
Platelet-Derived Growth Factor Induces SASP-Associated Gene Expression in Human Multipotent Mesenchymal Stromal Cells but Does Not Promote Cell Senescence

 , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
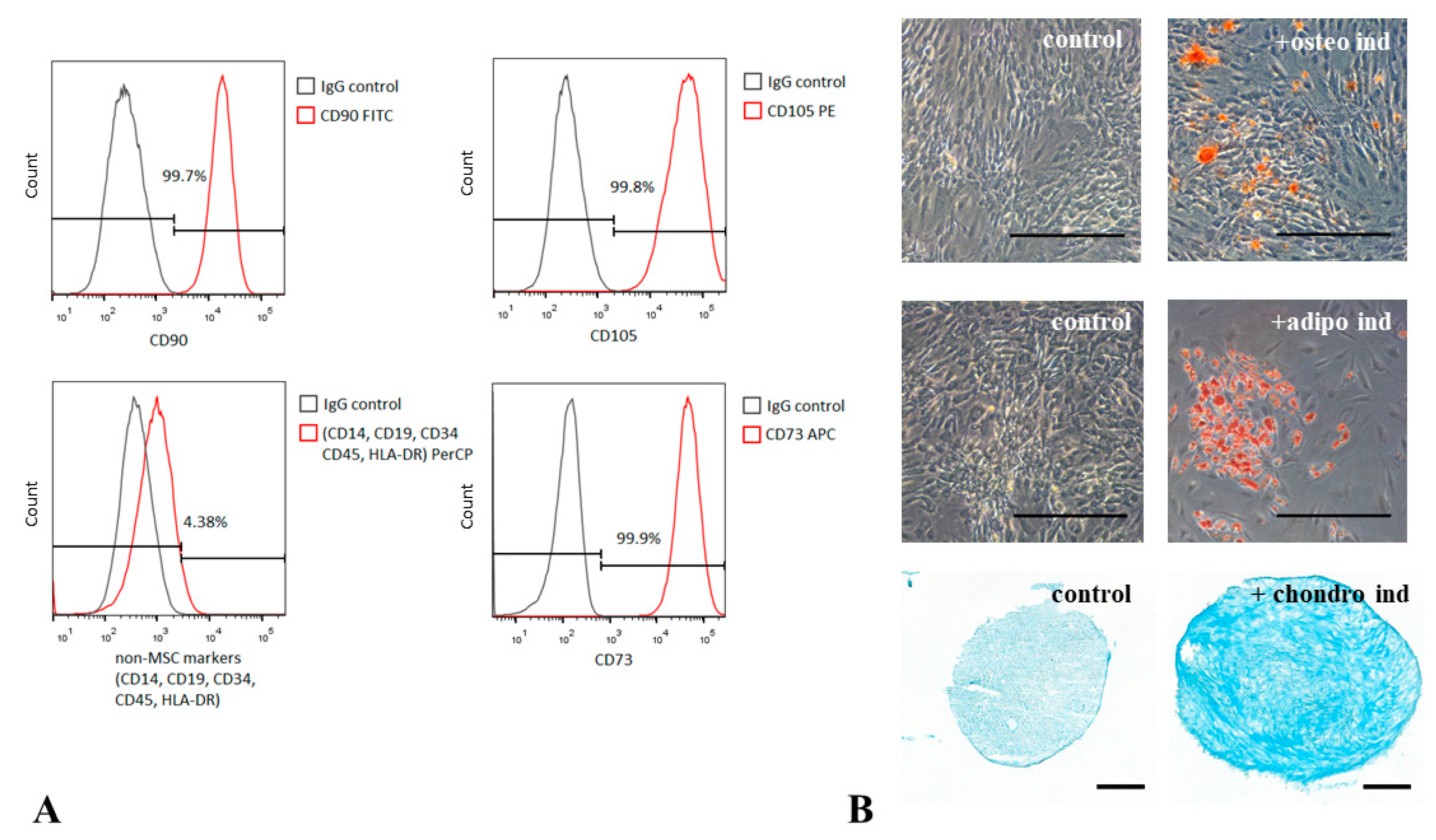
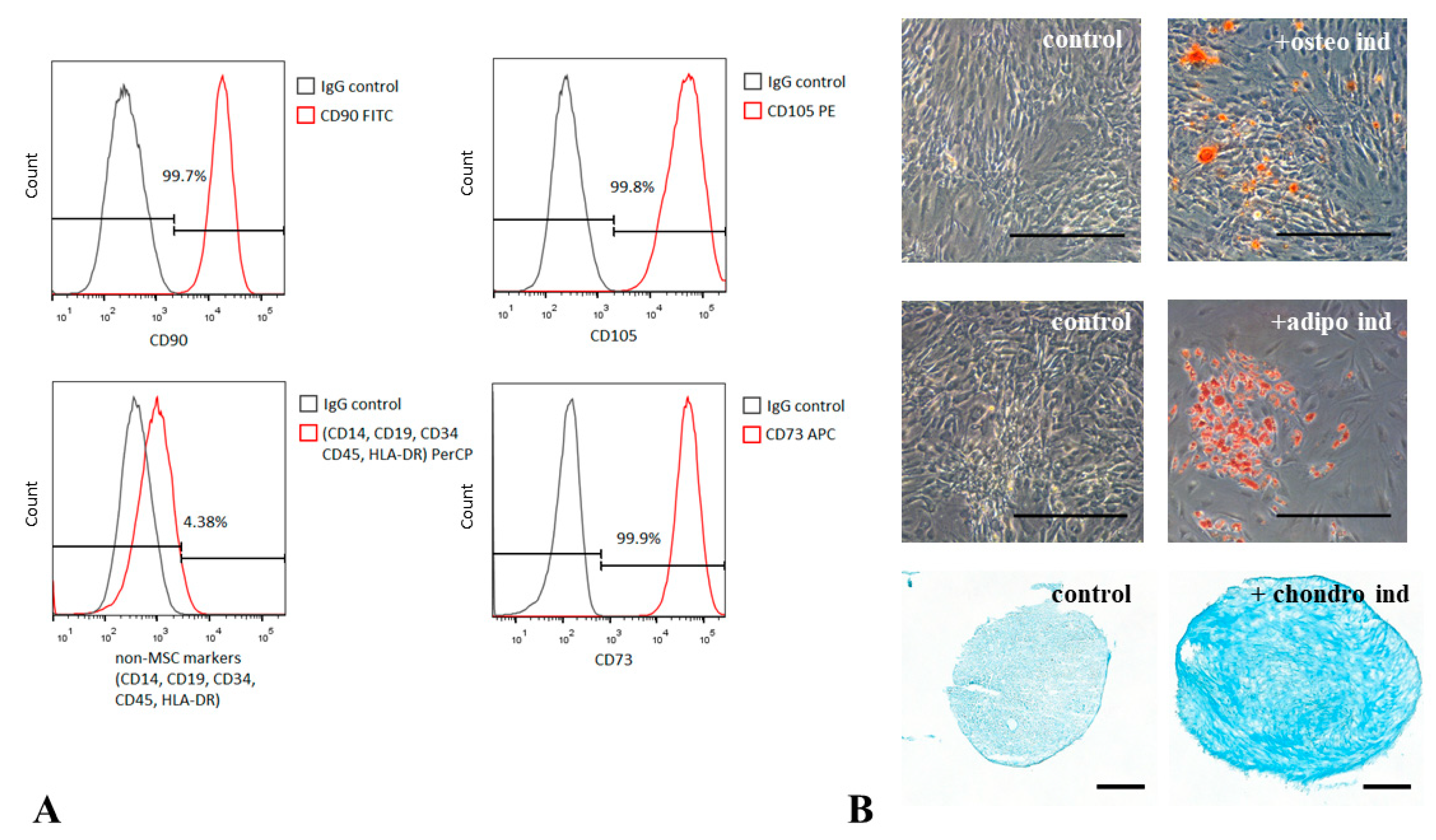
2.1. Human MSC Isolation and Culture
2.2. MSC Immunophenotyping
2.3. MSC Differentiation Assays
2.4. HUVEC Isolation and Culture
2.5. RNA Isolation and Transcriptome Analysis
2.6. Bead Array Hybridization
2.7. Data Preprocessing
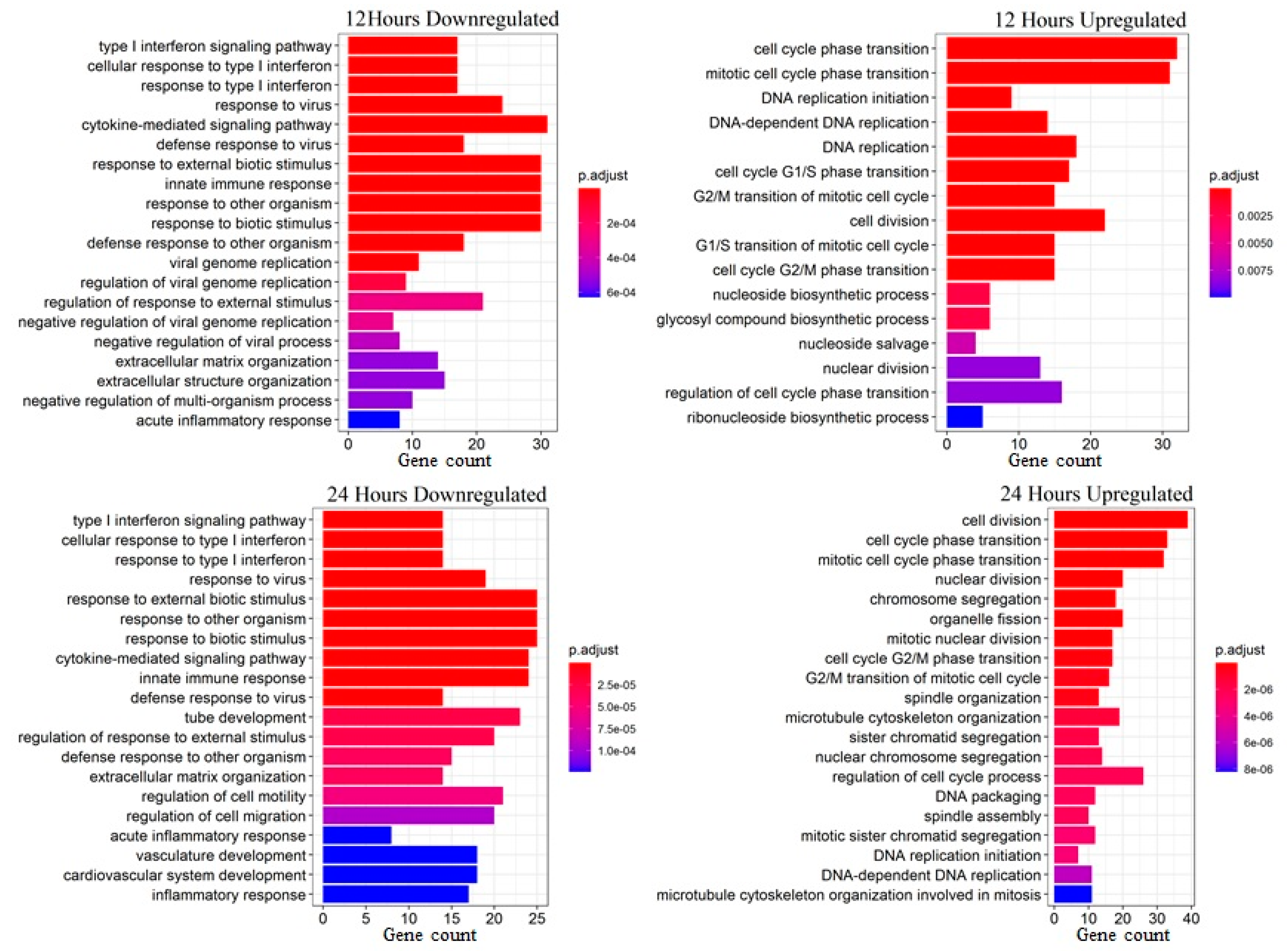
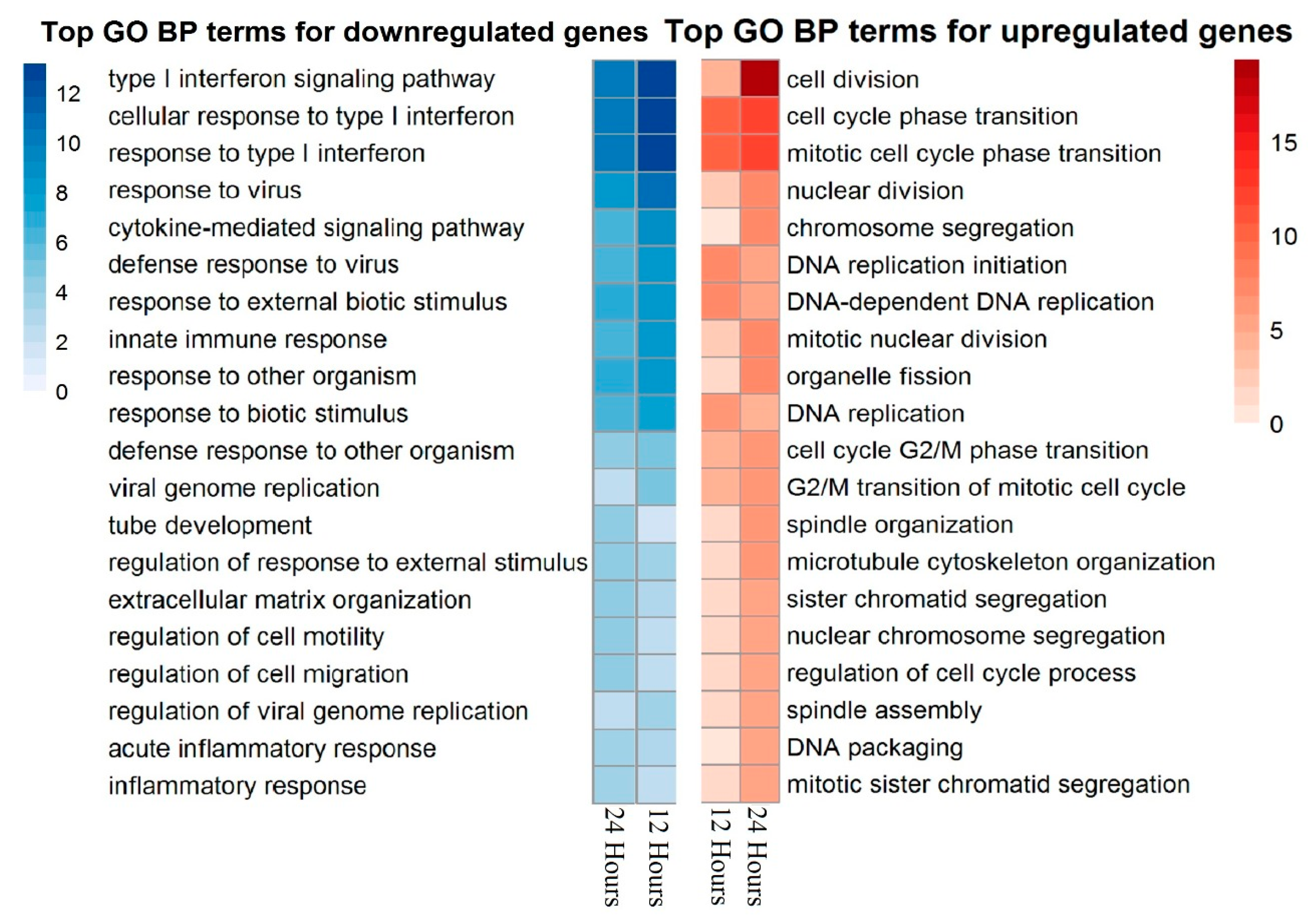
2.8. Enrichment Analysis
2.9. Data Records
2.10. Immunohistochemistry
2.11. β-Galactosidase Staining
2.12. ELISA Assay
2.13. Migration Assay
2.14. Statistical Analysis
3. Results
3.1. PDGF-BB Upregulates Genes Associated with Proliferation and Cell Cycle and Downregulates Immune-Response-Related Pathways in MSCs
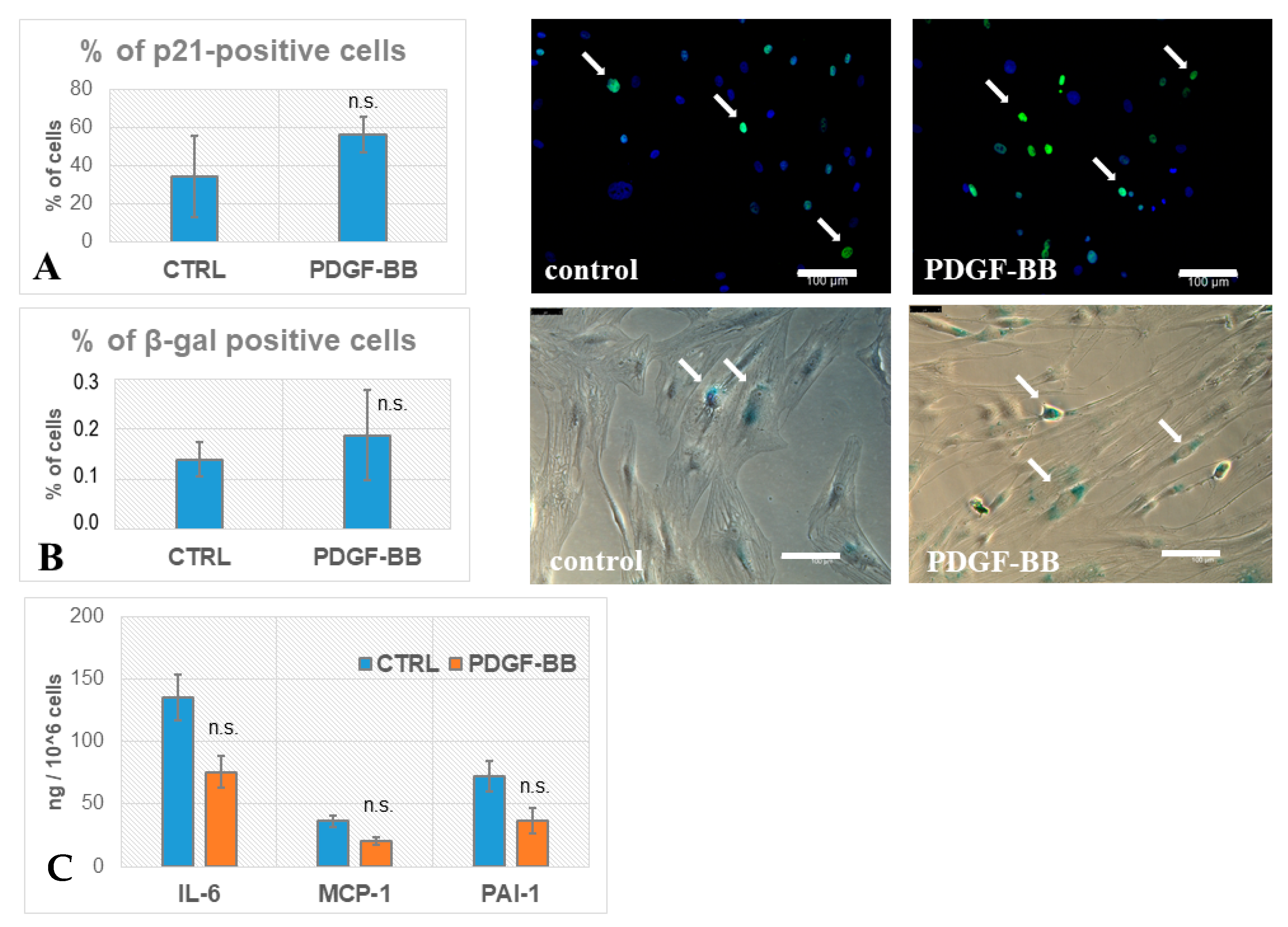
3.2. PDGF-BB-Treated MSCs Did Not Acquire Senescence Biomarkers
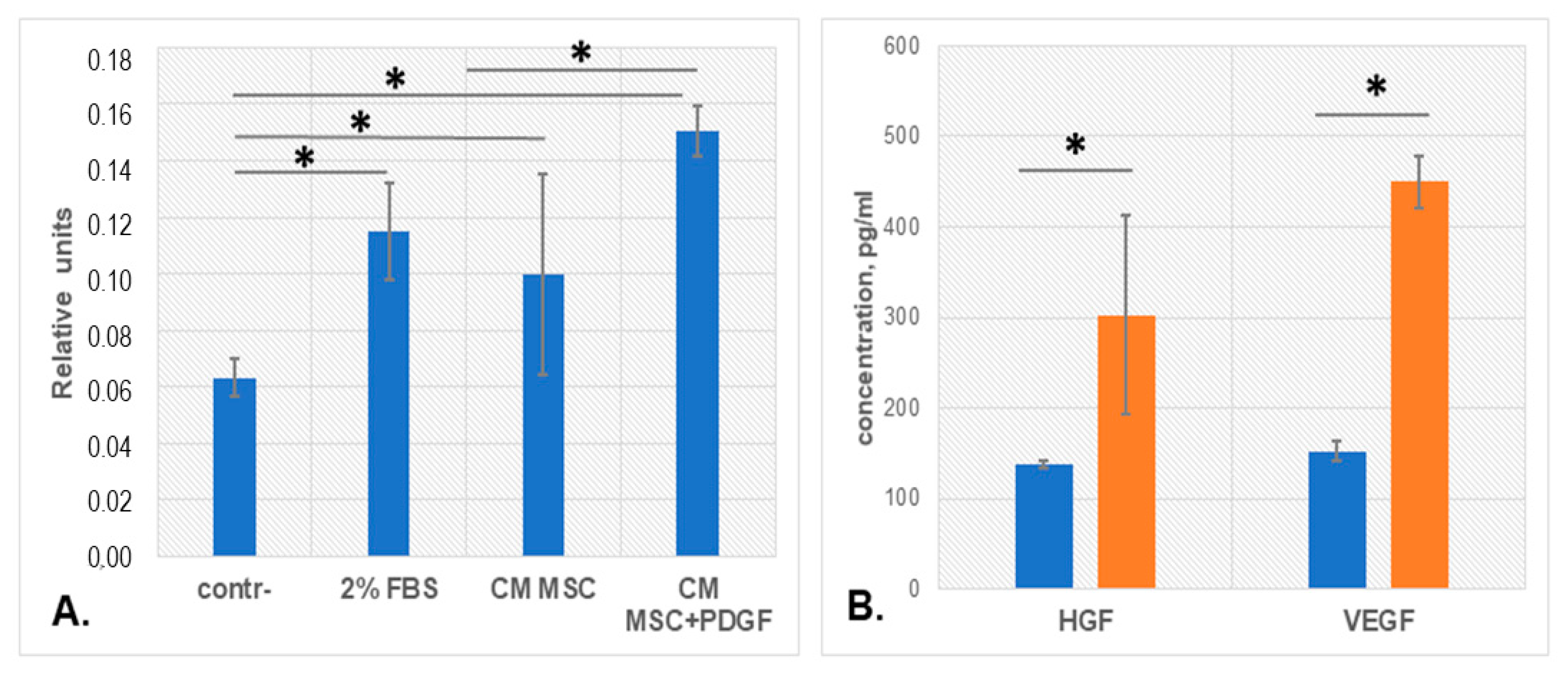
