
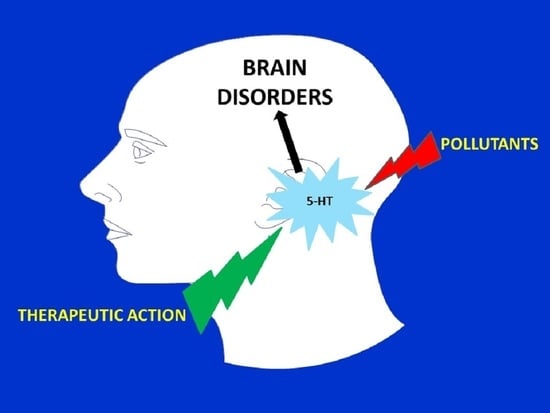
Is the Exposome Involved in Brain Disorders through the Serotoninergic System?




Abstract
{kind=link}
{kind=link}
1. Introduction
2. Effects of Environmental Chemical Pollutants on the Serotoninergic System
3. Neurodevelopmental Disorders
4. Neurodegenerative Disorders
5. Neurobehavioral Disorders
6. Cancer
7. Discussion and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Mohammad-Zadeh, L.F.; Moses, L.; Gwaltney-Brant, S.M. Serotonin: A review. J. Vet. Pharmacol. Ther. 2008, 31, 187–199. [Google Scholar] [CrossRef]
- Sarrouilhe, D.; Clarhaut, J.; Defamie, N.; Mesnil, M. Serotonin and cancer: What is the link? Curr. Mol. Med. 2015, 15, 62–77. [Google Scholar] [CrossRef]
- Gaspar, P.; Lillesaar, C. Probing the diversity of serotonin neurons. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 2382–2394. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, D.; Clarke, D.E.; Fozard, J.R.; Hartig, P.R.; Martin, G.R.; Mylecharane, E.J.; Saxena, P.R.; Humphrey, P.P., VII. International union of pharmacology classification of receptors for 5-Hydroxytryptamine (serotonin). Pharmacol. Rev. 1994, 46, 157–203. [Google Scholar] [PubMed]
- Noda, M.; Higashida, H.; Aoki, S.; Wada, K. Multiple signal transduction pathways mediated by 5-HT receptors. Mol. Neurobiol. 2004, 29, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Millan, M.J.; Marin, P.; Bockaert, J.; Mannoury la Cour, C. Signaling at G-protein-coupled serotonin receptors: Recent advances and future research directions. Trends Pharmacol. Sci. 2008, 29, 454–464. [Google Scholar] [CrossRef]
- Cowen, D.S. Serotonin and neuronal growth factors—A convergence of signaling pathways. J. Neurochem. 2007, 101, 1161–1171. [Google Scholar] [CrossRef]
- Yun, H.M.; Kim, S.; Kim, H.J.; Kostenis, E.; Kim, J.I.; Seong, J.Y.; Baik, J.H.; Rhim, H. The novel cellular mechanism of human 5-HT6 receptor through an interaction with Fyn. J. Biol. Chem. 2007, 282, 5496–5505. [Google Scholar] [CrossRef]
- Sarrouilhe, D.; Mesnil, M. Serotonin and human cancer: A critical view. Biochimie 2019, 161, 46–50. [Google Scholar] [CrossRef]
- Azmitia, E.C. Modern views on an ancient chemical: Serotonin effects on cell proliferation, maturation, and apoptosis. Brain Res. Bull. 2001, 56, 413–424. [Google Scholar] [CrossRef]
- Bonnin, A.; Goeden, N.; Chen, K.; Wilson, M.L.; King, J.; Shih, J.C.; Blakely, R.D.; Deneris, E.S.; Levitt, P. A transient placental source of serotonin for the fetal forebrain. Nature 2011, 472, 347–350. [Google Scholar] [CrossRef] [PubMed]
- Bonnin, A.; Levitt, P. Fetal, maternal, and placental sources of serotonin and new implications for developmental programming of the brain. Neuroscience 2011, 197, 1–7. [Google Scholar] [CrossRef]
- Xing, L.; Kalebic, N.; Namba, T.; Vaid, S.; Wimberger, P.; Huttner, V.B. Serotonin receptor 2A activation promotes evolutionarily relevant basal progenitor proliferation in the developing neocortex. Neuron 2020, 108, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Bonnin, A.; Torii, M.; Wang, L.; Rakic, P.; Levitt, P. Serotonin modulates the response of embryonic thalamocortical axons to netrin-1. Nat. Neurosci. 2007, 10, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Blue, M.E.; Johnston, M.V.; Moloney, C.B.; Hohmann, C.F. Serotonin dysfunction in autism. In Autism; Zimmerman, A.W., Ed.; Humana Press: Totowa, NJ, USA, 2008; pp. 111–132. [Google Scholar]
- Mandy, M.; Nyirenda, M. Developmental Origins of Health and Disease: The relevance to developing nations. Int. Health 2018, 10, 66–70. [Google Scholar] [CrossRef]
- Mesnil, M.; Defamie, N.; Naus, C.; Sarrouilhe, D. Brain disorders and chemical pollutants: A gap junction link? Biomolecules 2021, 11, 51. [Google Scholar] [CrossRef]
- Sun, Y.; Nakashima, M.N.; Takahashi, M.; Kuroda, N.; Nakashima, K. Determination of bisphenol A in rat brain by microdialysis and column switching high-performance liquid chromatography with fluorescence detection. Biomed. Chromatogr. 2002, 16, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Sarrouilhe, D.; Mesnil, M.; Dejean, C. The serotonin system in autism spectrum disorder. In Serotonin and Dopamine Receptors; Munoz, M., Mckinney, M., Eds.; Nova Publishers: New York, NY, USA, 2018; pp. 79–108. [Google Scholar]
- Honma, T.; Miyagawa, M.; Suda, M.; Wang, R.S.; Kobayashi, K.; Sekiguchi, S. Effects of perinatal exposure to bisphenol A on brain neurotransmitters in female rat offspring. Ind. Health 2006, 44, 510–524. [Google Scholar] [CrossRef][Green Version]
- Matsuda, S.; Saika, S.; Amano, K.; Shimizu, E.; Sajiki, J. Changes in brain monoamine levels in neonatal rats exposed to bisphenol A at low doses. Chemosphere 2010, 78, 894–906. [Google Scholar] [CrossRef]
- Nakamura, K.; Itoh, K.; Yoshimoto, K.; Sugimoto, T.; Fushiki, S. Prenatal and lactational exposure to low-doses of bisphenol A alters brain monoamine concentration in adult mice. Neurosci. Lett. 2010, 484, 66–70. [Google Scholar] [CrossRef]
- Matsuda, S.; Matsuzawa, D.; Ishii, D.; Tomizawa, H.; Sajiki, J.; Shimizu, E. Perinatal exposure to bisphenol A enhances contextual fear memory and affects the serotoninergic system in juvenile female mice. Horm. Behav. 2013, 63, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Goto, S.; Ogi, H.; Fushiki, S.; Itoh, K. Prenatal and lactational bisphenol A exposure does not alter serotonergic neurons morphologically in the murine dorsal raphe nucleus. Brain Dev. 2017, 39, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Saied, N.M.; Hassan, W.A. Long-term exposure to xenoestrogens alters some brain monoamines and both serum thyroid hormones and cortisol levels in adult male rats. J. Basic Appl. Zool. 2014, 67, 205–211. [Google Scholar] [CrossRef]
- Castro, B.; Sánchez, P.; Torres, J.M.; Ortega, E. Bisphenol A, bisphenol F and bisphenol S affect differently 5α-reductase expression and dopamine–serotonin systems in the prefrontal cortex of juvenile female rats. Environ. Res. 2015, 142, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Slotkin, T.A.; Tate, C.A.; Ryde, I.T.; Levin, E.D.; Seidler, F.J. Organophosphate insecticides target the serotonergic system in developing rat brain regions: Disparate effects of diazinon and parathion at doses spanning the threshold for cholinesterase inhibition. Environ. Health Perspect. 2006, 114, 1542–1546. [Google Scholar] [CrossRef]
- Slotkin, T.A.; Seidler, F.J. Developmental neurotoxicants target neurodifferentiation into the serotonin phenotype: Chlorpyrifos, diazinon, dieldrin and divalent nickel. Toxicol. Appl. Pharmacol. 2008, 233, 211–219. [Google Scholar] [CrossRef]
- Slotkin, T.A.; Skavicus, S.; Ko, A.; Levin, E.D.; Seidler, F.J. Perinatal diazinon exposure compromises the development of acetylcholine and serotonin systems. Toxicology 2019, 424, 152240. [Google Scholar] [CrossRef]
- Bist, R.; Bhatt, D.K. The evaluation of effect of alpha-lipoic acid and vitamin E on the lipid peroxidation, gamma-amino butyric acid and serotonin level in the brain of mice (Mus musculus) acutely intoxicated with lindane. J. Neurol. Sci. 2009, 276, 99–102. [Google Scholar] [CrossRef]
