
Reverse Engineering of the Pediatric Sepsis Regulatory Network and Identification of Master Regulators

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
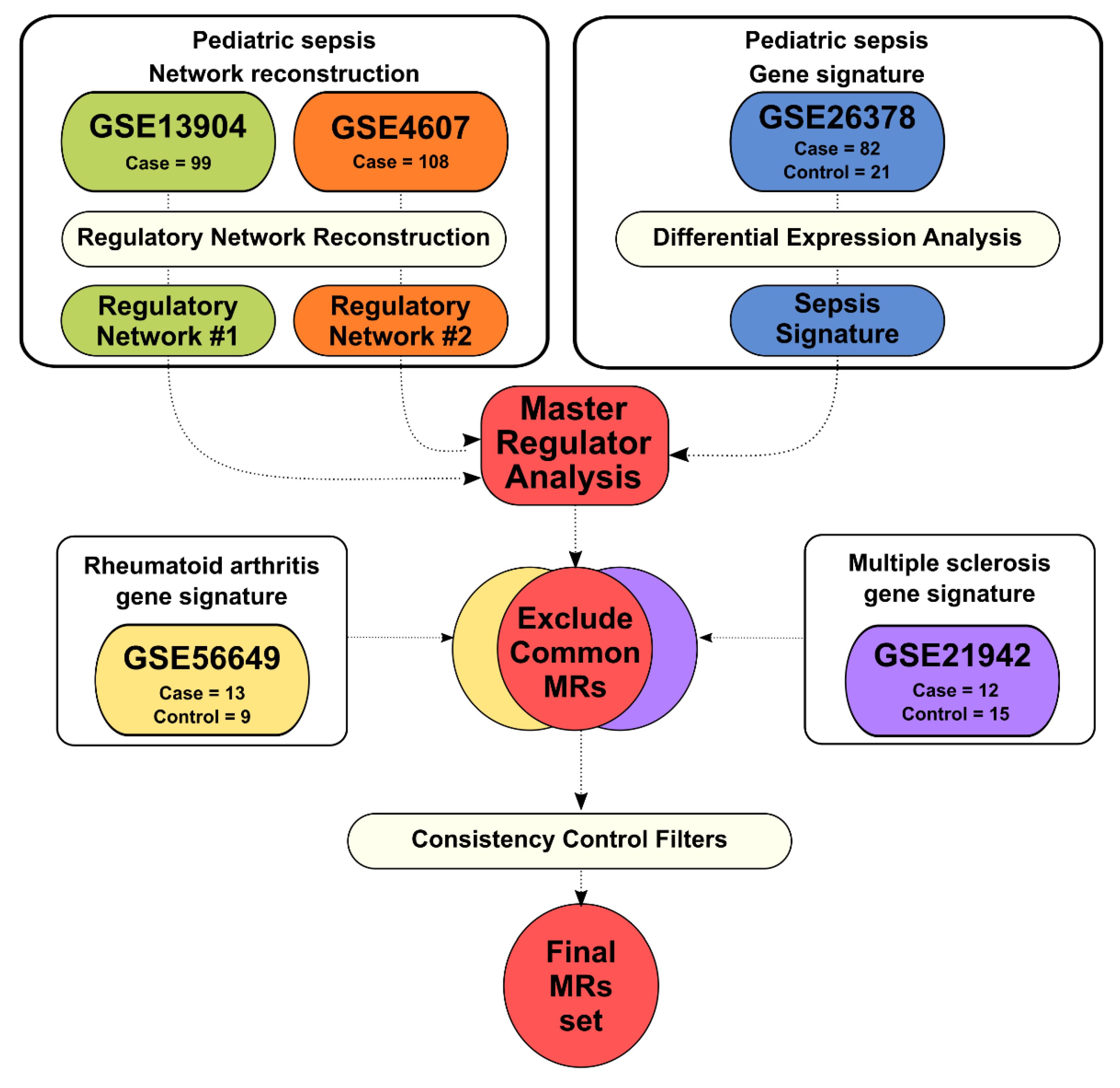
2. Materials and Methods
2.1. Pediatric Sepsis Regulatory Network Reconstruction
2.2. Gene Signatures from Sepsis, Rheumatoid Arthritis, and Multiple Sclerosis
2.3. Master Regulator Analysis
2.4. Network Visualizations
2.5. Regulon Activity Analysis
2.6. Gene Ontology Functional Enrichment
2.7. Master Regulator Expression
3. Results
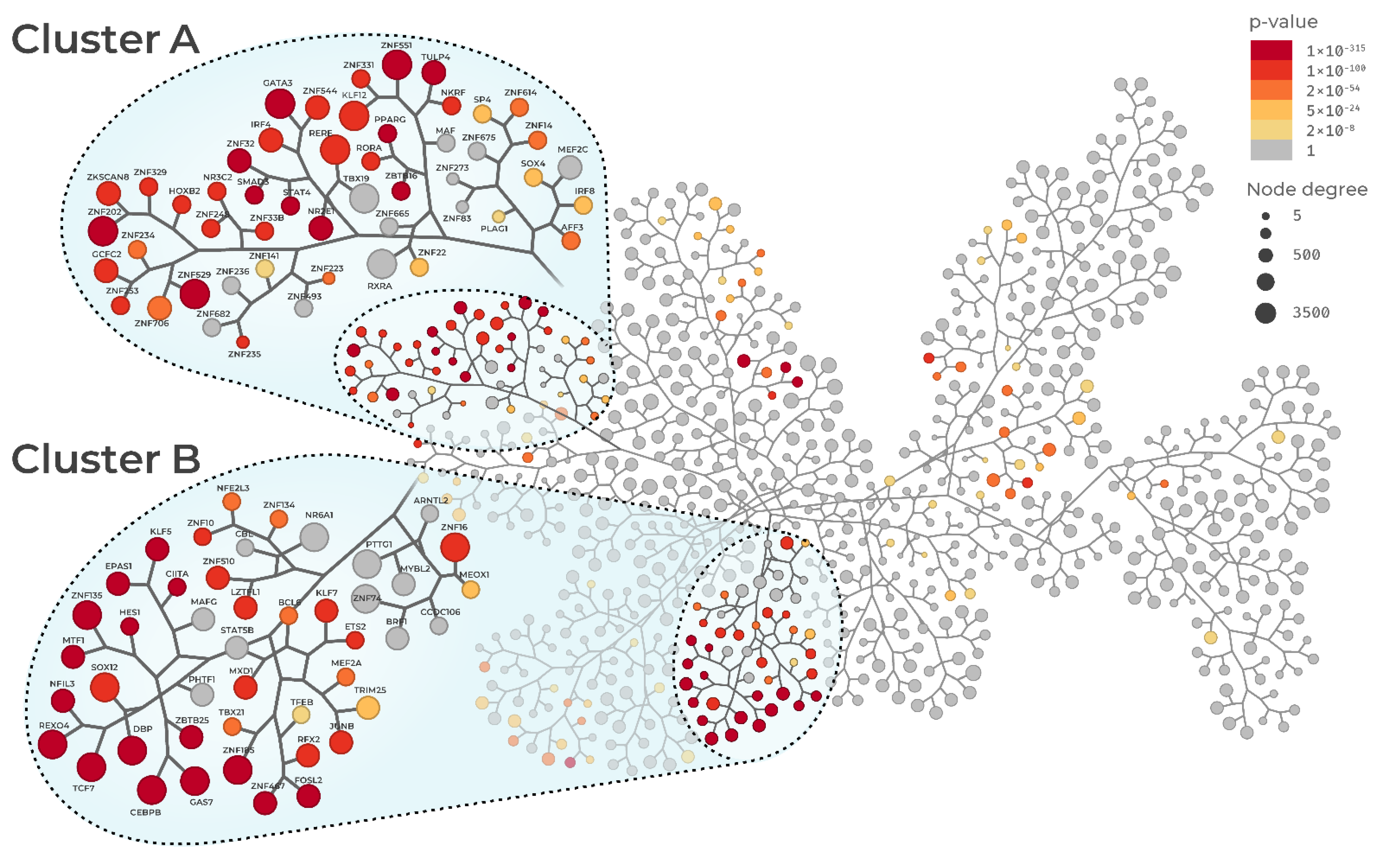
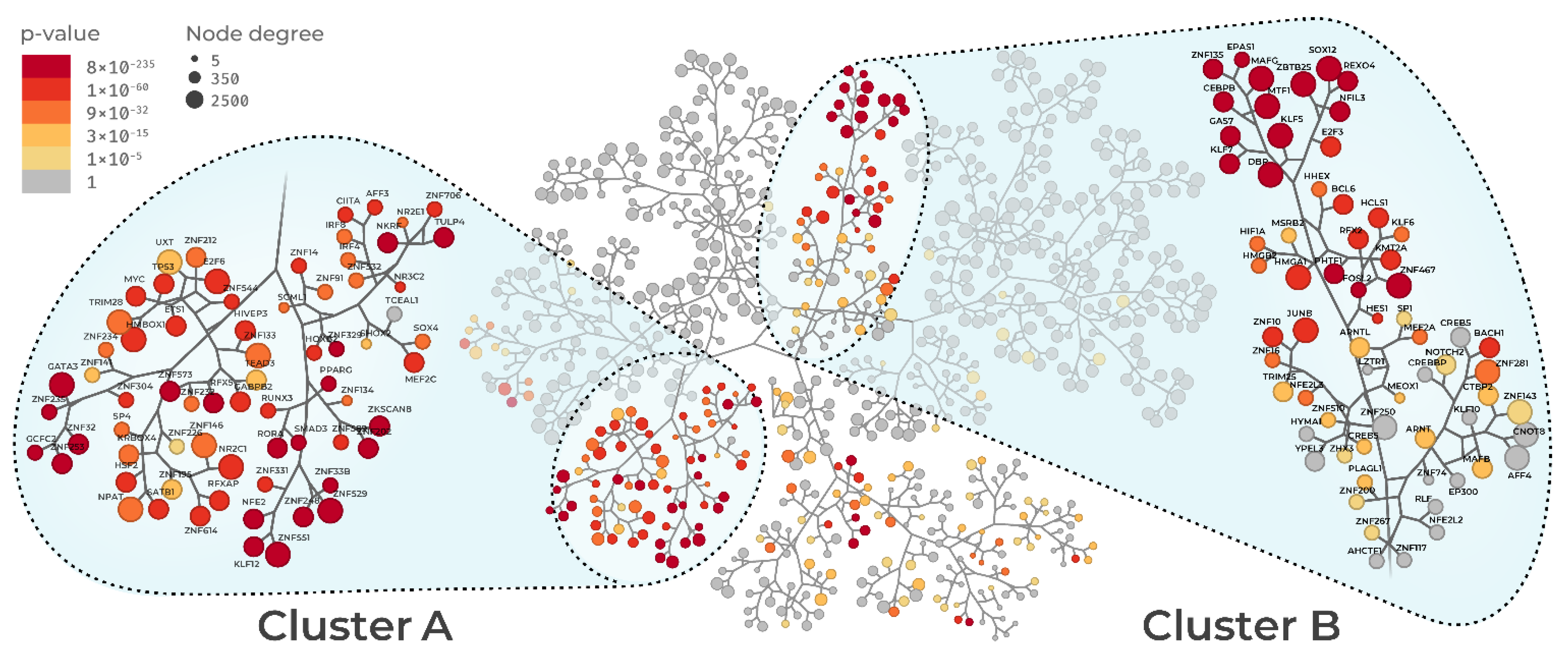
3.1. Sepsis Regulatory Networks and Master Regulator Analysis
3.2. Master Regulator Activity
3.3. Regulon Similarity
3.4. Regulon Functional Enrichment
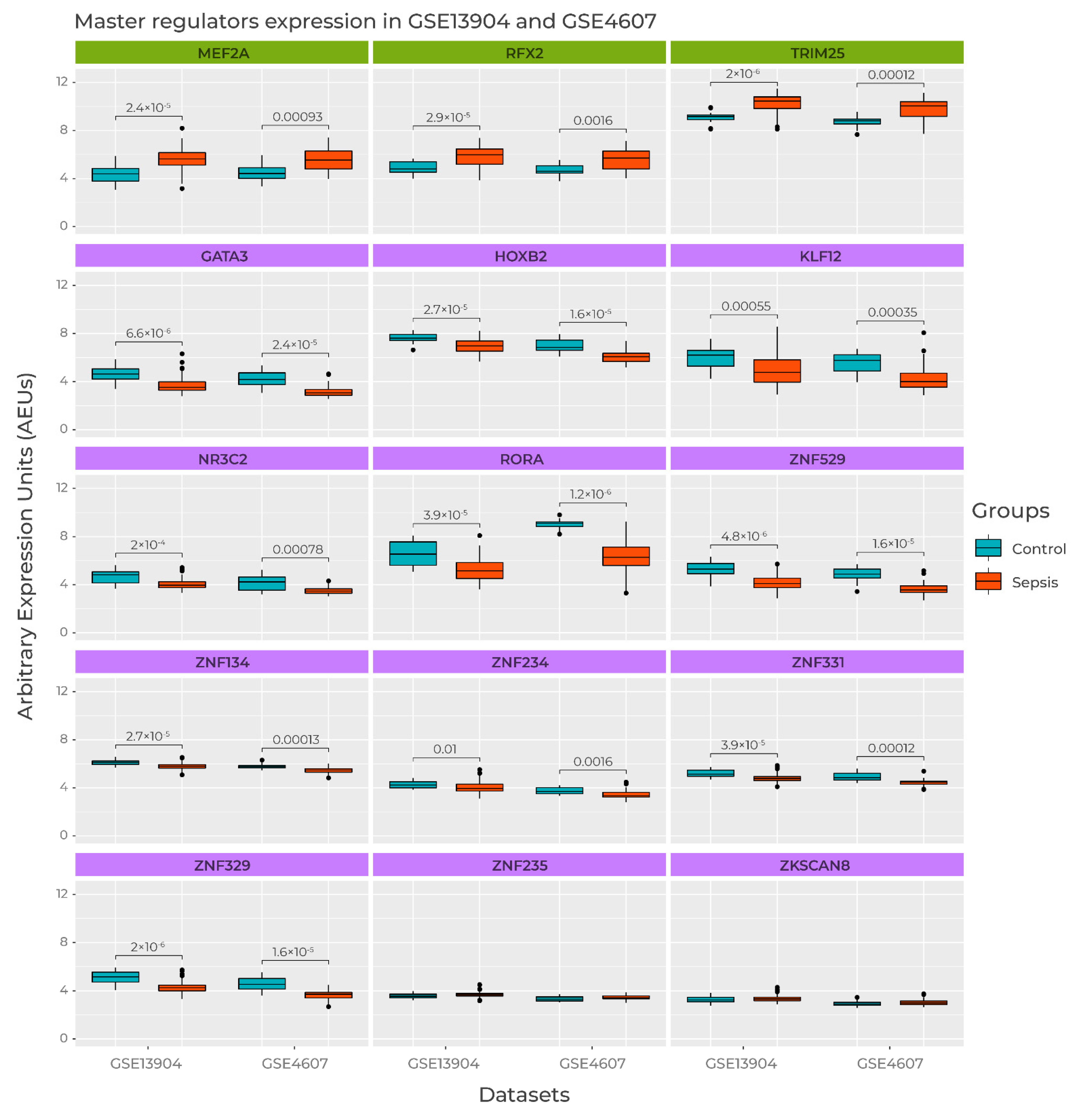
3.5. Master Regulators Expression in Sepsis Datasets
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ICUs | Intensive Care Units |
| MR | Master Regulator |
| MRA | Master Regulator Analysis |
| WBC | White Blood-cell Count |
| SIRS | Systemic Inflammatory Response Syndrome |
| CARS | Compensatory Anti-inflammatory Response Syndrome |
| TF | Transcription factor |
| GRN | Gene Regulatory Network |
| GEO | Gene Expression Omnibus |
| PBMC | Peripheral blood mononuclear cells |
| RA | Rheumatoid Arthritis |
| MS | Multiple Sclerosis |
| GPL | GEO Platform |
| RTN | Reconstruction of Transcriptional Networks |
| GSE | GEO Series |
| TNI | Transcription Network Inference |
| BH | Benjamini-Hochberg |
| GSEA2 | Two-Tailed Gene Set Enrichment Analysis |
References
- Singer, M.; Deutschman, C.S.; Seymour, C.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA J. Am. Med. Assoc. 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Mathias, B.; Mira, J.C.; Larson, S.D. Pediatric Sepsis. Curr. Opin. Pediatr. 2016, 28, 380–387. [Google Scholar] [CrossRef] [Green Version]
- Menon, K.; Schlapbach, L.J.; Akech, S.; Argent, A.; Chiotos, K.; Chisti, M.J.; Hamid, J.; Ishimine, P.; Kissoon, N.; Lodha, R.; et al. Pediatric Sepsis Definition—A Systematic Review Protocol by the Pediatric Sepsis Definition Taskforce. Crit. Care Explor. 2020, 2, e0123. [Google Scholar] [CrossRef]
