
Chemical Modification of Cysteine with 3-Arylpropriolonitrile Improves the In Vivo Stability of Albumin-Conjugated Urate Oxidase Therapeutic Protein


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Generation of pBAD_AgUox and pBAD_AgUox-196Amb Plasmids for Expression of AgUox Variants (Wild-Type AgUox and AgUox-frTet)
2.3. Expression and Purification of AgUox-WT and AgUox-frTet
2.4. MALDI-TOF Mass Spectrometry (MS) Analysis
2.5. Site-Specific Fluorescence Dye Labeling of AgUox-WT and AgUox-frTet
2.6. Generation of AgUox-HSA Conjugates (AgUox-MAL-HSA and AgUox-APN-HSA)
2.7. Enzymatic Activity and Stability Assays of AgUox-WT and AgUox-HSA Conjugates (AgUox-MAL-HSA and AgUox-APN-HSA)
2.8. In Vivo Stability Assays of AgUox Variants
3. Results and Discussion
3.1. Site-Specific Incorporation of frTet into AgUox
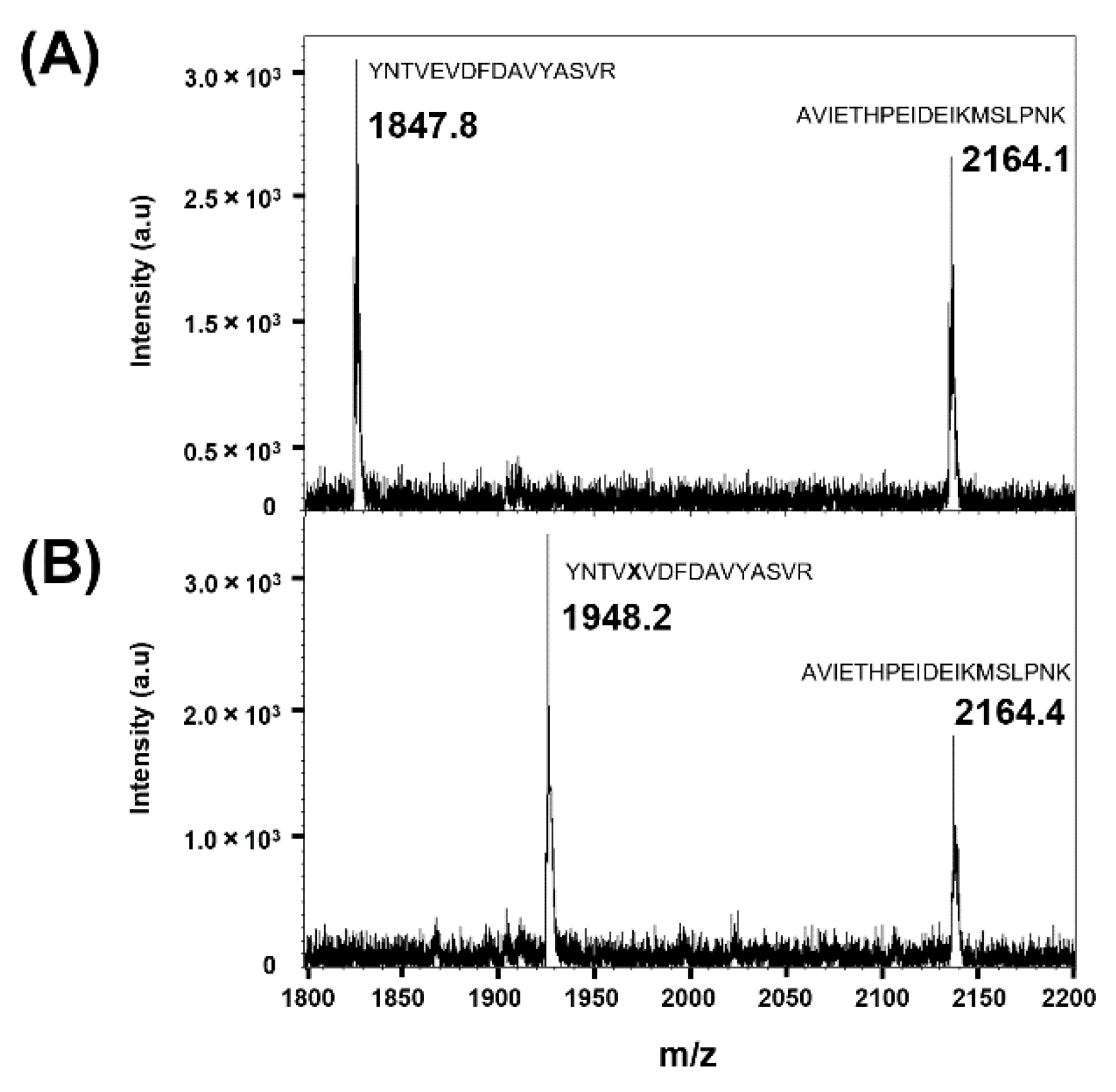
3.2. Confirmation of Site-Specific Incorporation of frTet into AgUox Using Fluorescence Dye Labeling and MALDI-TOF MS
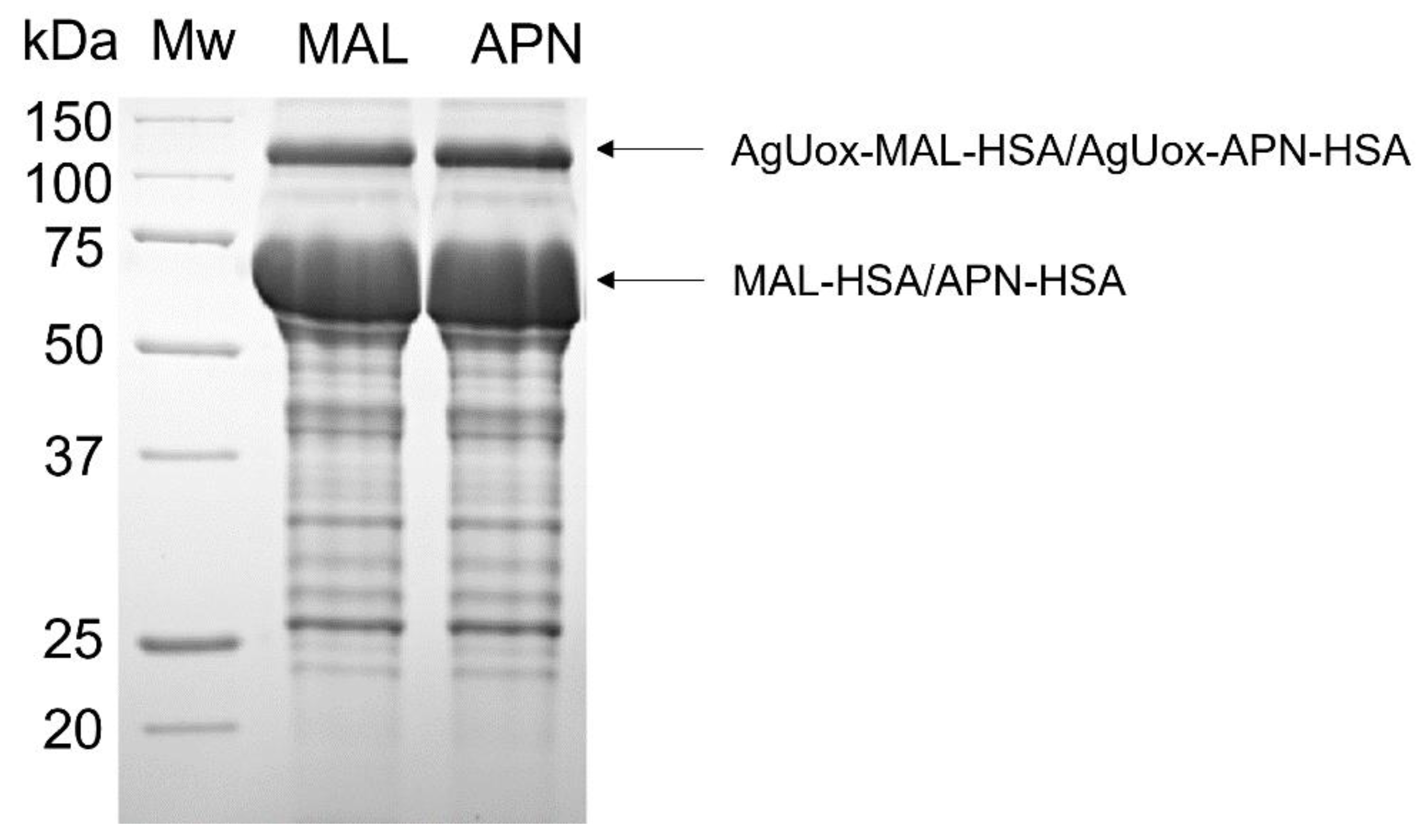
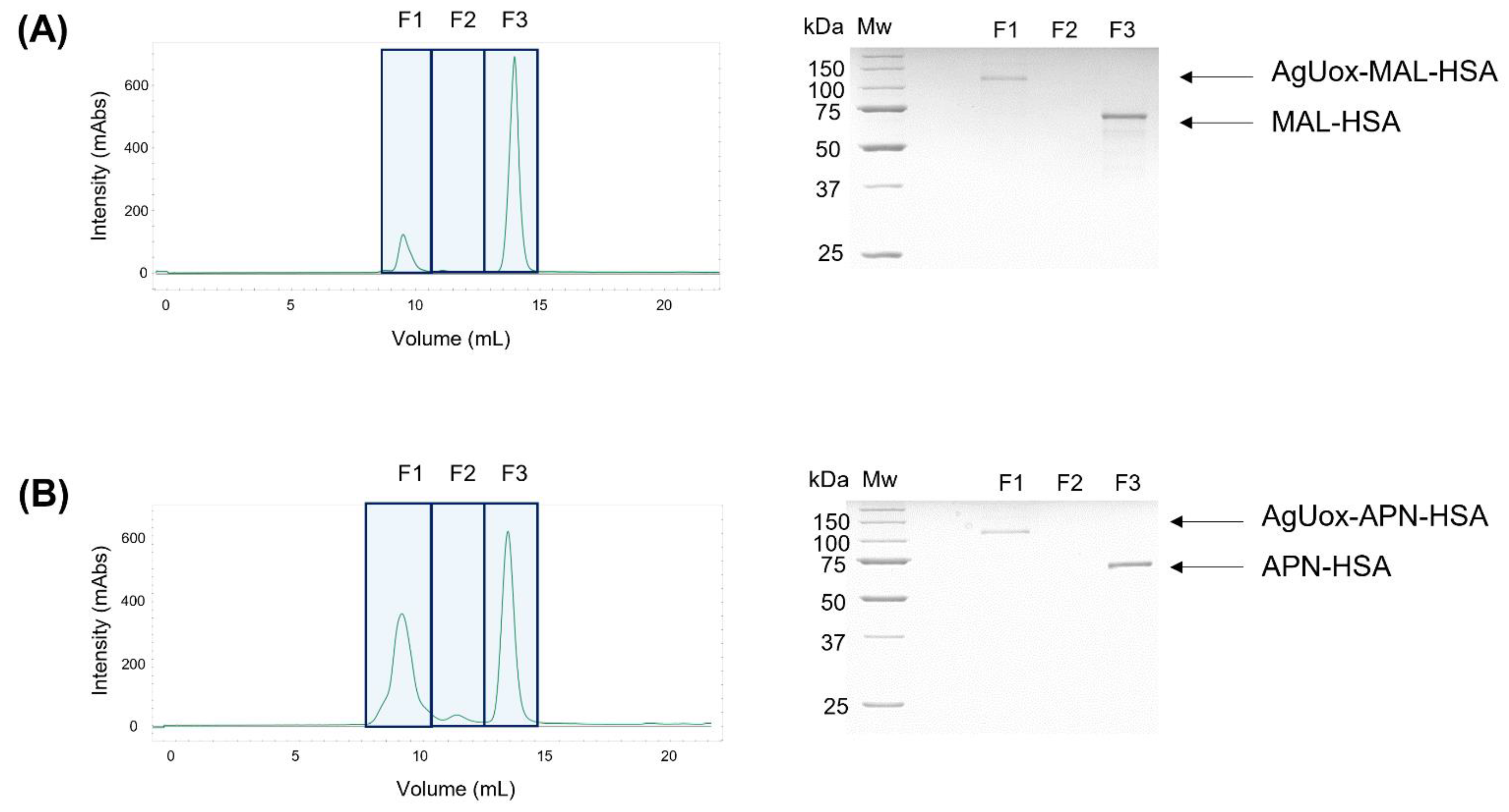
3.3. Site-Specific HSA Conjugation to AgUox and Purification of AgUox-HSA Conjugates
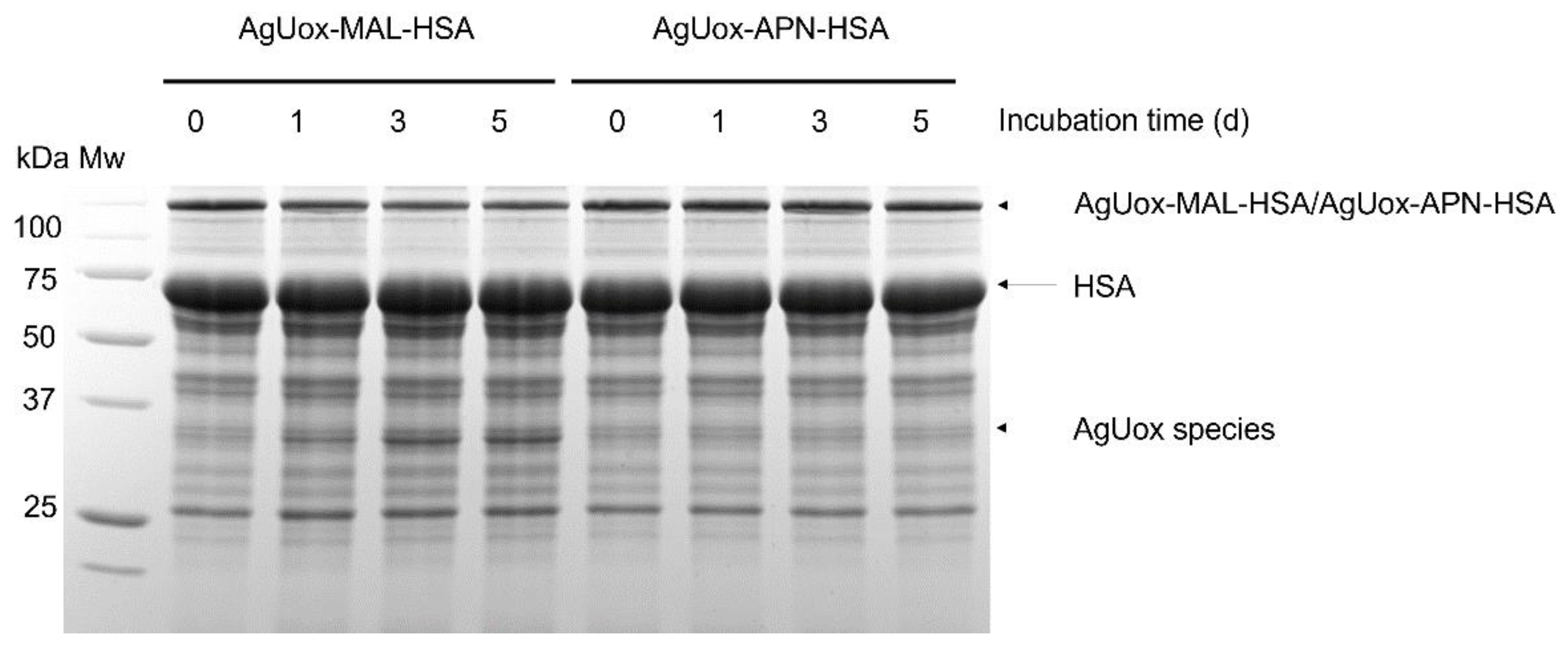
3.4. Enzymatic Activity Assays and In Vitro Stability Tests of AgUox-HSA Conjugates
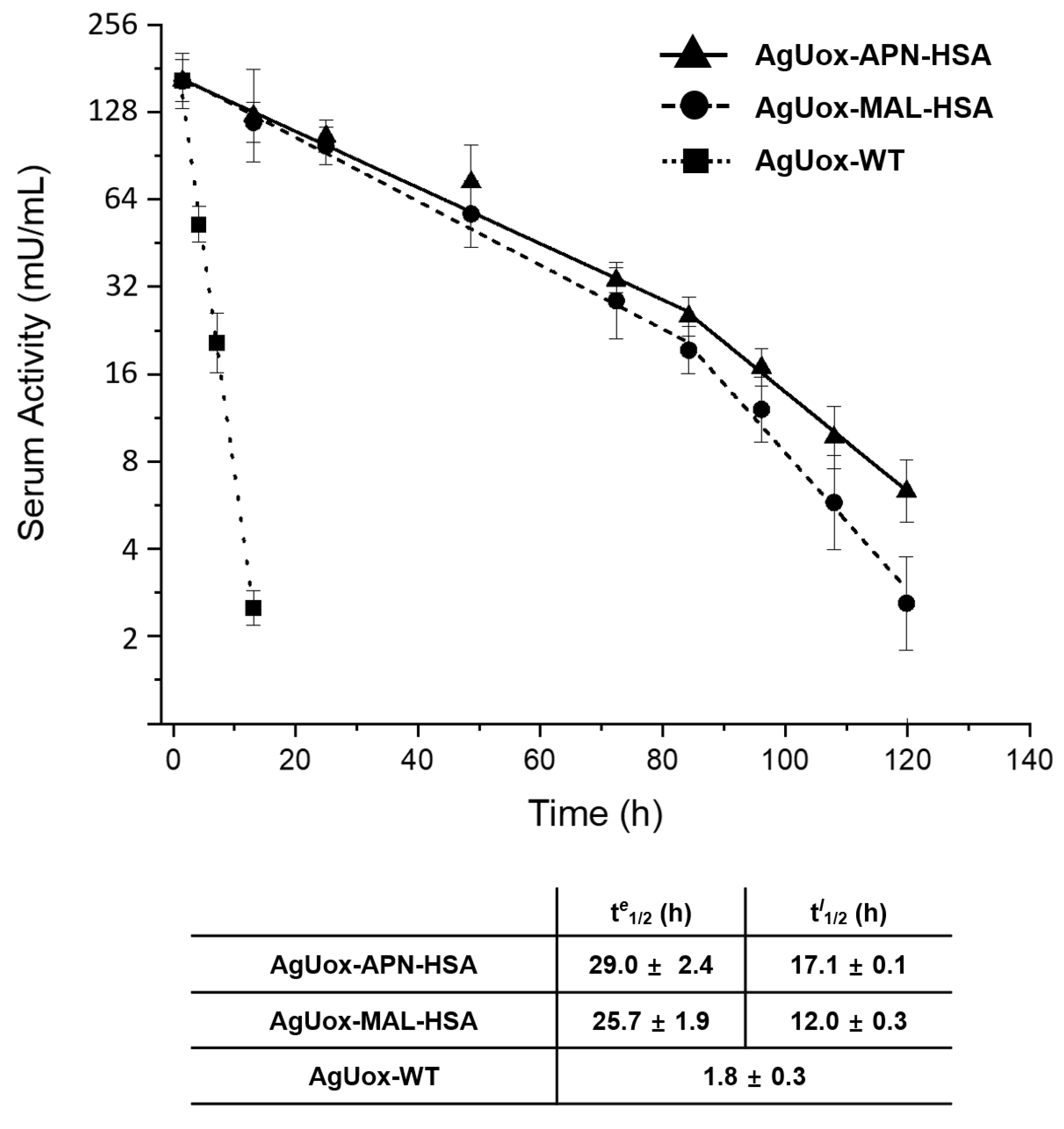
3.5. In Vivo Stability Tests of Uox-HSA Conjugates in Mice
