
Role of Lamin A/C as Candidate Biomarker of Aggressiveness and Tumorigenicity in Glioblastoma Multiforme

 ,
,  , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Analysis of Transcriptomic Data
2.2. Cell Cultures and Treatments
2.3. In Vivo Experiments
2.4. Ethics Statement
2.5. Western Blot Analysis
2.6. Colony Forming Assay
2.7. Confocal Microscopy
2.8. Total RNA Preparation
2.9. Real Time RT-PCR
2.10. Wound Healing Assay
2.11. Statistical Analysis
3. Results
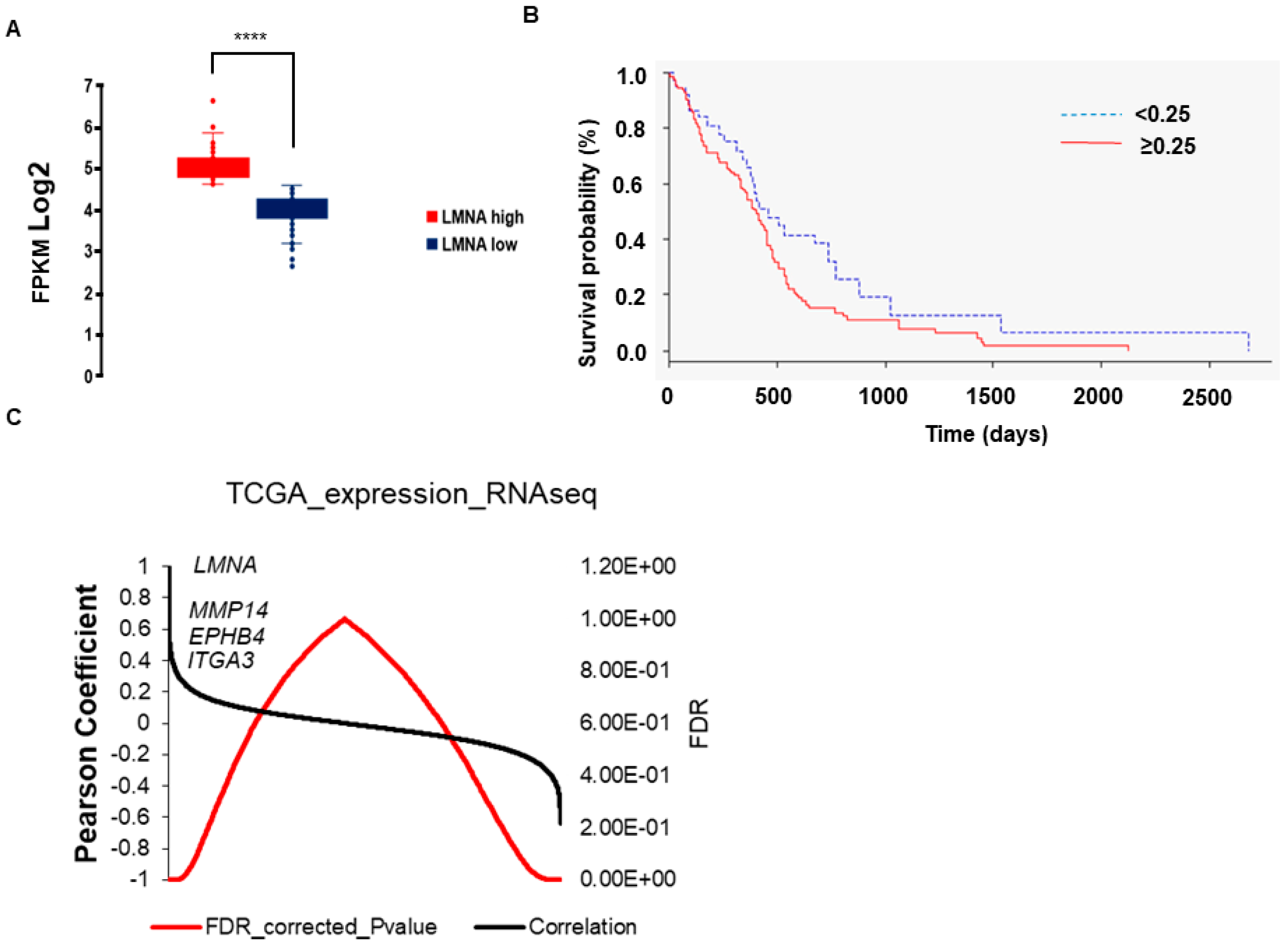
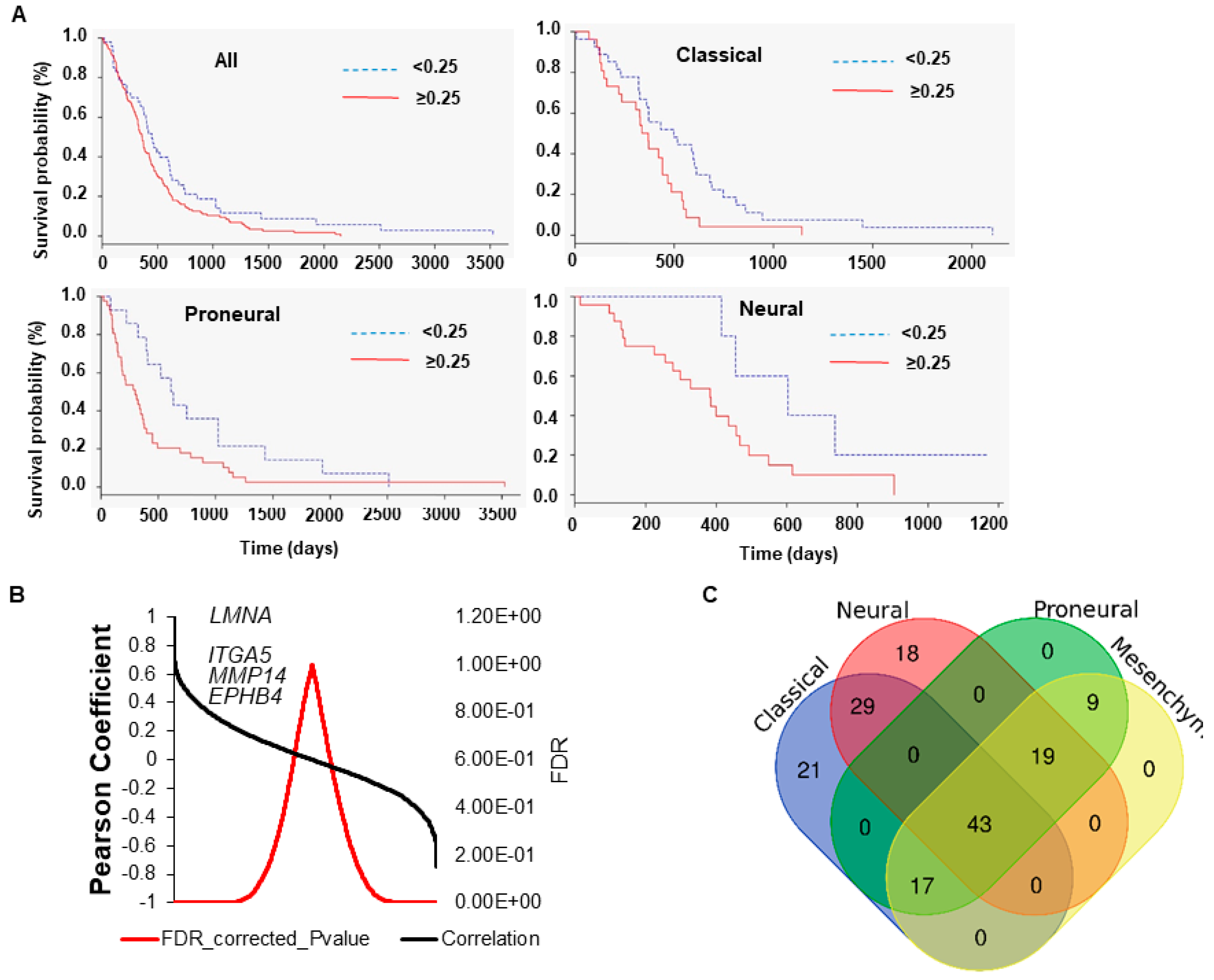
3.1. Expression of LMNA Gene Correlates with a Reduced Overall Survival and Tumorigenic Pathways in Glioblastoma Multiforme Patients
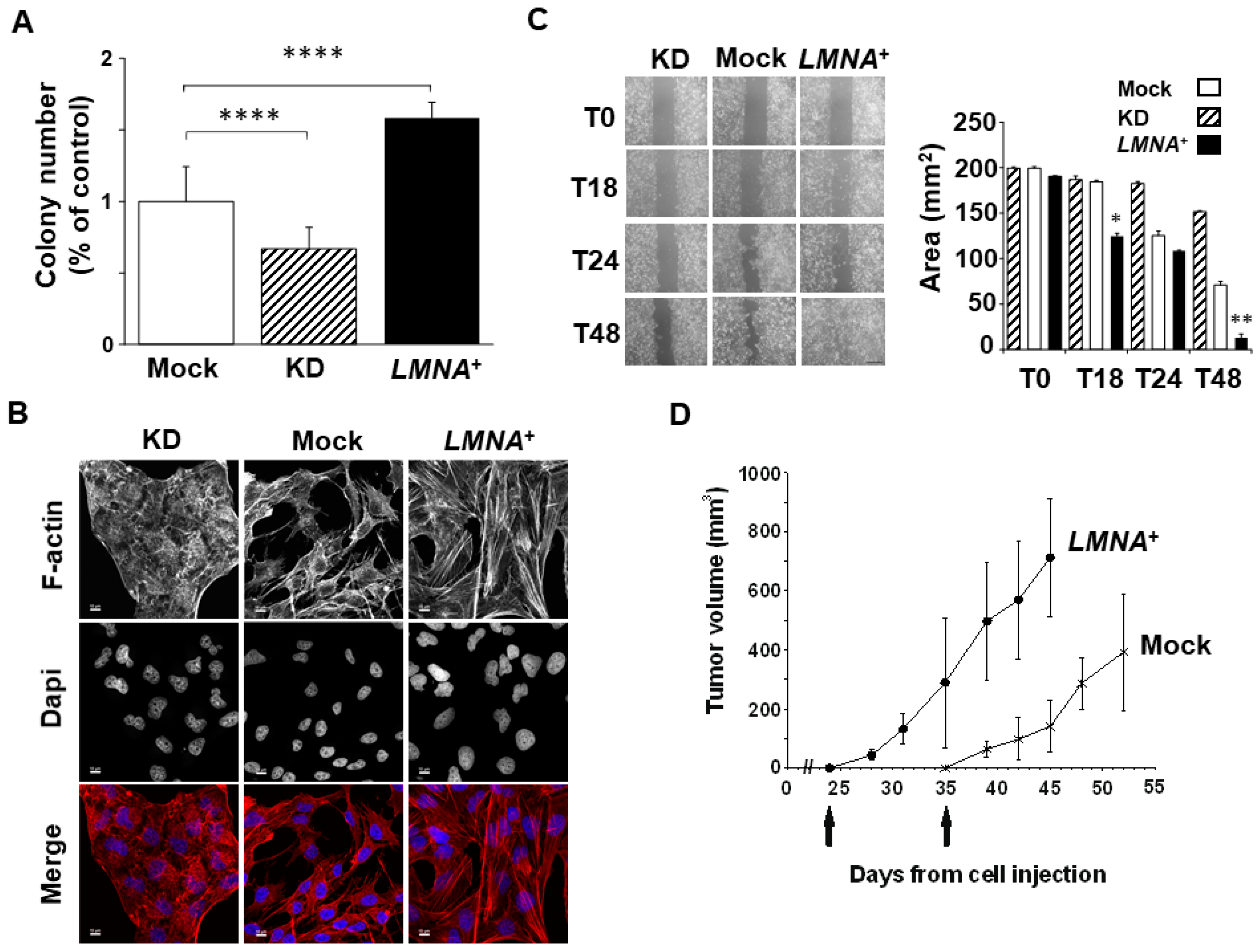
3.2. Modulation of LMNA Gene Expression Levels in a GBM Cell Model
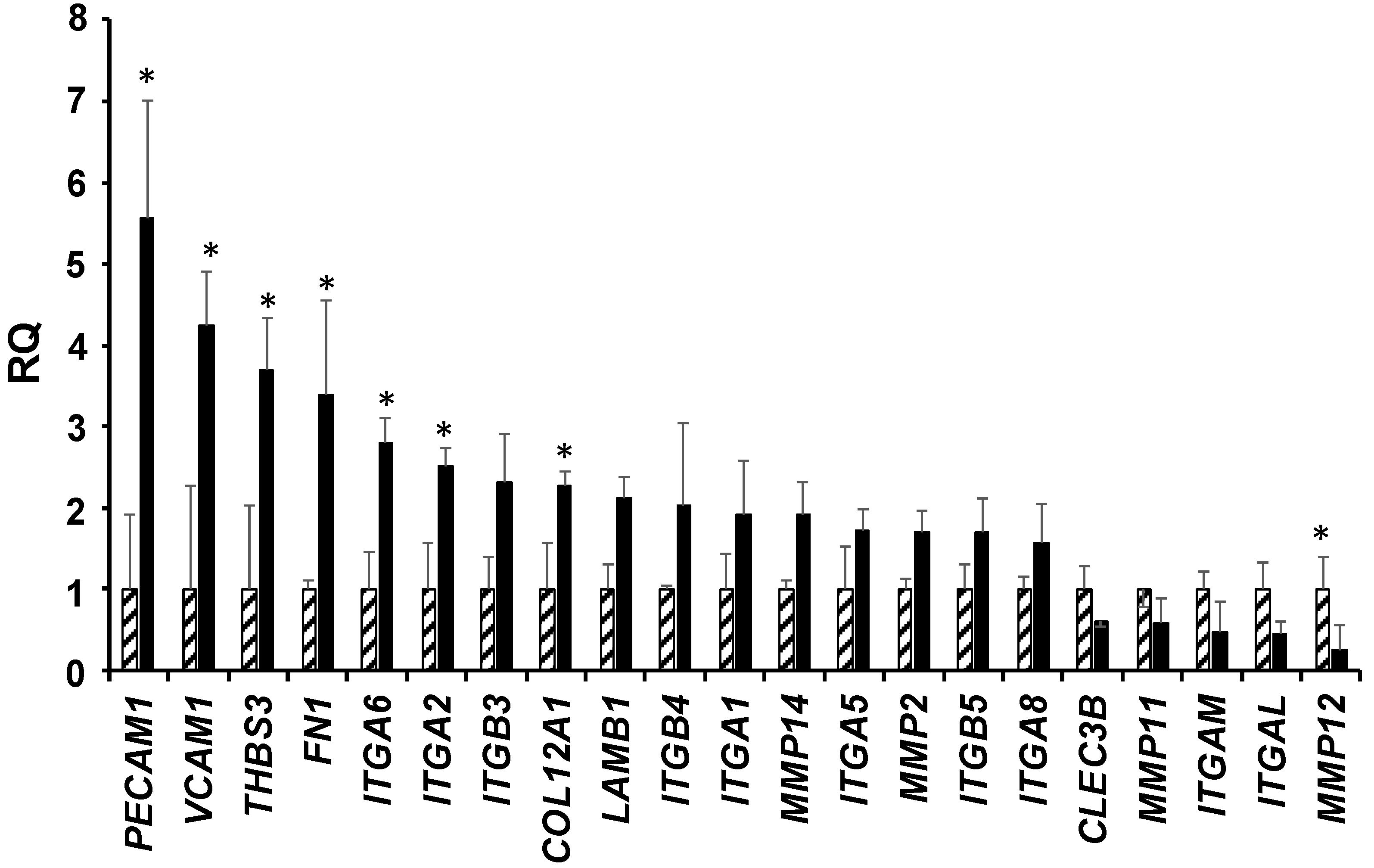
3.3. Upregulation of LMNA Gene Increase the Aggressiveness of T98G