- Sakr, Y.; Jaschinski, U.; Wittebole, X.; Szakmany, T.; Lipman, J.; Ñamendys-Silva, S.A.; Martin-Loeches, I.; Leone, M.; Lupu, M.-N.; Vincent, J.-L. Sepsis in Intensive Care Unit Patients: Worldwide Data from the Intensive Care over Nations Audit. Open Forum Infect. Dis. 2018, 5. [Google Scholar] [CrossRef]
- Cecconi, M.; Evans, L.; Levy, M.; Rhodes, A. Sepsis and Septic Shock. Lancet 2018, 392, 75–87. [Google Scholar] [CrossRef]
- Wong, H.R.; Cvijanovich, N.; Allen, G.L.; Lin, R.; Anas, N.; Meyer, K.; Freishtat, R.J.; Monaco, M.; Odoms, K.; Sakthivel, B.; et al. Genomic Expression Profiling across the Pediatric Systemic Inflammatory Response Syndrome, Sepsis, and Septic Shock Spectrum. Crit. Care Med. 2009, 37, 1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweeney, T.E.; Shidham, A.; Wong, H.R.; Khatri, P. A Comprehensive Time-Course-Based Multicohort Analysis of Sepsis and Sterile Inflammation Reveals a Robust Diagnostic Gene Set. Sci. Transl. Med. 2015, 7, 287ra71. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-C. Risk Stratification of Severe Sepsis Patients in the Emergency Department. Emerg. Med. J. 2006, 23, 281–285. [Google Scholar] [CrossRef] [Green Version]
- Quinten, V.M.; van Meurs, M.; Wolffensperger, A.E.; ter Maaten, J.C.; Ligtenberg, J.J.M. Sepsis Patients in the Emergency Department. Eur. J. Emerg. Med. 2018, 25, 328–334. [Google Scholar] [CrossRef] [Green Version]
- Mellhammar, L.; Linder, A.; Tverring, J.; Christensson, B.; Boyd, J.H.; Åkesson, P.; Kahn, F. Scores for Sepsis Detection and Risk Stratification—Construction of a Novel Score Using a Statistical Approach and Validation of RETTS. PLoS ONE 2020, 15, e0229210. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.; Vincent, J.-L.; Adhikari, N.K.J.; Machado, F.R.; Angus, D.C.; Calandra, T.; Jaton, K.; Giulieri, S.; Delaloye, J.; Opal, S.; et al. Sepsis: A Roadmap for Future Research. Lancet Infect. Dis. 2015, 15, 581–614. [Google Scholar] [CrossRef]
- Wong, H.R. Pediatric Sepsis Biomarkers for Prognostic and Predictive Enrichment. Pediatr. Res. 2021. [Google Scholar] [CrossRef]