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, X.; Zhou, Y.; Peng, X.; Yoon, J. Fluorescent and colorimetric probes for detection of thiols. Chem. Soc. Rev. 2010, 39, 2120–2135. [Google Scholar] [CrossRef]
- Hoogenboom, R. Thiol-yne chemistry: A powerful tool for creating highly functional materials. Angew. Chem. Int. Ed. Engl. 2010, 49, 3415–3417. [Google Scholar] [CrossRef]
- Giron, P.; Dayon, L.; Sanchez, J.C. Cysteine tagging for MS-based proteomics. Mass Spectrom. Rev. 2011, 30, 366–395. [Google Scholar] [CrossRef]
- Chan, A.O.-Y.; Tsai, J.L.-L.; Lo, V.K.-Y.; Li, G.-L.; Wong, M.-K.; Che, C.-M. Gold-Mediated selective cysteine modification of peptides using allenes. Chem. Commun. 2013, 49, 1428–1430. [Google Scholar] [CrossRef]
- Zhang, Y.; Zang, C.; An, G.; Shang, M.; Cui, Z.; Chen, G.; Xi, Z.; Zhou, C. Cysteine-specific protein multi-functionalization and disulfide bridging using 3-bromo-5-methylene pyrrolones. Nat. Commun. 2020, 11, 1015. [Google Scholar] [CrossRef] [Green Version]
- Brotzel, F.; Mayr, H. Nucleophilicities of amino acids and peptides. Org. Biomol. Chem. 2007, 5, 3814–3820. [Google Scholar] [CrossRef]
- Hackenberger, C.P.; Schwarzer, D. Chemoselective ligation and modification strategies for peptides and proteins. Angew. Chem. Int. Ed. Engl. 2008, 47, 10030–10074. [Google Scholar] [CrossRef]
- Kim, Y.; Ho, S.O.; Gassman, N.R.; Korlann, Y.; Landorf, E.V.; Collart, F.R.; Weiss, S. Efficient site-specific labeling of proteins via cysteines. Bioconjug. Chem. 2008, 19, 786–791. [Google Scholar] [CrossRef] [Green Version]