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Molinaro, A.M.; Taylor, J.W.; Wiencke, J.K.; Wrensch, M.R. Genetic and molecular epidemiology of adult diffuse glioma. Nat. Rev. Neurol. 2019, 15, 405–417. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG Island Methylator Phenotype that Defines a Distinct Subgroup of Glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.-A.; Jones, D.T.W.; Konermann, C.; Pfaff, E.; Tönjes, M.; Sill, M.; Bender, S.; et al. Hotspot Mutations in H3F3A and IDH1 Define Distinct Epigenetic and Biological Subgroups of Glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, N.I.; Guilhamon, P.; Desai, K.; McAdam, R.F.; Langille, E.; O’Connor, M.; Lan, X.; Whetstone, H.; Coutinho, F.J.; Vanner, R.J.; et al. ASCL1 Reorganizes Chromatin to Direct Neuronal Fate and Suppress Tumorigenicity of Glioblastoma Stem Cells. Cell Stem Cell 2017, 21, 209–224. [Google Scholar] [CrossRef] [PubMed]
- Park, C.-K.; Bae, J.M.; Park, S.-H. Long-term survivors of glioblastoma are a unique group of patients lacking universal characteristic features. Neuro-Oncol. Adv. 2020, 2, vdz056. [Google Scholar] [CrossRef] [Green Version]
- Bulstrode, H.; Johnstone, E.; Marques-Torrejon, M.A.; Ferguson, K.M.; Bressan, R.B.; Blin, C.; Grant, V.; Gogolok, S.; Gangoso, E.; Gagrica, S.; et al. Elevated FOXG1 and SOX2 in glioblastoma enforces neural stem cell identity through transcriptional control of cell cycle and epigenetic regulators. Genes Dev. 2017, 31, 757–773. [Google Scholar] [CrossRef] [Green Version]
- Verhaak, R.G.; Hoadley, K.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, T.E.; Liau, B.B.; Wallace, L.C.; Morton, A.; Xie, Q.; Dixit, D.; Factor, D.C.; Kim, L.J.Y.; Morrow, J.J.; Wu, Q.; et al. Transcription elongation factors represent in vivo cancer dependencies in glioblastoma. Nature 2017, 547, 355–359. [Google Scholar] [CrossRef]