- Hong, J.S.; Herr, D.W.; Hudson, P.M.; Tilson, H.A. Neurochemical effects of DDT in rat brain in vivo. Arch. Toxicol. Suppl. 1986, 9, 14–25. [Google Scholar] [CrossRef]
- Uphouse, L.; Eckols, K. Serotonin receptors in striatum after chlordecone treatment of adult female rats. Neurotoxicology 1986, 7, 25–32. [Google Scholar] [PubMed]
- Brown, H.E.; Salamanca, S.; Stewart, G.; Uphouse, L. Chlordecone (Kepone) on the night of proestrus inhibits female sexual behavior in CDF-344 rats. Toxicol. Appl. Pharmacol. 1991, 110, 97–106. [Google Scholar] [CrossRef]
- Bharatiya, R.; Chagraoui, A.; De Deurwaerdere, S.; Argiolas, A.; Melis, M.R.; Sanna, F.; De Deurwaerdere, P. Chronic administration of fipronil heterogeneously alters the neurochemistry of monoaminergic systems in the rat brain. Int. J. Mol. Sci. 2020, 21, 5711. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Larrañaga, M.R.; Anadón, A.; Martínez, M.A.; Martínez, M.; Castellano, V.J.; Díaz, M.J. 5-HT loss in rat brain by type II pyrethroid insecticides. Toxicol. Ind. Health. 2003, 19, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Honma, T.; Suda, M.; Miyagawa, M.; Wang, R.-S.; Kenichi Kobayashi, K.; Sekiguchi, S. Alteration of brain neurotransmitters in female rat offspring induced by prenatal administration of 16 and 64 mg/kg of 2,2′,4,4′,5,5′-hexachlorobiphenyl (PCB153). Ind. Health 2009, 47, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Kimber, I.; Dearman, R.J. An assessment of the ability of phthalates to influence immune and allergic responses. Toxicology 2010, 271, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Hlisníková, H.; Petrovičová, I.; Kolena, B.; Šidlovská, M.; Sirotkin, A. Effects and mechanisms of phthalates’ action on reproductive processes and reproductive health: A literature review. Int. J. Environ. Res. Public Health 2020, 17, 6811. [Google Scholar] [CrossRef]
- Ishido, M.; Masuo, Y.; Sayato-Suzuki, J.; Oka, S.; Niki, E.; Morita, M. Dicyclohexylphthalate causes hyperactivity in the rat concomitantly with impairment of tyrosine hydroxylase immunoreactivity. J. Neurochem. 2004, 91, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Deegan, A.M.; Steinhauer, R.B.; Feinn, R.S.; Moeller, M.C.; Pylypiw, H.M., Jr.; Nabel, M.; Kovelowsky, C.J.; Kaplan, L.A.E. Modulation of brain serotonin by benzyl butyl phthalate in Fundulus heteroclitus (mummichog). Ecotoxicology 2019, 28, 1038–1045. [Google Scholar] [CrossRef]
- Repouskou, A.; Papado-Poulou, A.K.; Panagiotidou, E.; Trichas, P.; Lindh, C.; Bergman, Å.; Gennings, C.; Bornehag, C.G.; Rüegg, J.; Kitraki, E.; et al. Long term transcriptional and behavioral effects in mice developmentally exposed to a mixture of endocrine disruptors associated with delayed human neurodevelopment. Sci. Rep. 2020, 10, 9367. [Google Scholar] [CrossRef]
- Carhart-Harris, R.; Nutt, D. Serotonin and brain function: A tale of two receptors. J. Psychopharmacol. 2017, 31, 1091–1120. [Google Scholar] [CrossRef]
- Block, M.L.; Calderon-Garciduenas, L. Air pollution: Mechanisms of neuroinflammation and CNS disease. Trends Neurosci. 2009, 32, 506–516. [Google Scholar] [CrossRef]
- Elder, A.; Gelein, R.; Silva, V.; Feikert, T.; Opanashuk, L.; Carter, J.; Potter, R.; Maynard, A.; Ito, Y.; Jacob Finkelstein, J.; et al. Translocation of inhaled ultrafine manganese oxide particles to the central nervous system. Environ. Health Perspect. 2006, 114, 1172–1178. [Google Scholar] [CrossRef]
- Block, M.L.; Elder, A.; Auten, R.L.; Bilbo, S.D.; Chen, H.; Chen, J.-C.; Cory-Slechta, D.A.; Costa, D.; Diaz-Sanchez, D.; Dorman, D.C.; et al. The outdoor air pollution and brain health workshop. Neuro Toxicol. 2012, 33, 972–984. [Google Scholar] [CrossRef]
- Tomei, F.; Rosati, M.V.; Ciarrocca, M.; Baccolo, T.P.; Caciari, T.; Tomao, E. Occupational exposure to urban pollutants and urinary 5-hydroxy-3-indoleacetic acid. J. Environ. Health 2004, 66, 38–42, 44. [Google Scholar] [PubMed]
- Yokota, S.; Oshio, S.; Moriya, N.; Takeda, K. Social isolation-induced territorial aggression in male offspring is enhanced by exposure to diesel exhaust during pregnancy. PLoS ONE 2016, 11, e0149737. [Google Scholar] [CrossRef] [PubMed]
- Rose, M.; Filiatreault, A.; Guénette, J.; Williams, A.; Thomson, E.M. Ozone increases plasma kynurenine-tryptophan ratio and impacts hippocampal serotonin receptor and neurotrophic factor expression: Role of stress hormones. Environ. Res. 2020, 185, 109483. [Google Scholar] [CrossRef] [PubMed]
- Mokoena, M.L.; Harvey, B.H.; Viljoen, F.; Ellis, S.M.; Brink, C.B. Ozone exposure of Flinders Sensitive Line rats is a rodent translational model of neurobiological oxidative stress with relevance for depression and antidepressant response. Psychopharmacology 2015, 232, 2921–2938. [Google Scholar] [CrossRef] [PubMed]
- Grandjean, P.; Landrigan, P.J. Neurobehavioural effects of developmental toxicity. Lancet Neurol. 2014, 13, 330–338. [Google Scholar] [CrossRef]
- Janusonis, S.; Anderson, G.M.; Shifrovich, I.; Rakic, P. Ontogeny of brain and blood serotonin levels in 5-HT receptor knockout mice: Potential relevance to the neurobiology of autism. J. Neurochem. 2006, 99, 1019–1031. [Google Scholar] [CrossRef]
- Leboyer, M.; Philippe, A.; Bouvard, M.; Guilloud-Bataille, M.; Bondoux, D.; Tabuteau, F.; Feingold, J.; Moure Si Meoni, M.C.; Launay, J.M. Whole blood serotonin and plasma beta-endorphin in autistic probands and their first-degree relatives. Biol. Psychiatry 1999, 45, 158–163. [Google Scholar] [CrossRef]
- Pagan, C.; Delorme, R.; Callebert, J.; Goubran-Botros, H.; Ansellem, F.; Drouot, X.; Boudebesse, C.; Le Dudal, K.; Ngo-Nguyen, N.; Laouamri, H.; et al. The serotonin-N-acetylserotonin-melatonin pathway as a biomarker for autism spectrum disorders. Transl. Psychiatry 2014, 4, e479. [Google Scholar] [CrossRef] [PubMed]
- Chandana, S.R.; Behen, M.E.; Juhász, C.; Muzik, O.; Rothermel, R.D.; Mangner, T.J.; Chakraborty, P.K.; Chugani, H.T.; Chugani, D.C. Significance of abnormalities in developmental trajectory and asymmetry of cortical serotonin synthesis in autism. Int. J. Dev. Neurosci. 2005, 23, 171–182. [Google Scholar] [CrossRef]
- Chugani, D.C. Application of PET and SPECT to the study of autism spectrum disorders. In PET and SPECT in Psychiatry; Dierckx, R.A.J.O., Otte, A., de Vries, E.F.J., van Waarde, A., den Boer, J.A., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 691–707. [Google Scholar] [CrossRef]
- Casanova, M.F.; van Kooten, I.A.; Switala, A.E.; van Engeland, H.; Heinsen, H.; Steinbusch, H.W.; Hof, P.R.; Trippe, J.; Stone, J.; Schmitz, C. Minicolumnar abnormalities in autism. Acta Neuropathol. 2006, 112, 287–303. [Google Scholar] [CrossRef] [PubMed]
- Nomura, Y.; Nagao, Y.; Kimura, K.; Hachimori, K.; Segawa, M. Epilepsy in autism: A pathophysiological consideration. Brain Dev. 2010, 32, 799–804. [Google Scholar] [CrossRef]
- Daly, E.M.; Deeley, Q.; Ecker, C.; Craig, M.; Hallahan, B.; Murphy, C.; Johnston, P.; Spain, D.; Gillan, N.; Brammer, M.; et al. Serotonin and the neural processing of facial emotions in adults with autism: An fMRI study using acute tryptophan depletion. Arch. Gen. Psychiatry 2012, 69, 1003–1013. [Google Scholar] [CrossRef]