- Chalker, J.M.; Bernardes, G.J.; Lin, Y.A.; Davis, B.G. Chemical modification of proteins at cysteine: Opportunities in chemistry and biology. Chem. Asian J. 2009, 4, 630–640. [Google Scholar] [CrossRef]
- Ren, H.; Xiao, F.; Zhan, K.; Kim, Y.P.; Xie, H.; Xia, Z.; Rao, J. A biocompatible condensation reaction for the labeling of terminal cysteine residues on proteins. Angew. Chem. Int. Ed. Engl. 2009, 48, 9658–9662. [Google Scholar] [CrossRef]
- Shen, B.Q.; Xu, K.; Liu, L.; Raab, H.; Bhakta, S.; Kenrick, M.; Parsons-Reponte, K.L.; Tien, J.; Yu, S.F.; Mai, E.; et al. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat. Biotechnol. 2012, 30, 184–189. [Google Scholar] [CrossRef]
- Lyon, R.P.; Setter, J.R.; Bovee, T.D.; Doronina, S.O.; Hunter, J.H.; Anderson, M.E.; Balasubramanian, C.L.; Duniho, S.M.; Leiske, C.I.; Li, F.; et al. Self-hydrolyzing maleimides improve the stability and pharmacological properties of antibody-drug conjugates. Nat. Biotechnol. 2014, 32, 1059–1062. [Google Scholar] [CrossRef] [PubMed]
- Ravasco, J.; Faustino, H.; Trindade, A.; Gois, P.M.P. Bioconjugation with Maleimides: A Useful Tool for Chemical Biology. Chemistry 2019, 25, 43–59. [Google Scholar] [CrossRef] [PubMed]
- Stehle, G.; Wunder, A.; Schrenk, H.H.; Hartung, G.; Heene, D.L.; Sinn, H. Albumin-based drug carriers: Comparison between serum albumins of different species on pharmacokinetics and tumor uptake of the conjugate. Anticancer Drugs 1999, 10, 785–790. [Google Scholar] [CrossRef]