- Liau, B.B.; Sievers, C.; Donohue, L.K.; Gillespie, S.; Flavahan, W.A.; Miller, T.E.; Venteicher, A.; Hebert, C.H.; Carey, C.D.; Rodig, S.J.; et al. Adaptive chromatin remodeling drives glioblastoma stem cell plasticity and drug tolerance. Cell Stem Cell 2017, 20, 233–246. [Google Scholar] [CrossRef] [Green Version]
- Dubik, N.; Mai, S. Lamin A/C: Function in Normal and Tumor Cells. Cancers 2020, 12, 3688. [Google Scholar] [CrossRef]
- Parnaik, V.K. Role of Nuclear Lamins in Nuclear Organization, Cellular Signaling, and Inherited Diseases. Int. Rev. Cell Mol. Biol. 2008, 266, 1937–6448. [Google Scholar]
- Sakthivel, K.M.; Sehgal, P. A Novel Role of Lamins from Genetic Disease to Cancer Biomarkers. Oncol. Rev. 2016, 10, 309. [Google Scholar] [CrossRef]
- Kong, L.; Schafer, G.; Bu, H.; Zhang, Y.; Zhang, Y.; Klocker, H. Lamin A/C protein is overexpressed in tissue-invading prostate cancer and promotes prostate cancer cell growth, migration and invasion through the PI3K/AKT/PTEN pathway. Carcinogenesis 2012, 33, 751–759. [Google Scholar] [CrossRef] [Green Version]
- Belt, E.J.; Fijneman, R.J.; Van den Berg, E.G.; Bril, H.; Diemen, P.D.-V.; Tijssen, M.; Van Essen, H.; Klerk, E.D.L.-D.; Belien, J.; Stockmann, H.; et al. Loss of lamin A/C expression in stage II and III colon cancer is associated with disease recurrence. Eur. J. Cancer 2011, 47, 1837–1845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Leeuw, R.; Gruenbaum, Y.; Medalia, O. Nuclear Lamins: Thin Filaments with Major Functions. Trends Cell Biol. 2018, 28, 34–45. [Google Scholar] [CrossRef] [PubMed]
- The R Development Core Team. R: A Language and Environment for Statistical Computing. In R Foundation for Statistical Computing; The R Development Core Team: Copenhagen, Denmark, 2018; Available online: https://www.R-project.org/ (accessed on 11 September 2017).