- Muller, C.L.; Anacker, A.M.J.; Veenstra-VanderWeele, J. The serotonin system in autism spectrum disorder: From biomarker to animal models. Neuroscience 2016, 321, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Harrington, R.A.; Lee, L.C.; Crum, R.M.; Zimmerman, A.W.; Hertz-Picciotto, I. Prenatal SSRI use and offspring with autism spectrum disorder or developmental delay. Pediatrics 2014, 133, e1241–e1248. [Google Scholar] [CrossRef]
- Ye, B.S.; Leung, A.O.W.; Wong, M.H. The association of environmental toxicants and autism spectrum disorders in children. Environ. Pollut. 2017, 227, 234–242. [Google Scholar] [CrossRef]
- Shelton, J.F.; Geraghty, E.M.; Tancredi, D.J.; Delwiche, L.D.; Schmidt, R.J.; Ritz, B.; Hansen, R.L.; Hertz-Picciotto, I. Neurodevelopmental disorders and prenatal residential proximity to agricultural pesticides: The CHARGE study. Environ. Health Perspect. 2014, 122, 10. [Google Scholar] [CrossRef]
- McGuinn, L.A.; Windham, G.C.; Kalkbrenner, A.E.; Bradley, C.; Di, Q.; Croen, L.A.; Fallin, M.D.; Hoffman, K.; Ladd-Acosta, C.; Schwartz, J.; et al. Early life exposure to air pollution and autism spectrum disorder: Findings from a multisite case-control study. Epidemiology 2020, 31, 103–114. [Google Scholar] [CrossRef]
- Stein, T.P.; Schluter, M.D.; Steer, R.A.; Guo, L.; Ming, X. Bisphenol A exposure in children with autism spectrum disorders. Autism Res. 2015, 8, 272–283. [Google Scholar] [CrossRef]
- Hsiao, E.Y.; McBride, S.W.; Hsien, S.; Sharon, G.; Hyde, E.R.; McCue, T.; Codelli, J.A.; Chow, J.; Reisman, S.E.; Petrosino, J.F.; et al. The micobiota modulates gut physiology and behavioral abnormalities associated with autism. Cell 2013, 155, 1451–1463. [Google Scholar] [CrossRef] [PubMed]
- Noakes, R. The aryl hydrocarbon receptor: A review of its role in the physiology and pathology of the integument and its relationship to the tryptophan metabolism. Int. J. Tryptophan Res. 2015, 8, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Sarrouilhe, D.; Dejean, C. Autism spectrum disorders and bisphenol A: Is serotonin the lacking link in the chain? L’Encéphale 2017, 43, 402–404. [Google Scholar] [CrossRef]
- Marks, A.R.; Harley, K.; Bradman, A.; Kogut, K.; Barr, D.B.; Johnson, C.; Eskenazi, B. Organophosphate pesticide exposure and attention in young Mexican-American children: The CHAMACOS study. Environ. Health Perspect. 2010, 118, 1768–1774. [Google Scholar] [CrossRef]
- Sagiv, S.K.; Thurston, S.W.; Bellinger, D.C.; Tolbert, P.E.; Altshul, L.M.; Korrick, S.A. Prenatal organochlorine exposure and behaviors associated with attention deficit hyperactivity disorder in school-aged children. Am. J. Epidemiol. 2010, 171, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Nigg, J.; Nikolas, M.; Mark Knowttnerus, G.; Kavanagh, K.; Friderici, K. Confirmation and extension of association of blood lead with attention-deficit/hyperactivity disorder (ADHD) and ADHD symptom domains at population typical exposure levels. J. Child Psychol. Psych. 2010, 51, 58–65. [Google Scholar] [CrossRef]
- Engel, S.M.; Villanger, G.D.; Nethery, R.C.; Thomsen, C.; Sakhi, A.K.; Drover, S.S.M.; Hoppin, J.A.; Zeiner, P.; Knudsen, G.P.; Reichborn-Kjennerud, T.; et al. Prenatal Phthalates, Maternal Thyroid Function, and Risk of Attention-Deficit Hyperactivity Disorder in the Norwegian Mother and Child Cohort. Environ. Health Perspect. 2018, 126, 5. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.M.; Yolton, K.; Dietrich, K.N.; Hornung, R.; Ye, X.; Calafat, A.M.; Lanphear, B.P. Prenatal bisphenol A exposure and early childhood behavior. Environ. Health Perspect. 2009, 117, 1945–1952. [Google Scholar] [CrossRef]
- Yorifuji, T.; Kashima, S.; Diez, M.H.; Kado, Y.; Sanada, S.; Doi, H. Prenatal exposure to outdoor air pollution and child behavioral problems at school age in Japan. Environ. Int. 2017, 99, 192–198. [Google Scholar] [CrossRef]
- Nigg, J.T. ADHD, lead exposure and prevention: How much lead or how much evidence is needed? Expert Rev. Neurother. 2008, 8, 519–521. [Google Scholar] [CrossRef]
- Rochester, J.R.; Bolden, A.L.; Kwiatkowski, C.F. Prenatal exposure to bisphenol A and hyperactivity in children: A systematic review and meta-analysis. Environ. Int. 2018, 114, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Thapar, A.; Cooper, M.; Eyre, O.; Langley, K. What have we learnt about the causes of ADHD? J. Child. Psychol. Psychiatry 2013, 54, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Banaschewski, T.; Becker, K.; Scherag, S.; Franke, B.; Coghill, D. Molecular genetics of attention-deficit/hyperactivity disorder: An overview. Eur. Child. Adolesc. Psychiatry 2010, 19, 237–257. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, E.; Nandagopal, K. Does serotonin deficit mediate susceptibility to ADHD? Neurochem. Int. 2015, 82, 52–68. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.J.; Yu, Y.H.; Fu, M.L.; Yeh, W.T.; Hsu, J.L.; Yang, Y.H.; Chen, W.J.; Chiang, B.L.; Pan, W.H. Attention deficit–hyperactivity disorder is associated with allergic symptoms and low levels of hemoglobin and serotonin. Sci. Rep. 2018, 8, 10229. [Google Scholar] [CrossRef] [PubMed]
- Petrucci, A.N.; Joyal, K.G.; Purnell, B.S.; Buchanan, G.F. Serotonin and sudden unexpected death in epilepsy. Exp. Neurol. 2020, 325, 113145. [Google Scholar] [CrossRef]
- Maia, G.H.; Brazete, C.S.; Soares, J.I.; Luz, L.L.; Lukoyanov, N.V. Serotonin depletion increases seizure susceptibility and worsens neuropathological outcomes in kainate model of epilepsy. Brain Res. Bull. 2017, 134, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Bagdy, G.; Kecskemeti, V.; Riba, P.; Jakus, R. Serotonin and epilepsy. J. Neurochem. 2007, 100, 857–873. [Google Scholar] [CrossRef] [PubMed]
- Richerson, G.B.; Buchanan, G.F. The serotonin axis: Shared mechanisms in seizures, depression, and SUDEP. Epilepsia 2011, 52, 28–38. [Google Scholar] [CrossRef]
- Schenkel, L.C.; Bragatti, J.A.; Torres, C.M.; Martin, K.C.; Gus-Manfro, G.; Leistner-Segal, S.; Bianchin, M.M. Serotonin transporter gene (5HTT) polymorphisms and temporal lobe epilepsy. Epilepsy Res. 2011, 95, 152–157. [Google Scholar] [CrossRef]
- Esmail, E.H.; Labib, D.M.; Rabie, W.A. Association of serotonin transporter gene (5HTT) polymorphism and juvenile myoclonic epilepsy: A case-control study. Acta Neurol. Belg. 2015, 115, 247–251. [Google Scholar] [CrossRef]
- Rocha, L.; Lorigados-Pedre, L.; Orozco-Suarez, S.; Morales-Chacon, L.; Alonso-Vanegas, M.; Garcia-Maeso, I.; Villeda-Hernández, J.; Osorio-Rico, L.; Estupiñán, B.; Quintana, C. Autoradiography reveals selective changes in serotonin binding in neocortex of patients with temporal lobe epilepsy. Prog. Neuropsychopharmacol. Biol. Psychiatry 2007, 31, 1208–1218. [Google Scholar] [CrossRef] [PubMed]
- Giroud, M.; Dumas, R.; Dauvergne, M.; D’Athis, P.; Rochette, L.; Beley, A.; Bralet, J. 5-Hydroxyindoleacetic acid and homovanillic acid in cerebrospinal fluid of children with febrile convulsions. Epilepsia 1990, 31, 178–181. [Google Scholar] [CrossRef] [PubMed]