- Sweeney, T.E.; Perumal, T.M.; Henao, R.; Nichols, M.; Howrylak, J.A.; Choi, A.M.; Bermejo-Martin, J.F.; Almansa, R.; Tamayo, E.; Davenport, E.E.; et al. A Community Approach to Mortality Prediction in Sepsis via Gene Expression Analysis. Nat. Commun. 2018, 9, 694. [Google Scholar] [CrossRef] [Green Version]
- Castro, M.A.A.; de Santiago, I.; Campbell, T.M.; Vaughn, C.; Hickey, T.E.; Ross, E.; Tilley, W.D.; Markowetz, F.; Ponder, B.A.J.; Meyer, K.B. Regulators of Genetic Risk of Breast Cancer Identified by Integrative Network Analysis. Nat. Genet. 2015, 48, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, M.N.C.; Castro, M.A.A.; Wang, X.; de Santiago, I.; O’Reilly, M.; Chin, S.-F.; Rueda, O.M.; Caldas, C.; Ponder, B.A.J.; Markowetz, F.; et al. Master Regulators of FGFR2 Signalling and Breast Cancer Risk. Nat. Commun. 2013, 4, 2464. [Google Scholar] [CrossRef] [PubMed]
- Albanus, R.D.O.; Dalmolin, R.J.S.; Castro, M.A.A.; De Bittencourt Pasquali, M.A.; De Miranda Ramos, V.; Gelain, D.P.; Moreira, J.C.F. Reverse Engineering the Neuroblastoma Regulatory Network Uncovers Max as One of the Master Regulators of Tumor Progression. PLoS ONE 2013, 8, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro-Dantas, M.D.C.; Oliveira Imparato, D.O.; Dalmolin, M.G.S.; de Farias, C.B.; Brunetto, A.T.; da Cunha Jaeger, M.; Roesler, R.; Sinigaglia, M.; Siqueira Dalmolin, R.J. Reverse Engineering of Ewing Sarcoma Regulatory Network Uncovers PAX7 and RUNX3 as Master Regulators Associated with Good Prognosis. Cancers 2021, 13, 1860. [Google Scholar] [CrossRef] [PubMed]
- Sartor, I.T.S.; Zeidán-Chuliá, F.; Albanus, R.D.; Dalmolin, R.J.S.; Moreira, J.C.F. Computational Analyses Reveal a Prognostic Impact of TULP3 as a Transcriptional Master Regulator in Pancreatic Ductal Adenocarcinoma. Mol. BioSyst. 2014, 10, 1461–1468. [Google Scholar] [CrossRef] [PubMed]
- Robertson, A.G.; Kim, J.; Al-Ahmadie, H.; Bellmunt, J.; Guo, G.; Cherniack, A.D.; Hinoue, T.; Laird, P.W.; Hoadley, K.A.; Akbani, R.; et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 2018, 174, 1033. [Google Scholar] [CrossRef] [Green Version]