3.3. PDGF-BB Stimulated MSC Pro-Angiogenic Activity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Neri, S.; Borzì, R.M. Molecular Mechanisms Contributing to Mesenchymal Stromal Cell Aging. Biomolecules 2020, 10, 340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalinina, N.; Kharlampieva, D.; Loguinova, M.; Butenko, I.; Pobeguts, O.; Efimenko, A.; Ageeva, L.; Sharonov, G.; Ischenko, D.; Alekseev, D.; et al. Characterization of secretomes provides evidence for adipose-derived mesenchymal stromal cells subtypes. Stem Cell Res. Ther. 2020, 6, 221. [Google Scholar] [CrossRef] [Green Version]
- Pittenger, M.F.; Discher, D.E.; Péault, B.M.; Phinney, D.G.; Hare, J.M.; Caplan, A.I. Mesenchymal stem cell perspective: Cell biology to clinical progress. NPJ Regen. Med. 2019, 4, 22. [Google Scholar] [CrossRef] [Green Version]
- Sagaradze, G.; Basalova, N.; Kirpatovsky, V.; Ohobotov, D.; Nimiritsky, P.; Grigorieva, O.; Popov, V.; Kamalov, A.; Tkachuk, V.; Efimenko, A. A magic kick for regeneration: Role of mesenchymal stromal cell secretome in spermatogonial stem cell niche recovery. Stem Cell Res. Ther. 2019, 10, 342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basalova, N.; Sagaradze, G.; Arbatskiy, M.; Evtushenko, E.; Kulebyakin, K.; Grigorieva, O.; Akopyan, Z.; Kalinina, N.; Efimenko, A. Secretome