- Pranzatelli, M.R.; Tate, E.; Huang, Y.; Haas, R.H.; Bodensteiner, J.; Ashwal, S.; Franz, D. Neuropharmacology of progressive myoclonus epilepsy: Response to 5-hydroxy-L-tryptophan. Epilepsia 1995, 36, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Marion, J.L.; Bigot, J.C.; Goas, J.Y. Alcoholic epilepsy—Low tryptophan levels in plasma and cerebrospinal-fluid. Presse Med. 1985, 14, 681–683. [Google Scholar] [PubMed]
- Ko, F.J.; Chiang, C.H.; Liu, W.J.; Chiang, W. Alteration of amino acid in plasma and cerebrospinal fluid of children with seizure disorders. Gaoxiong Yi Xue Ke Xue Za Zhi 1993, 9, 131–142. [Google Scholar] [PubMed]
- Vieira, É.L.M.; da Silva, M.C.M.; Gonçalves, A.P.; Martins, G.L.; Teixeira, A.L.; de Oliveira, A.C.P.; Reis, H.J. Serotonin and dopamine receptors profile on peripheral immune cells from patients with temporal lobe epilepsy. J. Neuroimmunol. 2021, 354, 577534. [Google Scholar] [CrossRef]
- Kopeikina, E.; Dukhinova, M.; Yung, A.W.Y.; Veremeyko, T.; Kuznetsova, I.S.; Lau, T.Y.B.; Levchuk, K.; Ponomarev, E.D. Platelets promote epileptic seizures by modulating brain serotonin level, enhancing neuronal electric activity, and contributing to neuroinflammation and oxidative stress. Prog. Neurobiol. 2020, 188, 101783. [Google Scholar] [CrossRef]
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Cookson, M.R.; Petrucelli, L.; La Spada, A.R. Converging pathways in neurodegeneration, from genetics to mechanisms. Nat. Neurosci. 2018, 21, 1300–1309. [Google Scholar] [CrossRef]
- Wyss-Coray, T. Ageing, neurodegeneration and brain rejuvenation. Nature 2016, 539, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Morais, L.H.; Lima, M.M.; Martynhak, B.J.; Santiago, R.; Takahashi, T.T.; Ariza, D.; Barbiero, J.K.; Andreatini, R.; Vital, M.A. Characterization of motor, depressive-like and neurochemical alterations induced by a short-term rotenone administration. Pharmacol. Rep. 2012, 64, 1081–1090. [Google Scholar] [CrossRef]
- Ren, Y.; Feng, J. Rotenone selectively kills serotonergic neurons through a microtubule-dependent mechanism. J. Neurochem. 2007, 103, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Morais, L.H.; Hara, D.B.; Bicca, M.A.; Poli, A.; Takahashi, R.N. Early signs of colonic inflammation, intestinal dysfunction, and olfactory impairments in the rotenone-induced mouse model of Parkinson’s disease. Behav. Pharmacol. 2018, 29, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Tanner, C.M.; Kamel, F.; Ross, G.W.; Hoppin, J.A.; Goldman, S.M.; Korell, M.; Marras, C.; Bhudhikanok, G.S.; Kasten, M.; Chade, A.R.; et al. Rotenone, paraquat, and Parkinson’s disease. Environ. Health Perspect. 2011, 119, 866–872. [Google Scholar] [CrossRef]
- Wang, A.; Costello, S.; Cockburn, M.; Zhang, X.; Bronstein, J.; Ritz, B. Parkinson’s disease risk from ambient exposure to pesticides. Eur. J. Epidemiol. 2011, 26, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Thiruchelvam, M.; Richfield, E.K.; Goodman, B.M.; Baggs, R.B.; Cory-Slechta, D.A. Developmental exposure to the pesticides paraquat and maneb and the Parkinson’s disease phenotype. Neurotoxicology 2002, 23, 621–633. [Google Scholar] [CrossRef]
- Kuter, K.; Smiałowska, M.; Wierońska, J.; Zieba, B.; Wardas, J.; Pietraszek, M.; Nowak, P.; Biedka, I.; Roczniak, W.; Konieczny, J.; et al. Toxic influence of subchronic paraquat administration on dopaminergic neurons in rats. Brain Res. 2007, 1155, 196–207. [Google Scholar] [CrossRef]
- Tinakoua, A.; Bouabid, S.; Faggiani, E.; De Deurwaerdère, P.; Lakhdar-Ghazal, N.; Benazzouz, A. The impact of combined administration of paraquat and maneb on motor and non-motor functions in the rat. Neuroscience 2015, 311, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Mack, J.M.; Moura, T.M.; Lanznaster, D.; Bobinski, F.; Massari, C.M.; Sampaio, T.B.; Schmitz, A.E.; Souza, L.F.; Walz, R.; Tasca, C.I.; et al. Intranasal administration of sodium dimethyldithiocarbamate induces motor deficits and dopaminergic dysfunction in mice. Neurotoxicology 2018, 66, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Avramopoulos, D. Genetics of Alzheimer’s disease: Recent advances. Genome Med. 2009, 1, 34. [Google Scholar] [CrossRef] [PubMed]
- Ballard, C.; Gauthier, S.; Corbett, A.; Brayne, C.; Aarsland, D.; Jones, E. Alzheimer’s disease. Lancet 2011, 377, 1019–1031. [Google Scholar] [CrossRef]
- Campbell, A. The potential role of aluminium in Alzheimer’s disease. Nephrol. Dial. Transplant. 2002, 17, 17–20. [Google Scholar] [CrossRef]
- Crapper, D.R.; Krishnan, S.S.; Dalton, A.J. Brain aluminum distribution in Alzheimer’s disease and experimental neurofibrillary degeneration. Science 1973, 180, 511–513. [Google Scholar] [CrossRef]
- Kawahara, M.; Kato-Negishi, M. Link between aluminum and the pathogenesis of Alzheimer’s disease: The integration of the aluminum and amyloid cascade hypotheses. Int. J. Alzheimer’s Dis. 2011, 2011, 276393. [Google Scholar] [CrossRef]
- Sparks, D.L.; Schreurs, B.G. Trace amounts of copper in water induce β-amyloid plaques and learning deficits in a rabbit model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 11065–11069. [Google Scholar] [CrossRef]
- Syme, C.D.; Nadal, R.C.; Rigby, S.E.J.; Viles, J.H. Copper binding to the amyloid-β (Aβ) peptide associated with Alzheimer’s disease: Folding, coordination geometry, pH dependence, stoichiometry, and affinity of Aβ-(1–28): Insights from a range of complementary spectroscopic techniques. J. Biol. Chem. 2004, 279, 18169–18177. [Google Scholar] [CrossRef]
- Singh, I.; Sagare, A.P.; Coma, M.; Perlmutter, D.; Gelein, R.; Bell, R.D.; Deane, R.J.; Zhong, E.; Parisi, M.; Ciszewski, J.; et al. Low levels of copper disrupt brain amyloid-β homeostasis by altering its production and clearance. Proc. Natl. Acad. Sci. USA 2013, 110, 14771–14776. [Google Scholar] [CrossRef]
- Baldi, I.; Lebailly, P.; Mohammed-Brahim, B.; Letenneur, L.; Dartigues, J.-F.; Brochard, P. Neurodegenerative diseases and exposure to pesticides in the elderly. Am. J. Epidemiol. 2003, 157, 409–414. [Google Scholar] [CrossRef]
- Chhillar, N.; Singh, N.K.; Banerjee, B.D.; Bala, K.; Sharma, D.; Mitrabasu, M. β-hexachlorocyclohexane as a risk for Alzheimer’s disease: A pilot study in North Indian population. Am. J. Alzheimer’s Dis. 2013, 1, 60–71. [Google Scholar] [CrossRef]
- Richardson, J.R.; Roy, A.; Shalat, S.L.; von Stein, R.T.; Hossain, M.M.; Buckley, B.; Gearing, M.; Levey, A.I.; German, D.C. Elevated serum pesticide levels and risk for alzheimer disease. JAMA Neurol. 2014, 71, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Zaganas, I.; Kapetanaki, S.; Mastorodemos, V.; Kanavouras, K.; Colosio, C.; Wilks, M.F.; Tsatsakis, A.M. Linking pesticide exposure and dementia: What is the evidence? Toxicology 2013, 307, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Hayden, K.M.; Norton, M.C.; Darcey, D.; Østbye, T.; Zandi, P.P.; Breitner, J.C.S.; Welsh-Bohmer, K.A.; Cache County Study Investigators. Occupational exposure to pesticides increases the risk of incident AD: The Cache County Study. Neurology 2010, 74, 1524–1530. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Chhillar, N.; Banerjee, B.; Bala, K.; Basu, M.; Mustafa, M. Organochlorine pesticide levels and risk of Alzheimer’s disease in north Indian population. Hum. Exp. Toxicol. 2013, 32, 24–30. [Google Scholar] [CrossRef]