- Szijj, P.A.; Bahou, C.; Chudasama, V. Minireview: Addressing the retro-Michael instability of maleimide bioconjugates. Drug Discov. Today Technol. 2018, 30, 27–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldwin, A.D.; Kiick, K.L. Tunable degradation of maleimide-thiol adducts in reducing environments. Bioconjug. Chem. 2011, 22, 1946–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahou, C.; Spears, R.J.; Aliev, A.E.; Maruani, A.; Fernandez, M.; Javaid, F.; Szijj, P.A.; Baker, J.R.; Chudasama, V. Use of pyridazinediones as extracellular cleavable Linkers through Reversible Cysteine Conjugation. Chem. Commun. 2019, 55, 14829–14832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyatzis, A.E.; Bringans, S.D.; Piggott, M.J.; Duong, M.N.; Lipscombe, R.J.; Arthur, P.G. Limiting the Hydrolysis and Oxidation of Maleimide-Peptide Adducts Improves Detection of Protein Thiol Oxidation. J. Proteome Res. 2017, 16, 2004–2015. [Google Scholar] [CrossRef]
- Koniev, O.; Leriche, G.; Nothisen, M.; Remy, J.S.; Strub, J.M.; Schaeffer-Reiss, C.; Van Dorsselaer, A.; Baati, R.; Wagner, A. Selective irreversible chemical tagging of cysteine with 3-arylpropiolonitriles. Bioconjug. Chem. 2014, 25, 202–206. [Google Scholar] [CrossRef]
- Fontaine, S.D.; Reid, R.; Robinson, L.; Ashley, G.W.; Santi, D.V. Long-Term Stabilization of Maleimide–Thiol Conjugates. Bioconjug. Chem. 2014, 26, 145–152. [Google Scholar] [CrossRef]