- Corces, M.R.; Granja, J.M.; Shams, S.; Louie, B.H.; Seoane, J.A.; Zhou, W.; Silva, T.C.; Groeneveld, C.; Wong, C.K.; Cho, S.W.; et al. The Chromatin Accessibility Landscape of Primary Human Cancers. Science 2018, 362, eaav1898. [Google Scholar] [CrossRef] [Green Version]
- Vargas, D.M.; De Bastiani, M.A.; Zimmer, E.R.; Klamt, F. Alzheimer’s Disease Master Regulators Analysis: Search for Potential Molecular Targets and Drug Repositioning Candidates. Alzheimers. Res. Ther. 2018, 10, 59. [Google Scholar] [CrossRef]
- Cazalis, M.; Lepape, A.; Venet, F.; Frager, F.; Mougin, B.; Vallin, H.; Paye, M.; Pachot, A.; Monneret, G. Early and Dynamic Changes in Gene Expression in Septic Shock Patients: A Genome-Wide Approach. Intensive Care Med. Exp. 2014, 2, 20. [Google Scholar] [CrossRef] [Green Version]
- Cvijanovich, N.; Shanley, T.P.; Lin, R.; Allen, G.L.; Thomas, N.J.; Checchia, P.; Anas, N.; Freishtat, R.J.; Monaco, M.; Odoms, K.; et al. Validating the Genomic Signature of Pediatric Septic Shock. Physiol. Genom. 2008, 34, 127–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, H.R.; Shanley, T.P.; Sakthivel, B.; Cvijanovich, N.; Lin, R.; Allen, G.L.; Thomas, N.J.; Doctor, A.; Kalyanaraman, M.; Tofil, N.M.; et al. Genome-Level Expression Profiles in Pediatric Septic Shock Indicate a Role for Altered Zinc Homeostasis in Poor Outcome. Physiol. Genom. 2007, 30, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.R.; Cvijanovich, N.Z.; Hall, M.; Allen, G.L.; Thomas, N.J.; Freishtat, R.J.; Anas, N.; Meyer, K.; Checchia, P.A.; Lin, R.; et al. Interleukin-27 Is a Novel Candidate Diagnostic Biomarker for Bacterial Infection in Critically Ill Children. Crit. Care 2012, 16, R213. [Google Scholar] [CrossRef] [Green Version]
- Sweeney, T.E.; Wong, H.R. Risk Stratification and Prognosis in Sepsis. Clin. Chest Med. 2016, 37, 209–218. [Google Scholar] [CrossRef] [Green Version]
- Margolin, A.A.; Wang, K.; Lim, W.K.; Kustagi, M.; Nemenman, I.; Califano, A. Reverse Engineering Cellular Networks. Nat. Protoc. 2006, 1, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for Functional Genomics Data Sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, H.R.; Cvijanovich, N.; Wheeler, D.S.; Bigham, M.T.; Monaco, M.; Odoms, K.; Macias, W.L.; Williams, M.D. Interleukin-8 as a Stratification Tool for Interventional Trials Involving Pediatric Septic Shock. Am. J. Respir. Crit. Care Med. 2008, 178, 276–282. [Google Scholar] [CrossRef]