- Huber, W.; Carey, V.J.; Gentleman, R.; Anders, S.; Carlson, M.; Carvalho, B.S.; Bravo, H.C.; Davis, S.; Gatto, L.; Girke, T.; et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 2015, 12, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Therneau, T. A Package for Survival Analysis. In S. R Package Version 2.37-7. 2014. Available online: http://CRAN.R-project.org/package=survival (accessed on 11 September 2017).
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Maresca, G.; Natoli, M.; Nardella, M.; Arisi, I.; Trisciuoglio, D.; Desideri, M.; Brandi, R.; D’Aguanno, S.; Nicotra, M.R.; D’Onofrio, M.; et al. LMNA knock-down affects differentiation and progression of human neuroblastoma cells. PLoS ONE 2012, 7, e45513. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Guglielmi, L.; Nardella, M.; Musa, C.; Iannetti, I.; Arisi, I.; D’Onofrio, M.; Storti, A.; Valentini, A.; Cacci, E.; Biagioni, S.; et al. Lamin A/C Is Required for ChAT-Dependent Neuroblastoma Differentiation. Mol. Neurobiol. 2017, 54, 3729–3744. [Google Scholar] [CrossRef] [PubMed]
- Nardella, M.; Guglielmi, L.; Musa, C.; Iannetti, I.; Maresca, G.; Amendola, D.; Porru, M.; Carico, E.; Sessa, G.; Camerlingo, R.; et al. Down-regulation of the Lamin A/C in neuroblastoma triggers the expansion of tumor initiating cells. Oncotarget 2015, 6, 32821–32840. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, M.; Shaw, M.; Mirochnik, Y.; Slobodskoy, L.; Glick, R.; Lichtor, T.; Chou, P.; Guinan, P. In Vivo Establishment of T98G Human Glioblastoma. Methods Find. Exp. Clin. Pharmacol. 1999, 21, 391–393. [Google Scholar] [CrossRef] [PubMed]
- Lattanzi, G.; Maggi, L.; Araujo-Vilar, D. Laminopathies. Nucleus 2018, 9, 543–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maraldi, N.M. The lamin code. Biosystems 2018, 164, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Evangelisti, C.; Paganelli, F.; Giuntini, G.; Mattioli, E.; Cappellini, A.; Ramazzotti, G.; Faenza, I.; Maltarello, M.C.; Martelli, A.M.; Scotlandi, K.; et al. Lamin A and Prelamin A Counteract Migration of Osteosarcoma Cells. Cells 2020, 9, 774. [Google Scholar] [CrossRef] [Green Version]
- Willis, N.D.; Cox, T.R.; Rahman-Casañs, S.F.; Smits, K.; Przyborski, S.A.; Brandt, P.V.D.; Van Engeland, M.; Weijenberg, M.; Wilson, R.G.; De Bruïne, A.; et al. Lamin A/C is a risk biomarker in colorectal cancer. PLoS ONE 2008, 3, e2988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wu, R.; Cho, K.R.; Thomas, D.G.; Gossner, G.; Liu, J.R.; Giordano, T.; Shedden, K.A.; Misek, D.E.; Lubman, D.M. Differential protein mapping of ovarian serous adenocarcinomas: Identification of potential markers for distinct tumor stage. J. Proteome Res. 2009, 8, 1452–1463. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Li, D.; Zhou, L.; Kan, S.; He, G.; Zhou, K.; Wang, L.; Chen, M.; Shu, W. LMNA functions as an oncogene in hepatocellular carcinoma by regulating the proliferation and migration ability. J. Cell Mol. Med. 2020, 24, 12008–12019. [Google Scholar] [CrossRef]
- Kim, Y.W.; Koul, D.; Kim, S.H.; Lucio-Eterovic, A.K.; Freire, P.R.; Yao, J.; Wang, J.; Almeida, J.S.; Aldape, K.; Yung, W.A. Identifcation of prognostic gene signatures of glioblastoma: A study based on TCGA data analysis. Neuro Oncol. 2013, 15, 829–839. [Google Scholar] [CrossRef] [Green Version]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [Green Version]