- St. Amant, A.H.; Lemen, D.; Florinas, S.; Mao, S.; Fazenbaker, C.; Zhong, H.; Wu, H.; Gao, C.; Christie, R.J.; de Alaniz, J.R. Tuning the Diels–Alder Reaction for Bioconjugation to Maleimide Drug-Linkers. Bioconjug. Chem. 2018, 29, 2406–2414. [Google Scholar] [CrossRef] [Green Version]
- Ochtrop, P.; Hackenberger, C.P.R. Recent advances of thiol-selective bioconjugation reactions. Curr. Opin. Chem. Biol. 2020, 58, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Kolodych, S.; Koniev, O.; Baatarkhuu, Z.; Bonnefoy, J.-Y.; Debaene, F.; Cianférani, S.; Van Dorsselaer, A.; Wagner, A. CBTF: New Amine-to-Thiol Coupling Reagent for Preparation of Antibody Conjugates with Increased Plasma Stability. Bioconjug. Chem. 2015, 26, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Padma, A.; Saara-Anne, A.M.; Freedy, A.D.; Cal, P.M.S.; Gois, P.M.L.; Bernardes, G.J. Construction of homogeneous antibody–drug conjugates using site-selective protein chemistry. Chem. Sci. 2016, 7, 2954–2963. [Google Scholar]
- Kratz, F. Albumin as a drug carrier: Design of prodrugs, drug conjugates and nanoparticles. J. Control Release 2008, 132, 171–183. [Google Scholar] [CrossRef]
- Bern, M.; Sand, K.M.; Nilsen, J.; Sandlie, I.; Andersen, J.T. The role of albumin receptors in regulation of albumin homeostasis: Implications for drug delivery. J. Control Release 2015, 211, 144–162. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.T.; Kang, T.H.; Kim, D. Engineering an Aglycosylated Fc Variant for Enhanced FcγRI Engagement and PH-Dependent Human FcRn Binding. Biotechnol. Bioprocess Eng. 2014, 19, 780–789. [Google Scholar] [CrossRef]