- Margolin, A.; Nemenman, I.; Basso, K.; Wiggins, C.; Stolovitzky, G.; Dalla Favera, R.; Califano, A. ARACNE: An Algorithm for the Reconstruction of Gene Regulatory Networks in a Mammalian Cellular Context. BMC Bioinformatics 2006, 7 (Suppl. 1), S7. [Google Scholar] [CrossRef] [Green Version]
- Gautier, L.; Cope, L.; Bolstad, B.M.; Irizarry, R.A. Affy—Analysis of Affymetrix GeneChip Data at the Probe Level. Bioinformatics 2004, 20, 307–315. [Google Scholar] [CrossRef]
- Wynn, J.L.; Cvijanovich, N.Z.; Allen, G.L.; Thomas, N.J.; Freishtat, R.J.; Anas, N.; Meyer, K.; Checchia, P.A.; Lin, R.; Shanley, T.P.; et al. The Influence of Developmental Age on the Early Transcriptomic Response of Children with Septic Shock. Mol. Med. 2011, 17, 1146–1156. [Google Scholar] [CrossRef]
- Ye, H.; Zhang, J.; Wang, J.; Gao, Y.; Du, Y.; Li, C.; Deng, M.; Guo, J.; Li, Z. CD4 T-Cell Transcriptome Analysis Reveals Aberrant Regulation of STAT3 and Wnt Signaling Pathways in Rheumatoid Arthritis: Evidence from a Case–Control Study. Arthritis Res. Ther. 2015, 17, 76. [Google Scholar] [CrossRef] [Green Version]
- Kemppinen, A.K.; Kaprio, J.; Palotie, A.; Saarela, J. Systematic Review of Genome-Wide Expression Studies in Multiple Sclerosis. BMJ Open 2011, 1, e000053. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.A.A.; Wang, X.; Fletcher, M.N.C.; Meyer, K.B.; Markowetz, F. RedeR: R/Bioconductor Package for Representing Modular Structures, Nested Networks and Multiple Levels of Hierarchical Associations. Genome Biol. 2012, 13, R29. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. ClusterProfiler: An R Package for Comparing Biological Themes among Gene Clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Zhou, Y.; Liu, Z.; Zhang, P. Interaction between ANXA1 and GATA-3 in Immunosuppression of CD4+ T Cells. Mediators Inflamm. 2016, 2016, 1701059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prucha, M.; Bellingan, G.; Zazula, R. Sepsis Biomarkers. Clin. Chim. Acta 2015. [Google Scholar] [CrossRef]