of Mesenchymal Stromal Cells Prevents Myofibroblasts Differentiation by Transferring Fibrosis-Associated microRNAs within Extracellular Vesicles. Cells 2020, 9, 1272. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- De Donatis, A.; Comito, G.; Buricchi, F.; Vinci, M.C.; Parenti, A.; Caselli, A.; Camici, G.; Manao, G.; Ramponi, G.; Cirri, P. Proliferation versus migration in platelet-derived growth factor signaling: The key role of endocytosis. J. Biol. Chem. 2008, 283, 19948–19956. [Google Scholar] [CrossRef] [Green Version]
- Fafián-Labora, J.A.; O’Loghlen, A. Classical and Nonclassical Intercellular Communication in Senescence and Ageing. Trends Cell Biol. 2020, 30, 628–639. [Google Scholar] [CrossRef]
- Heidaran, M.A.; Pierce, J.H.; Yu, J.C.; Lombardi, D.; Artrip, J.E.; Fleming, T.P.; Thomason, A.; Aaronson, S.A. Role of alpha beta receptor heterodimer formation in beta platelet-derived growth factor (PDGF) receptor activation by PDGF-AB. J. Biol. Chem. 1991, 266, 20232–20237. [Google Scholar] [CrossRef]
- Cheng, J.; Ye, H.; Liu, Z.; Xu, C.; Zhang, Z.; Liu, Y.; Sun, Y. Platelet-derived growth factor-BB accelerates prostate cancer growth by promoting the proliferation of mesenchymal stem cells. J. Cell. Biochem. 2013, 114, 1510–1518. [Google Scholar] [CrossRef]
- Klinkhammer, B.M.; Floege, J.; Boor, P. PDGF in organ fibrosis. Mol. Asp. Med. 2018, 62, 44–62. [Google Scholar] [CrossRef]
- Zuk, P.A.; Zhu, M.; Mizuno, H.; Huang, J.; Futrell, J.W.; Katz, A.J.; Benhaim, P.; Lorenz, H.P.; Hedrick, M.H. Multilineage cells from human adipose tissue: Implications for cell-based therapies. Tissue Eng. 2001, 7, 211–228. [Google Scholar] [CrossRef] [Green Version]
- Kuhbier, J.W.; Weyand, B.; Radtke, C.; Vogt, P.M.; Kasper, C.; Reimers, K. Isolation, characterization, differentiation, and application of adipose-derived stem cells. Adv. Biochem. Eng. Biotechnol. 2010, 123, 55–105. [Google Scholar] [CrossRef]
- Eremichev, R.; Kulebyakina, M.; Alexandrushkina, N.; Nimiritsky, P.; Basalova, N.; Grigorieva, O.; Egiazaryan, M.; Dyikanov, D.; Tkachuk, V.; Makarevich, P. Scar-Free Healing of Endometrium: Tissue-Specific Program of Stromal Cells and Its Induction by Soluble Factors Produced After Damage. Front. Cell Dev. Biol. 2021, 9, 616893. [Google Scholar] [CrossRef]