- Rahimizadeh, P.; Yang, S.; Lim, S.I. Albumin: An Emerging Opportunity in Drug Delivery. Biotechnol. Bioprocess Eng. 2020, 25, 985–995. [Google Scholar] [CrossRef]
- Pyzik, M.; Sand, K.M.K.; Hubbard, J.J.; Andersen, J.T.; Sandlie, I.; Blumberg, R.S. The Neonatal Fc Receptor (FcRn): A Misnomer? Front. Immunol. 2019, 10, 1540. [Google Scholar] [CrossRef]
- Lim, S.I.; Hahn, Y.S.; Kwon, I. Site-specific albumination of a therapeutic protein with multi-subunit to prolong activity in vivo. J. Control Release 2015, 207, 93–100. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Lim, S.I.; Kim, J.C.; Tae, G.; Kwon, I. Site-Specific Albumination as an Alternative to PEGylation for the Enhanced Serum Half-Life in Vivo. Biomacromolecules 2016, 17, 1811–1817. [Google Scholar] [CrossRef]
- Yang, B.; Kwon, K.; Jana, S.; Kim, S.; Avila-Crump, S.; Tae, G.; Mehl, R.A.; Kwon, I. temporal control of efficient in vivo bioconjugation using a genetically Encoded Tetrazine-Mediated Inverse-Electron-Demand Diels-Alder Reaction. Bioconjug. Chem. 2020, 31, 2456–2464. [Google Scholar] [CrossRef] [PubMed]