- Al-Mousa, F.; Michelangeli, F. Some Commonly Used Brominated Flame Retardants Cause Ca2+-ATPase Inhibition, Beta-Amyloid Peptide Release and Apoptosis in SH-SY5Y Neuronal Cells. PLoS ONE 2012, 7, e33059. [Google Scholar] [CrossRef]
- Hajszan, T.; Leranth, C. Bisphenol A interferes with synaptic remodeling. Front. Neuroendocrinol. 2010, 31, 519–530. [Google Scholar] [CrossRef]
- Sun, W.; Ban, J.-B.; Zhang, N.; Zu, Y.-K.; Sun, W.-X. Perinatal exposure to di-(2-ethylhexyl)-phthalate leads to cognitive dysfunction and phospho-tau level increase in aged rats. Environ. Toxicol. 2014, 29, 596–603. [Google Scholar] [CrossRef]
- Yegambaram, M.; Manivannan, B.; Beach, T.G.; Halden, R.U. Role of environmental contaminants in the etiology of Alzheimer’s disease: A review. Curr. Alzheimer Res. 2015, 12, 116–146. [Google Scholar] [CrossRef]
- Kilian, J.; Kitazawa, M. The emerging risk of exposure to air pollution on cognitive decline and Alzheimer’s disease—Evidence from epidemiological and animal studies. Biomed. J. 2018, 41, 141–162. [Google Scholar] [CrossRef]
- Wang, J.; Ma, T.; Ma, D.; Li, H.; Hua, L.; He, Q.; Deng, X. The Impact of Air Pollution on Neurodegenerative Diseases. Ther. Drug Monit. 2021, 43, 69–78. [Google Scholar] [CrossRef]
- Kioumourtzoglou, M.A.; Schwartz, J.D.; Weisskopf, M.G.; Melly, S.J.; Wang, Y.; Dominici, F.; Zanobetti, A. Long-term PM2.5 exposure and neurological hospital admissions in the northeastern United States. Environ. Health Perspect. 2016, 124, 23–29. [Google Scholar] [CrossRef]
- Jung, C.R.; Lin, Y.T.; Hwang, B.F. Ozone, particulate matter, andnewly diagnosed Alzheimer’s disease: A population-based cohort study in Taiwan. J. Alzheimer’s Dis. 2015, 44, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.C.; Lin, Y.C.; Yu, H.L.; Chen, J.H.; Chen, T.F.; Sun, Y.; Wen, L.-L.; Yip, R.-K.; Chu, Y.-M.; Chen, Y.-C. Association between air pollutants and dementia risk in the elderly. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2015, 1, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Correia, A.S.; Cardoso, A.; Vale, N. Highlighting Immune System and Stress in Major Depressive Disorder, Parkinson’s, and Alzheimer’s Diseases, with a Connection with Serotonin. Int. J. Mol. Sci. 2021, 22, 8525. [Google Scholar] [CrossRef] [PubMed]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef]
- Calderon-Garciduenas, L.; Kavanaugh, M.; Block, M.L.; D’Angiulli, A.; Delgado-Chavez, R.; Torres-Jardon, R.; González-Maciel, A.; Reynoso-Robles, R.; Osnaya, N.; Villarreal-Calderon, R.; et al. Neuroinflammation, hyperphosphorilated tau, diffuse amyloid plaques and down-regulation of the cellular prion protein in air pollution exposed children and adults. J. Alzheimer’s Dis. 2012, 28, 93–107. [Google Scholar] [CrossRef]
- Wang, W.Y.; Tan, M.S.; Yu, J.T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Smith, J.A.; Das, A.; Ray, S.K.; Banik, N.L. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res. Bull. 2012, 87, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Cribbs, D.H.; Berchtold, N.C.; Perreau, V.; Coleman, P.D.; Rogers, J.; Tenner, A.J.; Cotman, C.W. Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: A microarray study. J. Neuroinflamm. 2012, 9, 1–18. [Google Scholar] [CrossRef]
- Ojala, J.; Alafuzoff, I.; Herukka, S.K.; van Groen, T.; Tanila, H.; Pirttilä, T. Expression of interleukin-18 is increased in the brains of Alzheimer’s disease patients. Neurobiol. Aging 2009, 30, 198–209. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Mielke, M.L.; Gómez-Isla, T.; Betensky, R.A.; Growdon, J.H.; Frosch, M.P.; Hyman, B.T. Reactive glia not only associates with plaques but also parallels tangles in Alzheimer’s disease. Am. J. Pathol. 2011, 179, 1373–1384. [Google Scholar] [CrossRef]
- Calderon-Garciduenas, L.; Reed, W.; Maronpot, R.R.; Henriquez-Roldan, C.; Delgado-Chavez, R.; Calderon-Garciduenas, A.; Dragustinovis, I.; Franco-Lira, M.; Aragón-Flores, M.; Solt, A.C.; et al. Brain inflammation and Alzheimer’s-like pathology in individuals exposed to severe air pollution. Toxicol. Pathol. 2004, 32, 650–658. [Google Scholar] [CrossRef]
- Calderon-Garciduenas, L.; Solt, A.C.; Henriquez-Roldan, C.; Torres-Jardon, R.; Nuse, B.; Herritt, L.; Villarreal-Calderón, R.; Osnaya, N.; Stone, I.; García, R.; et al. Long-term air pollution exposure is associated with neuroinflammation, an altered innate immune response, disruption of the blood-brain barrier, ultrafine particulate deposition, and accumulation of amyloid β-42 and α-synuclein in children and young adults. Toxicol. Pathol. 2008, 36, 289–310. [Google Scholar] [CrossRef]
- Calderon-Garciduenas, L.; Mora-Tiscareno, A.; Styner, M.; Gomez-Garza, G.; Zhu, H.; Torres-Jardon, R.; Carlos, E.; Solorio-López, E.; Medina-Cortina, H.; Kavanaugh, M.; et al. White matter hyperintensities, systemic inflammation, brain growth, and cognitive functions in children exposed to air pollution. J. Alzheimer’s Dis. 2012, 31, 183–191. [Google Scholar] [CrossRef]
- Calderon-Garciduenas, L.; Franco-Lira, M.; Henriquez-Roldan, C.; Osnaya, N.; Gonzalez-Maciel, A.; Reynoso-Robles, R.; Villarreal-Calderon, R.; Herritt, L.; Brooks, D.; Keefe, S.; et al. Urban air pollution: Influences on olfactory function and pathology in exposed children and young adults. Exp. Toxicol. Pathol. 2010, 62, 91–102. [Google Scholar] [CrossRef]
- Calderón-Garcidueñas, L.; Azzarelli, B.; Acuna, H.; Garcia, R.; Gambling, T.; Osnaya, N. Air pollution and brain damage. Toxicol. Pathol. 2002, 30, 373–389. [Google Scholar] [CrossRef]
- Calderon-Garciduenas, L.; Maronpot, R.R.; Torres-Jardon, R.; Henriquez-Roldan, C.; Schoonhoven, R.; Acuna-Ayala, H. DNA damage in nasal and brain tissues of canines exposed to air pollutants is associated with evidence of chronic brain inflammation and neurodegeneration. Toxicol. Pathol. 2003, 31, 524–538. [Google Scholar] [CrossRef]
- Calderón-Garcidueñas, L.; Mora-Tiscareño, A.; Ontiveros, E.; Gómez-Garza, G.; Barragán-Mejía, G.; Broadway, J. Air pollution, cognitive deficits and brain abnormalities: A pilot study with children and dogs. Brain Cognit. 2008, 68, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Saffari, A.; Sioutas, C.; Forman, H.J.; Morgan, T.E.; Finch, C.E. Nanoscale particulate matter from urban traffic rapidly induces oxidative stress and inflammation in olfactory epithelium with concomitant effects on brain. Environ. Health Perspect. 2016, 124, 1537–1546. [Google Scholar] [CrossRef] [PubMed]
- Guerra, R.; Vera-Aguilar, E.; Uribe-Ramirez, M.; Gookin, G.; Camacho, J.; Osornio-Vargas, A.R. Exposure to inhaled particulate matter activates early markers of oxidative stress, inflammation and unfolded protein response in rat striatum. Toxicol. Lett. 2013, 222, 146–154. [Google Scholar] [CrossRef]
- Kleinman, M.T.; Araujo, J.; Nel, A.; Sioutas, C.; Campbell, A.; Cong, P.Q. Inhaled ultrafine particulate matter affects CNS inflammatory processes and may act via MAP kinase signaling pathways. Toxicol. Lett. 2008, 178, 127–130. [Google Scholar] [CrossRef]
