutrophils contribute to the first line of defense in the acute inflammatory response. Blood 2016, 128, 2327–2337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pescatori, M.; Broccolini, A.; Minetti, C.; Bertini, E.; Bruno, C.; D’amico, A.; Bernardini, C.; Mirabella, M.; Silvestri, G.; Giglio, V.; et al. Gene expression profiling in the early phases of DMD: A constant molecular signature characterizes DMD muscle from early postnatal life throughout disease progression. FASEB J. 2007, 21, 1210–1226. [Google Scholar] [CrossRef]
- Kranig, S.A.; Tschada, R.; Braun, M.; Pöschl, J.; Frommhold, D.; Hudalla, H. Dystrophin deficiency promotes leukocyte recruitment in mdx mice. Pediatr. Res. 2020, 87, 798. [Google Scholar] [CrossRef] [PubMed]
- Vidal, B.; Ardite, E.; Suelves, M.; Ruiz-Bonilla, V.; Janué, A.; Flick, M.J.; Degen, J.L.; Serrano, A.L.; Muñoz-Cánoves, P. Amelioration of Duchenne muscular dystrophy in mdx mice by elimination of matrix-associated fibrin-driven inflammation coupled to the αβ leukocyte integrin receptor. Hum. Mol. Genet. 2012, 21, 1989–2004. [Google Scholar] [CrossRef] [Green Version]
- Vidal, B.; Serrano, A.L.; Tjwa, M.; Suelves, M.; Ardite, E.; De Mori, R.; Baeza-Raja, B.; Martínez de Lagrán, M.; Lafuste, P.; Ruiz-Bonilla, V.; et al. Fibrinogen drives dystrophic muscle fibrosis via a TGFβ/alternative macrophage activation pathway. Genes Dev. 2008, 22, 1747–1752. [Google Scholar] [CrossRef] [Green Version]
- Rubel, C.; Fernández, G.C.; Dran, G.; Bompadre, M.B.; Isturiz, M.A.; Palermo, M.S. Fibrinogen promotes neutrophil activation and delays apoptosis. J. Immunol. 2001, 166, 2002. [Google Scholar] [CrossRef] [Green Version]
- Mortaz, E.; Alipoor, S.D.; Adcock, I.M.; Mumby, S.; Koenderman, L. Update on neutrophil function in severe inflammation. Front. Immunol. 2018, 9, 2171. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tulangekar, A.; Sztal, T.E. Inflammation in Duchenne Muscular Dystrophy–Exploring the Role of Neutrophils in Muscle Damage and Regeneration. Biomedicines 2021, 9, 1366. https://doi.org/10.3390/biomedicines9101366
Tulangekar A, Sztal TE. Inflammation in Duchenne Muscular Dystrophy–Exploring the Role of Neutrophils in Muscle Damage and Regeneration. Biomedicines. 2021; 9(10):1366. https://doi.org/10.3390/biomedicines9101366
Chicago/Turabian StyleTulangekar, Ankita, and Tamar E. Sztal. 2021. "Inflammation in Duchenne Muscular Dystrophy–Exploring the Role of Neutrophils in Muscle Damage and Regeneration" Biomedicines 9, no. 10: 1366. https://doi.org/10.3390/biomedicines9101366
APA StyleTulangekar, A., & Sztal, T. E. (2021). Inflammation in Duchenne Muscular Dystrophy–Exploring the Role of Neutrophils in Muscle Damage and Regeneration. Biomedicines, 9(10), 1366. https://doi.org/10.3390/biomedicines9101366