- Coiffier, B.; Mounier, N.; Bologna, S.; Fermé, C.; Tilly, H.; Sonet, A.; Christian, B.; Casasnovas, O.; Jourdan, E.; Belhadj, K.; et al. Efficacy and safety of rasburicase (recombinant urate oxidase) for the prevention and treatment of hyperuricemia during induction chemotherapy of aggressive non-Hodgkin’s lymphoma: Results of the GRAAL1 (Grouped’Etude des Lymphomes de l’Adulte Trial on Rasburicase Activity in Adult Lymphoma) study. J. Clin. Oncol. 2003, 21, 4402–4406. [Google Scholar] [PubMed]
- Ryu, J.K.; Kim, H.S.; Nam, D.H. Current status and perspectives of biopharmaceutical drugs. Biotechnol. Bioprocess Eng. 2012, 17, 900–911. [Google Scholar] [CrossRef]
- Schlesinger, N.; Yasothan, U.; Kirkpatrick, P. Pegloticase. Nat. Rev. Drug Discov. 2011, 10, 17–18. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Kwon, I. Thermostable and long-circulating albumin-conjugated Arthrobacter globiformis urate oxidase. Pharmaceutics 2021, 13, 1298. [Google Scholar] [CrossRef]
- Caddick, S.; Wilden, J.D.; Judd, D.B. Observations on the reactivity of pentafluorophenyl sulfonate esters. Chem. Commun. 2005, 21, 2727–2728. [Google Scholar] [CrossRef]
- Nyborg, A.C.; Ward, C.; Zacco, A.; Chacko, B.; Grinberg, L.; Geoghegan, J.C.; Bean, R.; Wendeler, M.; Bartnik, F.; O’Connor, E.; et al. A Therapeutic Uricase with Reduced Immunogenicity Risk and Improved Development Properties. PLoS ONE 2016, 11, e0167935. [Google Scholar]
- Shi, Y.; Wang, T.; Zhou, X.E.; Liu, Q.F.; Jiang, Y.; Xu, H.E. Structure-based design of a hyperthermostable AgUricase for hyperuricemia and gout therapy. Acta Pharmacol. Sin. 2019, 40, 1364–1372. [Google Scholar] [CrossRef]