- Lin, S.M.; Du, P.; Huber, W.; Kibbe, W.A. Model-based variance-stabilizing transformation for Illumina microarray data. Nucleic Acids Res. 2008, 36, gkm1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, P.; Kibbe, W.A.; Lin, S.M. lumi: A pipeline for processing Illumina microarray. Bioinformatics 2008, 24, 1547–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falcon, S.; Gentleman, R. Using GOstats to test gene lists for GO term association. Bioinformatics 2007, 23, 257–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consortium, G.O. The Gene Ontology (GO) project in 2006. Nucl. Acids Res. 2006, 34 (Suppl. 1), D322–D326. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [Green Version]
- Macip, S.; Igarashi, M.; Fang, L.; Chen, A.; Pan, Z.Q.; Lee, S.W.; Aaronson, S.A. Inhibition of p21-mediated ROS accumulation can rescue p21-induced senescence. EMBO J. 2002, 21, 2180–2188. [Google Scholar] [CrossRef] [PubMed]
- Lopatina, T.; Favaro, E.; Grange, C.; Cedrino, M.; Ranghino, A.; Occhipinti, S.; Fallo, S.; Buffolo, F.; Gaykalova, D.A.; Zanone, M.M.; et al. PDGF enhances the protective effect of adipose stem cell-derived extracellular vesicles in a model of acute hindlimb ischemia. Sci. Rep. 2018, 8, 17458. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Ouyang, G.; Bao, S. The activation of Akt/PKB signaling pathway and cell survival. J. Cell. Mol. Med. 2005, 9, 59–71. [Google Scholar] [CrossRef]
- Pierce, G.F.; Mustoe, T.A.; Altrock, B.W.; Deuel, T.F.; Thomason, A. Role of platelet-derived growth factor in wound healing. J. Cell. Biochem. 1991, 45, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, D.E.; Rai, R.; Khan, S.S.; Eren, M.; Ghosh, A.K. Plasminogen Activator Inhibitor-1 Is a Marker and a Mediator of Senescence. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1446–1452. [Google Scholar] [CrossRef] [Green Version]
- Efimenko, A.; Dzhoyashvili, N.A.; Kalinina, N.I.; Kochegura, T.N.; Akchurin, R.S.; Tkachuk, V.A.; Parfyonova, Y.V. Adipose-derived stromal cells (ADSC) from aged patients with coronary artery disease keep MSC properties but exhibit age markers and have an impaired angiogenic potential. Stem Cells Transl. Med. 2014, 3, 32–41. [Google Scholar] [CrossRef]
- Amor, C.; Feucht, J.; Leibold, J.; Ho, Y.J.; Zhu, C.; Alonso-Curbelo, D.; Mansilla-Soto, J.; Boyer, J.A.; Li, X.; Giavridis, T.; et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature 2020, 583, 127–132. [Google Scholar] [CrossRef]