- Campbell, A.; Oldham, M.; Becaria, A.; Bondy, S.C.; Meacher, D.; Sioutas, C. Particulate matter in polluted air may increase biomarkers of inflammation in mouse brain. Neurotoxicology 2005, 26, 133–140. [Google Scholar] [CrossRef]
- Fonken, L.K.; Xu, X.; Weil, Z.M.; Chen, G.; Sun, Q.; Rajagopalan, S. Air pollution impairs cognition, provokes depressive-like behaviors and alters hippocampal cytokine expression and morphology. Mol. Psychiatr. 2011, 16, 987–995. [Google Scholar] [CrossRef]
- Bhatt, D.P.; Puig, K.L.; Gorr, M.W.; Wold, L.E.; Combs, C.K. A pilot study to assess effects of long-term inhalation of airborne particulate matter on early Alzheimer-like changes in the mouse brain. PLoS ONE 2015, 10, e0127102. [Google Scholar] [CrossRef]
- Durga, M.; Devasena, T.; Rajasekar, A. Determination of LC50 and sub-chronic neurotoxicity of diesel exhaust nanoparticles. Environ. Toxicol. Pharmacol. 2015, 40, 615–625. [Google Scholar] [CrossRef]
- Levesque, S.; Surace, M.J.; McDonald, J.; Block, M.L. Air pollution & the brain: Subchronic diesel exhaust exposure causes neuroinflammation and elevates early markers of neurodegenerative disease. J. Neuroinflamm. 2011, 8, 105. [Google Scholar] [CrossRef]
- Kim, S.H.; Knight, E.M.; Saunders, E.L.; Cuevas, A.K.; Popovech, M.; Chen, L.C. Rapid doubling of Alzheimer’s amyloid-β40 and 42 levels in brains of mice exposed to a nickel nanoparticle model of air pollution. F1000Research 2012, 1, 70. [Google Scholar] [CrossRef]
- Calderón-Garcidueñas, L.; Jewells, V.; Galaz-Montoya, C.; van Zundert, B.; Pérez-Calatayud, A.; Ascencio-Ferrel, E. Interactive and additive influences of gender, BMI and Apolipoprotein 4 on cognition in children chronically exposed to high concentrations of PM2.5 and ozone. APOE 4 females are at highest risk in Mexico City. Environ. Res. 2016, 150, 411–422. [Google Scholar] [CrossRef]
- Schikowski, T.; Vossoughi, M.; Vierkötter, A.; Schulte, T.; Teichert, T.; Sugir, D. Association of air pollution with cognitive functions and its modification by APOE gene variants in elderly women. Environ. Res. 2015, 142, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s Dementia Transl. Res. Clin. Interv. 2018, 4, 575–590. [Google Scholar] [CrossRef]
- Morgan, B.P. Complement in the pathogenesis of Alzheimer’s disease. Semin. Immunopathol. 2018, 40, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Chowdhury, S.; Ma, R.; Le, K.X.; Hong, S.; Caldarone, B.J.; Stevens, B.; Lemere, C.A. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci. Transl. Med. 2017, 9, eaaf6295. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.B.; Blakely, R.D.; Hewlett, W.A. The proinflammatory cytokines interleukin-1beta and tumor necrosis factor-alpha activate serotonin transporters. Neuropsychopharmacology 2006, 31, 2121–2131. [Google Scholar] [CrossRef]
- Metaxas, A.; Anzalone, M.; Vaitheeswaran, R.; Petersen, S.; Landau, A.M.; Finsen, B. Neuroinflammation and amyloid-beta 40 are associated with reduced serotonin transporter (SERT) activity in a transgenic model of familial Alzheimer’s disease. Alzheimer’s Res. Ther. 2019, 11, 1–13. [Google Scholar] [CrossRef]
- Ledo, J.H.; Azevedo, E.P.; Beckman, D.; Ribeiro, F.C.; Santos, L.E.; Razolli, D.S.; Kincheski, G.C.; Melo, H.M.; Bellio, M.; Teixeira, A.L.; et al. Cross talk between brain innate immunity and serotonin signaling underlies depressive-like behavior induced by Alzheimer’s amyloid-β oligomers in mice. J. Neurosci. 2016, 36, 12106–12116. [Google Scholar] [CrossRef] [PubMed]
- Elsworthy, R.J.; Aldred, S. Depression in Alzheimer’s disease: An alternative role for selective serotonin reuptake inhibitors? J. Alzheimer’s Dis. 2019, 69, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Caraci, F.; Copani, A.; Nicoletti, F.; Drago, F. Depression and Alzheimer’s disease: Neurobiological links and common pharmacological targets. Eur. J. Pharmacol. 2010, 626, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Cirrito, J.R.; Disabato, B.M.; Restivo, J.L.; Verges, D.K.; Goebel, W.D.; Sathyan, A.; Hayreh, D.; D’Angelo, G.; Benzinger, T.; Yoon, H.; et al. Serotonin signaling is associated with lower amyloid-β levels and plaques in transgenic mice and humans. Proc. Natl. Acad. Sci. USA 2011, 108, 14968–14973. [Google Scholar] [CrossRef] [PubMed]
- Claeysen, S.; Bockaert, J.; Giannoni, P. Serotonin: A new hope in Alzheimer’s disease? ACS Chem. Neurosci. 2015, 6, 940–943. [Google Scholar] [CrossRef] [PubMed]
- Šimić, G.; Babić Leko, M.; Wray, S.; Harrington, C.R.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; de Silva, R.; Di Giovanni, G.; et al. Monoaminergic neuropathology in Alzheimer’s disease. Prog. Neurobiol. 2017, 151, 101–138. [Google Scholar] [CrossRef]
- Smith, G.S.; Barrett, F.S.; Joo, J.H.; Nassery, N.; Savonenko, A.; Sodums, D.J.; Marano, C.M.; Munro, C.A.; Brandt, J.; Kraut, M.A.; et al. Molecular imaging of serotonin degeneration in mild cognitive impairment. Neurobiol. Dis. 2017, 105, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Zasler, N.D.; Martelli, M.F.; Jacobs, H.E. Neurobehavioral disorders. In Handbook of Clinical Neurology; Barnes, M.P., Good, D.C., Eds.; Elsevier: Amsterdam, The Netherlands, 2013; Volume 110, pp. 377–388. [Google Scholar] [CrossRef]
- Kim, J.S.; Choi-Kwon, S. Poststroke depression and emotional incontinence: Correlation with lesion location. Neurology 2000, 54, 1805–1810. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S. Serotonin and stroke. In Handbook of Behavioral Neuroscience; Müller, C.P., Cunningham, K.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; Volume 31, pp. 989–1000. [Google Scholar] [CrossRef]
- Lee, E.; Oh, M.; Kim, J.S.; Chang, D.I.; Park, J.H.; Cha, J.K.; Heo, J.H.; Sohn, S.I.; Kim, D.E.; Kim, H.Y.; et al. Serotonin transporter gene polymorphisms may be associated with poststroke neurological recovery after escitalopram use. J. Neurol. Neurosurg. Psychiatry 2018, 89, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Monaco, F.; Fumero, S.; Mondino, A.; Mutani, R. Plasma and cerebrospinal fluid tryptophan in multiple sclerosis and degenerative diseases. J. Neurol. Neurosurg. Psychiatry 1979, 42, 640–641. [Google Scholar] [CrossRef]
- Tagliamonte, A.; Biggio, G.; Vargiu, L.; Gessa, G.L. Free tryptophan in serum controls brain tryptophan level and serotonin synthesis. Life Sci. II 1973, 12, 277–287. [Google Scholar] [CrossRef]
- Andersen, O.; Johansson, B.B.; Svennerholm, L. Monoamine metabolites in successive samples of spinal fluid. Acta Neurol. Scand. 1981, 63, 247–254. [Google Scholar] [CrossRef]
- Mostert, J.P.; Admiraal-Behloul, F.; Hoogduin, J.M.; Luyendijk, J.; Heersema, D.J.; van Buchem, M.A.; De Keyser, J. Effects of fluoxetine on disease activity in relapsing multiple sclerosis: A double-blind, placebo-controlled, exploratory study. J. Neurol. Neurosurg. Psychiatry 2008, 79, 1027–1031. [Google Scholar] [CrossRef]
- Mohr, D.C.; Boudewyn, A.C.; Goodkin, D.E.; Bostrom, A.; Epstein, L. Comparative outcomes for individual cognitive-behavior therapy, supportive-expressive group psychotherapy, and sertraline for the treatment of depression in multiple sclerosis. J. Consult. Clin. Psychol. 2001, 69, 942–949. [Google Scholar] [CrossRef]