- Chen, X.-H.; Yin, Y.-J.; Zhang, J.-X. Sepsis and Immune Response. World J. Emerg. Med. 2011, 2, 88–92. [Google Scholar] [CrossRef]
- Van Der Poll, T.; Van De Veerdonk, F.L.; Scicluna, B.P.; Netea, M.G. The Immunopathology of Sepsis and Potential Therapeutic Targets. Nat. Rev. Immunol. 2017, 17, 407–420. [Google Scholar] [CrossRef]
- Xue, M.; Xie, J.; Liu, L.; Huang, Y.; Guo, F.; Xu, J.; Yang, Y.; Qiu, H. Early and Dynamic Alterations of Th2/Th1 in Previously Immunocompetent Patients with Community-Acquired Severe Sepsis: A Prospective Observational Study. J. Transl. Med. 2019, 17, 57. [Google Scholar] [CrossRef]
- Takahashi, J.S. Transcriptional Architecture of the Mammalian Circadian Clock. Nat. Rev. Genet. 2017, 18, 164–179. [Google Scholar] [CrossRef] [Green Version]
- Truong, K.K.; Lam, M.T.; Grandner, M.A.; Sassoon, C.S.; Malhotra, A. Timing Matters: Circadian Rhythm in Sepsis, Obstructive Lung Disease, Obstructive Sleep Apnea, and Cancer. Ann. Am. Thorac. Soc. 2016, 13, 1144–1154. [Google Scholar] [CrossRef]
- Arriza, J.; Weinberger, C.; Cerelli, G.; Glaser, T.; Handelin, B.; Housman, D.; Evans, R. Cloning of Human Mineralocorticoid Receptor Complementary DNA: Structural and Functional Kinship with the Glucocorticoid Receptor. Science 1987, 237, 268–275. [Google Scholar] [CrossRef]
- Plieger, T.; Felten, A.; Splittgerber, H.; Duke, É.; Reuter, M. The Role of Genetic Variation in the Glucocorticoid Receptor (NR3C1) and Mineralocorticoid Receptor (NR3C2) in the Association between Cortisol Response and Cognition under Acute Stress. Psychoneuroendocrinology 2018, 87, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Hannibal, K.E.; Bishop, M.D. Chronic Stress, Cortisol Dysfunction, and Pain: A Psychoneuroendocrine Rationale for Stress Management in Pain Rehabilitation. Phys. Ther. 2014, 94, 1816–1825. [Google Scholar] [CrossRef]