- Ohgaki, H.; Kleihues, P. The definition of primary and secondary glioblastoma. Clin. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef] [Green Version]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; De Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [Green Version]
- Gong, G.; Lin, T.; Yuan, Y. Integrated analysis of gene expression and DNA methylation profiles in ovarian cancer. J. Ovarian Res. 2020, 13, 30. [Google Scholar] [CrossRef] [Green Version]
- Ranamukhaarachchi, S.K.; Modi, R.N.; Han, A.; Velez, D.O.; Kumar, A.; Engler, A.J.; Fraley, S.I. Macromolecular crowding tunes 3D collagen architecture and cell morphogenesis. Biomater. Sci. 2019, 7, 618–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuan, Z.; Zhou, Q.; Wu, X.; Xu, S.; Hu, X.; Tao, X.; Li, B.; Peng, J.; Li, D.; Shen, L.; et al. VCAM-1 secreted from cancer-associated fibroblasts enhances the growth and invasion of lung cancer cells through AKT and MAPK signalling. Cancer Lett. 2020, 473, 62–73. [Google Scholar]
- Li, B.; Shen, W.; Peng, H.; Li, Y.; Chen, F.; Zheng, L.; Xu, J.; Jia, L. Fibronectin 1 promotes melanoma proliferation and metastasis by inhibiting apoptosis and regulating EMT. Onco Targets Ther. 2019, 12, 3207–3221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Wu, M.; Xu, X.; Zhang, L.; Huang, Y.; Xu, Z.; He, K.; Wang, H.; Wang, H.; Teng, L. COL12A1, a novel potential prognostic factor and therapeutic target in gastric cancer. Mol. Med. Rep. 2019, 20, 3103–3112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Kim, W.-J.; Kang, H.-G.; Jang, J.-H.; Choi, I.J.; Chun, K.-H.; Kim, S.-J. Upregulation of LAMB1 via ERK/c-Jun Axis Promotes Gastric Cancer Growth and Motility. Int. J. Mol. Sci. 2021, 22, 626. [Google Scholar] [CrossRef] [PubMed]
- Gobin, E.; Bagwell, K.; Wagner, J.; Mysona, D.; Sandirasegarane, S.; Smith, N.; Bai, S.; Sharma, A.; Schleifer, R.; She, J.-X. A pan-cancer perspective of matrix metalloproteases (MMP) gene expression profile and their diagnostic/prognostic potential. BMC Cancer 2019, 19, 581. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Huang, S.; Guo, J.; Zhou, L.; You, L.; Zhang, T.; Zhao, Y. Insights into the distinct roles of MMP-11 in tumor biology and future therapeutics (Review). Int. J. Oncol. 2016, 48, 1783–1793. [Google Scholar] [CrossRef] [Green Version]
- Decock, J.; Thirkettle, S.; Wagstaff, L.; Edwards, D.R. Matrix metalloproteinases: Protective roles in cancer. J. Cell. Mol. Med. 2011, 15, 1254–1265. [Google Scholar] [CrossRef] [Green Version]
- Ellert-Miklaszewska, A.; Poleszak, K.; Pasierbinska, M.; Kaminska, B. Integrin Signaling in Glioma Pathogenesis: From Biology to Therapy. Int. J. Mol. Sci. 2020, 21, 888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Gibbons, R.M.; Harrington, S.M.; Krco, C.J.; Markovic, S.N.; Kwon, E.D.; Dong, H. Endogenous tumor-reactive CD8+ T cells are differentiated effector cells expressing high levels of CD11a and PD-1 but are unable to control tumor growth. OncoImmunology 2013, 6, e23972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, M.C.; Khan, S.Q.; Kaneda, M.M.; Pathria, P.; Shepard, R.; Louis, T.L.; Anand, S.; Woo, G.; Leem, C.; Faridi, M.H.; et al. Integrin CD11b activation drives anti-tumor innate immunity. Nat. Commun. 2018, 9, 5379. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Shao, X.; He, Q.; Wang, C.; Xia, L.; Yue, D.; Qin, G.; Jia, C.; Chen, R. Quantitative Proteomics Implicates Rictor/mTORC2 in Cell Adhesion. J. Proteome Res. 2018, 17, 3360–3369. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.-H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged Rapamycin Treatment Inhibits mTORC2 Assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef]