- Grech, L.B.; Butler, E.; Stuckey, S.; Hester, R. Neuroprotective benefits of antidepressants in multiple sclerosis: Are we missing the mark? J. Neuropsy. Clin. Neurosci. 2019, 31, 289–297. [Google Scholar] [CrossRef]
- Mdawar, B.; Ghossoub, E.; Khoury, R. Selective serotonin reuptake inhibitors and Alzheimer’s disease. Neural Regen. Res. 2020, 15, 41–46. [Google Scholar] [CrossRef]
- Caragher, S.P.; Hall, R.R.; Ahsan, R.; Ahmed, A.U. Monoamines in glioblastoma: Complex biology with therapeutic potential. Neuro-Oncology 2018, 20, 1014–1025. [Google Scholar] [CrossRef]
- Mugge, L.; Mansour, T.R.; Crippen, M.; Alam, Y.; Schroeder, J. Depression and glioblastoma, complicated concomitant diseases: A systemic review of published literature. Neurosurg. Rev. 2020, 43, 497–511. [Google Scholar] [CrossRef]
- Butterworth, R.F. Metabolic Encephalopathies. In Basic Neurochemistry: Molecular, Cellular and Medical Aspects; Siegel, G.J., Agranoff, B.W., Albers, R.W., Fisher, S.K., Uhler, M.D., Eds.; Lippincott-Raven: Philadelphia, PA, USA, 1999; Available online: https://www.ncbi.nlm.nih.gov/books/NBK20383/ (accessed on 10 September 2021).
- Weissenborn, K.; Bokemeyer, M.; Krause, J.; Ennen, J.; Ahl, B. Neurological and neuropsychiatric syndromes associated with liver disease. AIDS 2005, 19, S93–S98. [Google Scholar] [CrossRef]
- Borg, J.; Warter, J.M.; Schlienger, J.L.; Imler, M.; Marescaux, C.; Mack, G. Neurotransmitter modifications in human cerebrospinal fluid and serum during hepatic encephalopathy. J. Neurol. Sci. 1982, 57, 343–356. [Google Scholar] [CrossRef]
- Dhanda, S.; Sandhir, R. Role of dopaminergic and serotonergic neurotransmitters in behavioral alterations observed in rodent model of hepatic encephalopathy. Behav. Brain Res. 2015, 286, 222–235. [Google Scholar] [CrossRef]
- Honoré, P.J.; Deianova, N.; Loret, G.; Hemelsoet, D. An easily overlooked cause of toxic encephalopathy: Methylene blue in a patient on an SSRI. Acta Neurol. Belg. 2018, 118, 121–122. [Google Scholar] [CrossRef]
- Otte, C.; Gold, S.M.; Penninx, B.W.; Pariante, C.M.; Etkin, A.; Mohr, D.C.; Schatzberg, A.F. Major depressive disorder. Nat. Rev. Dis. Primers 2016, 2, 16065. [Google Scholar] [CrossRef]
- Hirschfeld, R.M. History and evolution of the monoamine hypothesis of depression. J. Clin. Psychiatry 2000, 61, 4–6. [Google Scholar] [PubMed]
- Underwood, M.D.; Kassir, S.A.; Bakalian, M.J.; Galfalvy, H.; Dwork, A.J.; Mann, J.J.; Arango, V. Serotonin receptors and suicide, major depression, alcohol use disorder and reported early life adversity. Transl. Psychiatry 2018, 8, 279. [Google Scholar] [CrossRef] [PubMed]
- Yohn, C.N.; Gergues, M.M.; Samuels, B.A. The role of serotonin receptors in depression. Mol. Brain 2017, 10, 28. [Google Scholar] [CrossRef] [PubMed]
- Van den Bosch, M.; Meyer-Lindenberg, A. Environmental exposures and depression: Biological mechanisms and epidemiological evidence. Annu. Rev. Public Health 2019, 40, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Lim, Y.H.; Kim, K.N.; Choi, Y.H.; Hong, Y.C.; Lee, N. Urinary phthalate metabolites concentrations and symptoms of depression in an elderly population. Sci. Total Environ. 2018, 625, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Shiue, I. Urinary heavy metals, phthalates and polyaromatic hydrocarbons independent of health events are associated with adult depression: USA NHANES, 2011–2012. Environ. Sci. Pollut. Res. 2015, 22, 17095–17103. [Google Scholar] [CrossRef]
- Roberts, S.; Arseneault, L.; Barratt, B.; Beevers, S.; Danese, A.; Odgers, C.L.; Moffitt, T.E.; Reubeng, A.; Kelly, F.J.; Fisher, H.L. Exploration of NO2 and PM2.5 air pollution and mental health problems using high-resolution data in London-based children from a UK longitudinal cohort study. Psychiatry Res. 2019, 272, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Xin, F.; Fischer, E.; Krapp, C.; Krizman, E.N.; Lan, Y.; Mesaros, C.; Snyder, N.W.; Bansal, A.; Robinson, M.B.; Simmons, R.A.; et al. Mice exposed to bisphenol A exhibit depressive-like behavior with neurotransmitter and neuroactive steroid dysfunction. Horm. Behav. 2018, 102, 93–104. [Google Scholar] [CrossRef]
- Hamel, E. Serotonin and migraine: Biology and clinical implication. Cephalalgia 2007, 27, 1295–1300. [Google Scholar] [CrossRef] [PubMed]
- Deen, M.; Christensen, C.E.; Hougaard, A.; Hansen, H.D.; Knudsen, G.M.; Ashina, M. Serotonergic mechanisms in the migraine brain—A systematic review. Cephalalgia 2017, 37, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, A.H.; Kogelman, L.J.A.; Kristensen, D.M.; Chalmer, M.A.; Olesen, J.; Folkmann Hansen, T. Functional gene networks reveal distinct mechanisms segregating in migraine families. Brain 2020, 143, 2945–2956. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Bertisch, S.M.; Mostofsky, E.; Buettner, C.; Mittleman, M.A. Weather, ambient air pollution, and risk of migraine headache onset among patients with migraine. Environ. Int. 2019, 132, 105100. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Myung, W.; Cheong, H.K.; Yi, S.M.; Hong, Y.C.; Cho, S.I.; Kim, H. Ambient air pollution exposure and risk of migraine: Synergistic effect with high temperature. Environ. Int. 2018, 121, 383–391. [Google Scholar] [CrossRef]
- Steinemann, A. Ten questions concerning air fresheners and indoor built environments. Build. Environ. 2017, 111, 279–284. [Google Scholar] [CrossRef]
- Badiye, A.; Kapoor, N.; Khajuria, H. Copper toxicity: A comprehensive study. Res. J. Recent Sci. 2013, 2, 58–67. [Google Scholar]
- Wöber, C.; Wöber-Bingöl, C. Triggers of migraine and tension-type headache. In Handbook of Clinical Neurology, Headache, 3rd ed.; Nappi, G., Moskowitz, M.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2011; Volume 97, pp. 161–172. [Google Scholar] [CrossRef]
- Vermeer, L.M.D.; Gregory, E.; Winter, M.K.; McCarson, K.E.; Berman, N.E. Exposure to BPA exacerbates migraine-like behaviors in a multibehavior model of rat migraine. Toxicol. Sci. 2014, 137, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Pouchieu, C.; Baldi, I.; Gruber, A.; Berteaud, E.; Carles, C.; Loiseau, H. Descriptive epidemiology and risk factors of primary central nervous system tumors: Current knowledge. Rev. Neurol. 2016, 172, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Piel, C.; Pouchieu, C.; Carles, C.; Béziat, B.; Boulanger, M.; Bureau, M.; Busson, A.; Grüber, A.; Lecluse, Y.; Migault, L.; et al. Agricultural exposures to carbamate herbicides and fungicides and central nervous system tumour incidence in the cohort AGRICAN. Environ. Int. 2019, 130, 104876. [Google Scholar] [CrossRef] [PubMed]
- Rivière, S.; Catelinois, O.; Mouly, D. Suspicion D’Excès de Cas de Glioblastomes Dans les Communes Gardoises de Salindres et Rousson: Mise à Jour des Données de Surveillance et Premières Investigations de L’Environnement; Rapport D’étape; Santé Publique France: Saint-Maurice, QC, Canada, 2020; Available online: https://www.santepubliquefrance.fr (accessed on 1 April 2021).