- Pavón, M.A.; Arroyo-Solera, I.; Céspedes, M.V.; Casanova, I.; León, X.; Mangues, R. uPA/uPAR and SERPINE1 in head and neck cancer: Role in tumor resistance, metastasis, prognosis and therapy. Oncotarget 2016, 7, 57351–57366. [Google Scholar] [CrossRef] [Green Version]
- Veevers-Lowe, J.; Ball, S.G.; Shuttleworth, A.; Kielty, C.M. Mesenchymal stem cell migration is regulated by fibronectin through α5β1-integrin-mediated activation of PDGFR-β and potentiation of growth factor signals. J. Cell Sci. 2011, 24 Pt 8, 1288–1300. [Google Scholar] [CrossRef] [Green Version]
- Guillon, J.; Petit, C.; Moreau, M.; Toutain, B.; Henry, C.; Roché, H.; Bonichon-Lamichhane, N.; Salmon, J.P.; Lemonnier, J.; Campone, M.; et al. Regulation of senescence escape by TSP1 and CD47 following chemotherapy treatment. Cell Death Dis. 2019, 10, 199. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grigorieva, O.; Arbatskiy, M.; Novoseletskaya, E.; Dyachkova, U.; Ishkin, A.; Kalinina, N.; Efimenko, A. Platelet-Derived Growth Factor Induces SASP-Associated Gene Expression in Human Multipotent Mesenchymal Stromal Cells but Does Not Promote Cell Senescence. Biomedicines 2021, 9, 1290. https://doi.org/10.3390/biomedicines9101290