- Henriquez-Camacho, C.; Losa, J. Biomarkers for Sepsis. Biomed. Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Ozato, K.; Shin, D.M.; Chang, T.H.; Morse, H.C. TRIM Family Proteins and Their Emerging Roles in Innate Immunity. Nat. Rev. Immunol. 2008, 8, 849–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gack, M.U.; Shin, Y.C.; Joo, C.H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; et al. TRIM25 RING-Finger E3 Ubiquitin Ligase Is Essential for RIG-I-Mediated Antiviral Activity. Nature 2007, 446, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E. Nuclear Factor–ΚB and Its Role in Sepsis-Associated Organ Failure. J. Infect. Dis. 2003, 187, S364–S369. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, J.; Li, H.; Wang, G.; Wang, Q.; Zhang, X.; Li, B.; Xu, H. LncRNA MIR155HG Accelerates the Progression of Sepsis via Upregulating MEF2A by Sponging MiR-194-5p. DNA Cell Biol. 2021, 40, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Pon, J.R.; Marra, M.A. MEF2 Transcription Factors: Developmental Regulators and Emerging Cancer Genes. Oncotarget 2016, 7, 2297–2312. [Google Scholar] [CrossRef] [Green Version]
- Clark, R.I.; Tan, S.W.S.; Péan, C.B.; Roostalu, U.; Vivancos, V.; Bronda, K.; Pilátová, M.; Fu, J.; Walker, D.W.; Berdeaux, R.; et al. XMEF2 Is an in Vivo Immune-Metabolic Switch. Cell 2013. [Google Scholar] [CrossRef] [Green Version]
- Kaneki, M. Metabolic Inflammatory Complex in Sepsis. SHOCK 2017, 48, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson, K.; Tjäder, I.; Keller, P.; Petrovic, N.; Ahlman, B.; Schéele, C.; Wernerman, J.; Timmons, J.A.; Rooyackers, O. Dysregulation of Mitochondrial Dynamics and the Muscle Transcriptome in ICU Patients Suffering from Sepsis Induced Multiple Organ Failure. PLoS ONE 2008, 3, e3686. [Google Scholar] [CrossRef]
- Van den Berghe, G.; Wilmer, A.; Hermans, G.; Meersseman, W.; Wouters, P.J.; Milants, I.; Van Wijngaerden, E.; Bobbaers, H.; Bouillon, R. Intensive Insulin Therapy in the Medical ICU. N. Engl. J. Med. 2006, 354, 449–461. [Google Scholar] [CrossRef] [Green Version]
- Iwama, A.; Pan, J.; Zhang, P.; Reith, W.; Mach, B.; Tenen, D.G.; Sun, Z. Dimeric RFX Proteins Contribute to the Activity and Lineage Specificity of the Interleukin-5 Receptor α Promoter through Activation and Repression Domains. Mol. Cell. Biol. 1999, 19, 3940–3950. [Google Scholar] [CrossRef] [Green Version]