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K. (Eds.) WHO Classification of Tumours of the Central Nervous System; IARC: Lyon, France, 2016; ISBN 978-92-832-2430-3. [Google Scholar]
- Brezun, J.M.; Daszuta, A. Depletion in serotonin decreases neurogenesis in the dentate gyrus and the subventricular zone of adult rats. Neuroscience 1999, 89, 999–1002. [Google Scholar] [CrossRef]
- Radley, J.J.; Jacobs, B.L. 5-HT1A receptor antagonist administration decreases cell proliferation in the dentate gyrus. Brain Res. 2002, 955, 264–267. [Google Scholar] [CrossRef]
- Schmitt, A.; Benninghoff, J.; Moessner, R.; Rizzi, M.; Paizanis, E.; Doenitz, C.; Gross, S.; Hermann, M.; Gritti, A.; Lanfumey, L.; et al. Adult neurogenesis in serotonin transporter deficient mice. J. Neural Trans. 2007, 114, 1107–1119. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Yuan, X.; Zeng, Z.; Tunici, P.; Ng, H.; Abdulkadir, I.R.; Lu, L.; Irvin, D.; Black, K.L.; Yu, J.S. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol. Cancer 2006, 5, 67. [Google Scholar] [CrossRef] [PubMed]
- Cohen, Z.; Bouchelet, I.; Olivier, A.; Villemure, J.G.; Ball, R.; Stanimirovic, D.B.; Hamel, E. Multiple microvascular and astroglial 5-hydroxytryptamine receptor subtypes in human brain: Molecular and pharmacologic characterization. Blood Flow Metab. 1999, 19, 908–917. [Google Scholar] [CrossRef]
- Mahé, C.; Bernhard, M.; Bobirnac, I.; Keser, C.; Loetscher, E.; Feuerbach, D.; Dev, K.K.; Schoeffter, P. Functional expression of the serotonin 5-HT7 receptor in human glioblastoma cell lines. Br. J. Pharmacol. 2004, 143, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Merzak, A.; Koochekpour, S.; Fillion, M.P.; Fillion, G.; Pilkington, G.J. Expression of serotonin receptors in human fetal astrocytes and glioma cell lines: A possible role in glioma cell proliferation and migration. Brain Res. Mol. Brain Res. 1996, 41, 1–7. [Google Scholar] [CrossRef]
- Siddiqui, E.J.; Thompson, C.S.; Mikhailidis, D.P.; Mumtaz, F.H. The role of serotonin in tumour growth. Oncol. Rep. 2005, 14, 1593–1597. [Google Scholar] [CrossRef]
- Lu, D.Y.; Leung, Y.M.; Cheung, C.W.; Chen, Y.R.; Wong, K.L. Glial cell line-derived neurotrophic factor induces cell migration and matrix metalloproteinase-13 expression in glioma cells. Biochem. Pharmacol. 2010, 80, 1201–1209. [Google Scholar] [CrossRef]
- Spies, M.; Knudsen, G.M.; Lanzenberger, R.; Kasper, S. The serotonin transporter in psychiatric disorders: Insights from PET imaging. Lancet Psychiatry 2015, 2, 743–755. [Google Scholar] [CrossRef]
- Kamson, D.O.; Lee, T.J.; Varadarajan, K.; Robinette, N.L.; Muzik, O.; Chakraborty, P.K.; Snyder, M.; Barger, G.R.; Mittal, S.; Juhász, C. Clinical significance of tryptophan metabolism in the nontumoral hemisphere in patients with malignant glioma. J. Nucl. Med. 2014, 55, 1605–1610. [Google Scholar] [CrossRef]
- Roy, A.; Zamani, A.; Ananthan, S.; Qu, Z.C. Serotonin: A neurotransmitter as well as a potent angiokine. In Proceedings of the AACR Annual Meeting, Denver, CO, USA, 18–22 April 2009; Volume 69. Abstract 4035. [Google Scholar]
- Zamani, A.; Qu, Z.C. Serotonin activates angiogenic phosphorylation signaling in human endothelial cells. FEBS Lett. 2012, 586, 2360–2365. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights Intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed]
- Pollard, S.M.; Yoshikawa, K.; Clarke, I.D.; Danovi, D.; Stricker, S.; Russell, R.; Bayani, J.; Head, R.; Lee, M.; Bernstein, M.; et al. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell 2009, 4, 568–580. [Google Scholar] [CrossRef] [PubMed]
- GBD 2016 Neurology Collaborators. Global, regional, and national burden of neurological disorders, 1990–2016: A systematic analysis for the global burden of disease study 2016. Lancet Neurol. 2019, 18, 459–480. [Google Scholar] [CrossRef]
- Woodley, M.A.; Te Nijenhuis, J.; Murphy, R. Were the Victorians cleverer than us? The decline in general intelligence estimated from a meta-analysis of the slowing of simple reaction time. Intelligence 2013, 41, 843–850. [Google Scholar] [CrossRef]
- Mazarati, A.M.; Lewis, M.L.; Pittman, Q.J. Neurobehavioral comorbidities of epilepsy: Role of inflammation. Epilepsia 2017, 58, 48–56. [Google Scholar] [CrossRef]
- Sarrouilhe, D.; Dejean, C.; Mesnil, M. Connexin43- and Pannexin-Based Channels in Neuroinflammation and Cerebral Neuropathies. Front. Mol. Neurosci. 2017, 10, 320. [Google Scholar] [CrossRef]
- Sarrouilhe, D.; Mesnil, M.; Dejean, D. Targeting Gap Junctions: New insights into the treatment of major depressive disorder. Curr. Med. Chem. 2019, 26, 3775–3791. [Google Scholar] [CrossRef]
- Chiu, K.; Warner, G.; Nowak, R.A.; Flaws, J.A.; Mei, W. The impact of environmental chemicals on the gut microbiome. Toxicol. Sci. 2020, 176, 253–284. [Google Scholar] [CrossRef]
- O’Mahony, S.M.; Clarke, G.; Borre, Y.E.; Dinan, T.G.; Cryan, J.F. Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. Behav. Brain Res. 2015, 277, 32–48. [Google Scholar] [CrossRef]
- Dales, R.; Smith-Doiron, M.; Stieb, D.M.; Brook, J.R. Air pollution and sudden infant death syndrome. Pediatrics 2004, 113, e628–e631. [Google Scholar] [CrossRef] [PubMed]
- Haynes, R.L.; Frelinger, A.L., 3rd; Giles, E.K.; Goldstein, R.D.; Tran, H.; Kozakewich, H.P.; Haas, E.A.; Gerrits, A.J.; Mena, O.J.; Trachtenberg, F.L.; et al. High serum serotonin in sudden infant death syndrome. Proc. Natl. Acad. Sci. USA 2017, 114, 7695–7700. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarrouilhe, D.; Defamie, N.; Mesnil, M. Is the Exposome Involved in Brain Disorders through the Serotoninergic System? Biomedicines 2021, 9, 1351. https://doi.org/10.3390/biomedicines9101351
Sarrouilhe D, Defamie N, Mesnil M. Is the Exposome Involved in Brain Disorders through the Serotoninergic System? Biomedicines. 2021; 9(10):1351. https://doi.org/10.3390/biomedicines9101351
Chicago/Turabian StyleSarrouilhe, Denis, Norah Defamie, and Marc Mesnil. 2021. "Is the Exposome Involved in Brain Disorders through the Serotoninergic System?" Biomedicines 9, no. 10: 1351. https://doi.org/10.3390/biomedicines9101351
APA StyleSarrouilhe, D., Defamie, N., & Mesnil, M. (2021). Is the Exposome Involved in Brain Disorders through the Serotoninergic System? Biomedicines, 9(10), 1351. https://doi.org/10.3390/biomedicines9101351