Grigorieva O, Arbatskiy M, Novoseletskaya E, Dyachkova U, Ishkin A, Kalinina N, Efimenko A. Platelet-Derived Growth Factor Induces SASP-Associated Gene Expression in Human Multipotent Mesenchymal Stromal Cells but Does Not Promote Cell Senescence. Biomedicines. 2021; 9(10):1290. https://doi.org/10.3390/biomedicines9101290
Chicago/Turabian StyleGrigorieva, Olga, Mikhail Arbatskiy, Ekaterina Novoseletskaya, Uliana Dyachkova, Alexander Ishkin, Natalia Kalinina, and Anastasia Efimenko. 2021. "Platelet-Derived Growth Factor Induces SASP-Associated Gene Expression in Human Multipotent Mesenchymal Stromal Cells but Does Not Promote Cell Senescence" Biomedicines 9, no. 10: 1290. https://doi.org/10.3390/biomedicines9101290
APA StyleGrigorieva, O., Arbatskiy, M., Novoseletskaya, E., Dyachkova, U., Ishkin, A., Kalinina, N., & Efimenko, A. (2021). Platelet-Derived Growth Factor Induces SASP-Associated Gene Expression in Human Multipotent Mesenchymal Stromal Cells but Does Not Promote Cell Senescence. Biomedicines, 9(10), 1290. https://doi.org/10.3390/biomedicines9101290