- Yang, B.; Kwon, I. Multivalent Albumin-Neonatal Fc Receptor Interactions Mediate a Prominent Extension of the Serum Half-Life of a Therapeutic Protein. Mol. Pharm. 2021, 18, 2397–2405. [Google Scholar] [CrossRef]
- Deck, M.B.; Sjölin, P.; Unanue, E.R.; Kihlberg, J. MHC-Restricted, Glycopeptide-Specific T Cells Show Specificity for Both Carbohydrate and Peptide Residues. J. Immunol. 1999, 162, 4740–4744. [Google Scholar]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, B.; Kwon, I. Chemical Modification of Cysteine with 3-Arylpropriolonitrile Improves the In Vivo Stability of Albumin-Conjugated Urate Oxidase Therapeutic Protein. Biomedicines 2021, 9, 1334. https://doi.org/10.3390/biomedicines9101334
Yang B, Kwon I. Chemical Modification of Cysteine with 3-Arylpropriolonitrile Improves the In Vivo Stability of Albumin-Conjugated Urate Oxidase Therapeutic Protein. Biomedicines. 2021; 9(10):1334. https://doi.org/10.3390/biomedicines9101334
Chicago/Turabian StyleYang, Byungseop, and Inchan Kwon. 2021. "Chemical Modification of Cysteine with 3-Arylpropriolonitrile Improves the In Vivo Stability of Albumin-Conjugated Urate Oxidase Therapeutic Protein" Biomedicines 9, no. 10: 1334. https://doi.org/10.3390/biomedicines9101334
APA StyleYang, B., & Kwon, I. (2021). Chemical Modification of Cysteine with 3-Arylpropriolonitrile Improves the In Vivo Stability of Albumin-Conjugated Urate Oxidase Therapeutic Protein. Biomedicines, 9(10), 1334. https://doi.org/10.3390/biomedicines9101334