- Gkountakos, A.; Pilotto, S.; Mafficini, A.; Vicentini, C.; Simbolo, M.; Milella, M.; Tortora, G.; Scarpa, A.; Bria, E.; Corbo, V. Unmasking the impact of Rictor in cancer: Novel insights of mTORC2 complex. Carcinogenesis 2018, 39, 971–980. [Google Scholar] [CrossRef] [Green Version]
- Masri, J.; Bernath, A.; Martin, J.; Jo, O.D.; Vartanian, R.; Funk, A.; Gera, J. mTORC2 activity is elevated in gliomas and promotes growth and cell motility via overexpression of rictor. Cancer Res. 2007, 67, 11712–11720. [Google Scholar] [CrossRef] [Green Version]
- Rosner, M.; Hengstschlager, M. Cytoplasmic and nuclear distribution of the protein complexes mTORC1 and mTORC2: Rapamycin triggers dephosphorylation and delocalization of the mTORC2 components rictor and sin1. Hum. Mol. Genet. 2008, 17, 2934–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarenga, A.W.; Machado, L.E.; Rodrigues, B.R.; Lupinacci, F.C.S.; Sanemastu, P.; Matta, E.; Roffé, M.; Torres, L.F.B.; Da Cunha, I.W.; Martins, V.R.; et al. Evaluation of Akt and RICTOR Expression Levels in Astrocytomas of All Grades. J. Histochem. Cytochem. 2017, 65, 93–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, S.-J.; Zhan, S.-K.; Pan, Y.-X.; Liu, W.; Bian, L.-G.; Sun, B.; Sun, Q.-F. Tetraspanin 8-Rictor-Integrin α3 Complex Is Required for Glioma Cell Migration. Int. J. Mol. Sci. 2015, 16, 5363–5374. [Google Scholar] [CrossRef] [Green Version]
- Chantaravisoot, N.; Wongkongkathep, P.; Loo, J.A.; Mischel, P.S.; Tamanoi, F. Significance of filamin A in mTORC2 function in glioblastoma. Mol. Cancer 2015, 14, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gatti, G.; Vilardo, L.; Musa, C.; Di Pietro, C.; Bonaventura, F.; Scavizzi, F.; Torcinaro, A.; Bucci, B.; Saporito, R.; Arisi, I.; et al. Role of Lamin A/C as Candidate Biomarker of Aggressiveness and Tumorigenicity in Glioblastoma Multiforme. Biomedicines 2021, 9, 1343. https://doi.org/10.3390/biomedicines9101343
Gatti G, Vilardo L, Musa C, Di Pietro C, Bonaventura F, Scavizzi F, Torcinaro A, Bucci B, Saporito R, Arisi I, et al. Role of Lamin A/C as Candidate Biomarker of Aggressiveness and Tumorigenicity in Glioblastoma Multiforme. Biomedicines. 2021; 9(10):1343. https://doi.org/10.3390/biomedicines9101343
Chicago/Turabian StyleGatti, Giuliana, Laura Vilardo, Carla Musa, Chiara Di Pietro, Fabrizio Bonaventura, Ferdinando Scavizzi, Alessio Torcinaro, Barbara Bucci, Raffaele Saporito, Ivan Arisi, and et al. 2021. "Role of Lamin A/C as Candidate Biomarker of Aggressiveness and Tumorigenicity in Glioblastoma Multiforme" Biomedicines 9, no. 10: 1343. https://doi.org/10.3390/biomedicines9101343
APA StyleGatti, G., Vilardo, L., Musa, C., Di Pietro, C., Bonaventura, F., Scavizzi, F., Torcinaro, A., Bucci, B., Saporito, R., Arisi, I., De Santa, F., Raspa, M., Guglielmi, L., & D’Agnano, I. (2021). Role of Lamin A/C as Candidate Biomarker of Aggressiveness and Tumorigenicity in Glioblastoma Multiforme. Biomedicines, 9(10), 1343. https://doi.org/10.3390/biomedicines9101343