- Linch, S.N.; Danielson, E.T.; Kelly, A.M.; Tamakawa, R.A.; Lee, J.J.; Gold, J.A. Interleukin 5 Is Protective during Sepsis in an Eosinophil-Independent Manner. Am. J. Respir. Crit. Care Med. 2012, 186, 246–254. [Google Scholar] [CrossRef] [Green Version]
- Carr, T.M.; Wheaton, J.D.; Houtz, G.M.; Ciofani, M. JunB Promotes Th17 Cell Identity and Restrains Alternative CD4+ T-Cell Programs during Inflammation. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.-N.; Li, D.; Xia, J.; Wu, Q.-J.; Wen, R.; Yang, N.; Liu, C.-F. Non-Coding RNA: A Potential Biomarker and Therapeutic Target for Sepsis. Oncotarget 2017, 8, 91765–91778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.; Leung, K.-S.; Wong, M.-H.; Cheng, L. Long Non-Coding RNA Pairs to Assist in Diagnosing Sepsis. BMC Genom. 2021, 22, 275. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliveira, R.A.d.C.; Imparato, D.O.; Fernandes, V.G.S.; Cavalcante, J.V.F.; Albanus, R.D.; Dalmolin, R.J.S. Reverse Engineering of the Pediatric Sepsis Regulatory Network and Identification of Master Regulators. Biomedicines 2021, 9, 1297. https://doi.org/10.3390/biomedicines9101297
Oliveira RAdC, Imparato DO, Fernandes VGS, Cavalcante JVF, Albanus RD, Dalmolin RJS. Reverse Engineering of the Pediatric Sepsis Regulatory Network and Identification of Master Regulators. Biomedicines. 2021; 9(10):1297. https://doi.org/10.3390/biomedicines9101297
Chicago/Turabian StyleOliveira, Raffael Azevedo de Carvalho, Danilo Oliveira Imparato, Vítor Gabriel Saldanha Fernandes, João Vitor Ferreira Cavalcante, Ricardo D’Oliveira Albanus, and Rodrigo Juliani Siqueira Dalmolin. 2021. "Reverse Engineering of the Pediatric Sepsis Regulatory Network and Identification of Master Regulators" Biomedicines 9, no. 10: 1297. https://doi.org/10.3390/biomedicines9101297
APA StyleOliveira, R. A. d. C., Imparato, D. O., Fernandes, V. G. S., Cavalcante, J. V. F., Albanus, R. D., & Dalmolin, R. J. S. (2021). Reverse Engineering of the Pediatric Sepsis Regulatory Network and Identification of Master Regulators. Biomedicines, 9(10), 1297. https://doi.org/10.3390/biomedicines9101297