Modulatory Effects of Autophagy on APP Processing as a Potential Treatment Target for Alzheimer’s Disease

 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  , ,
, ,  and
and
Abstract
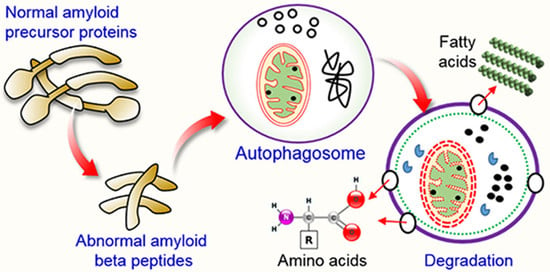
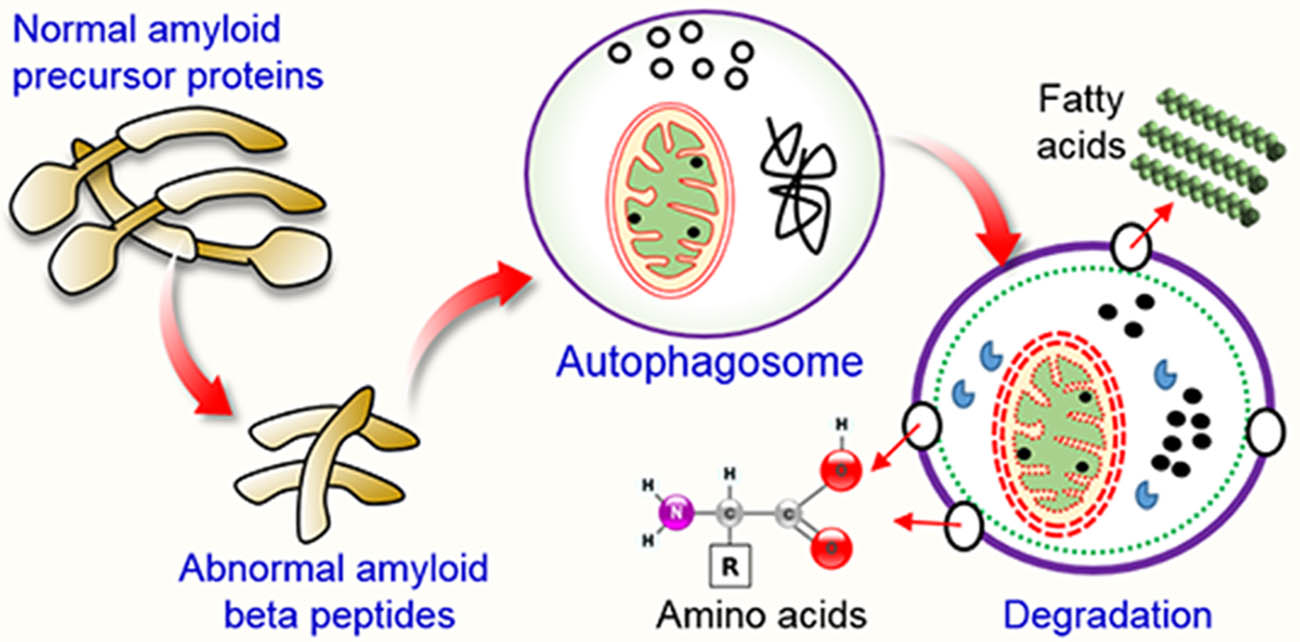
1. Introduction
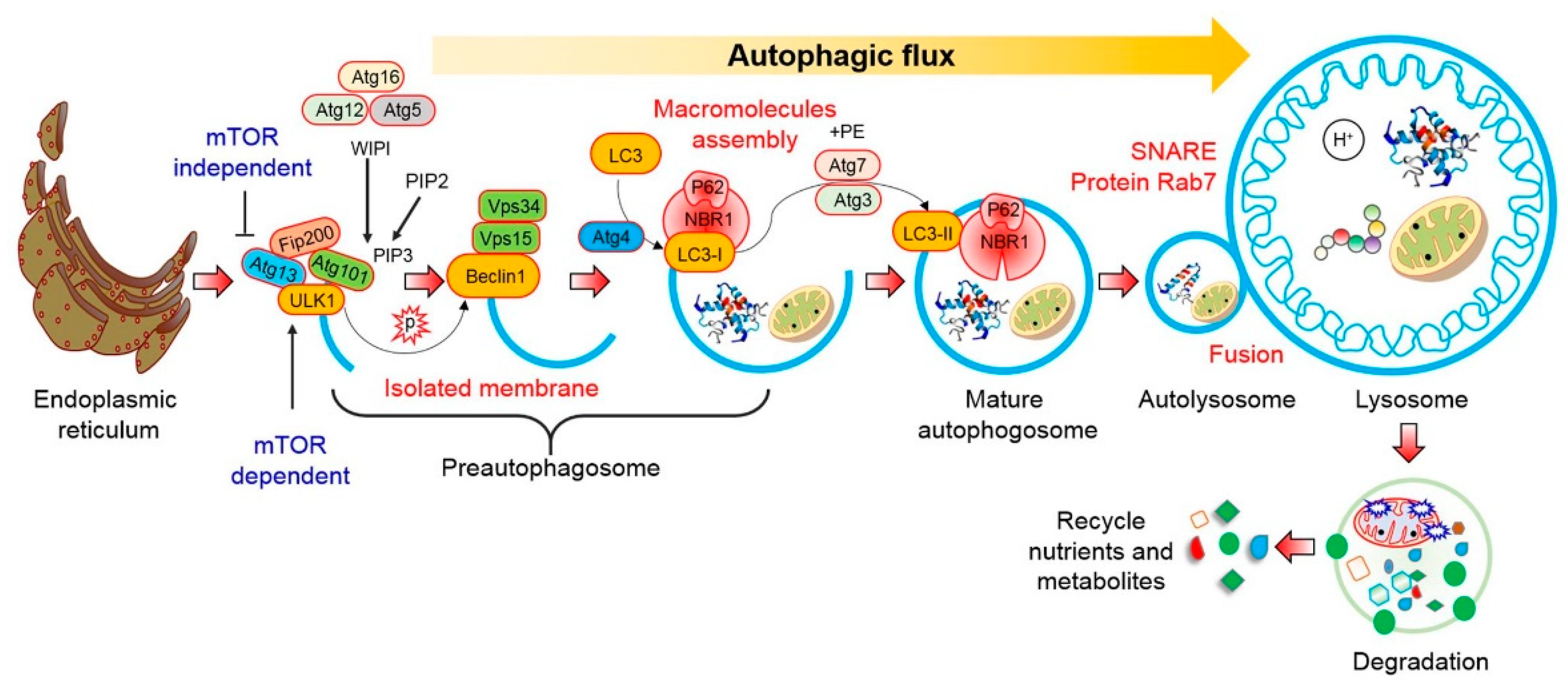
2. Autophagy Pathway
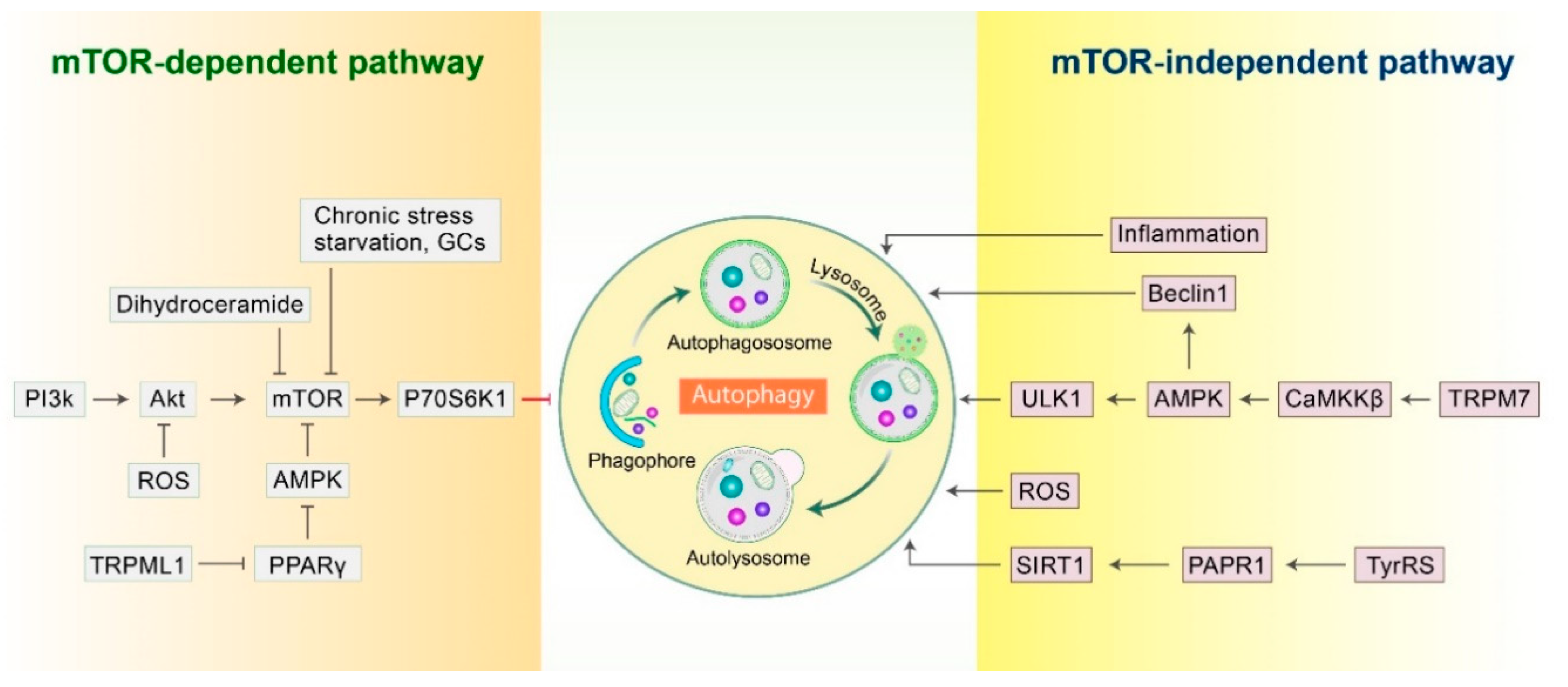
2.1. mTOR-Dependent Autophagy Pathway
2.2. mTOR-Independent Autophagy Pathway
3. Neuronal Roles of APP
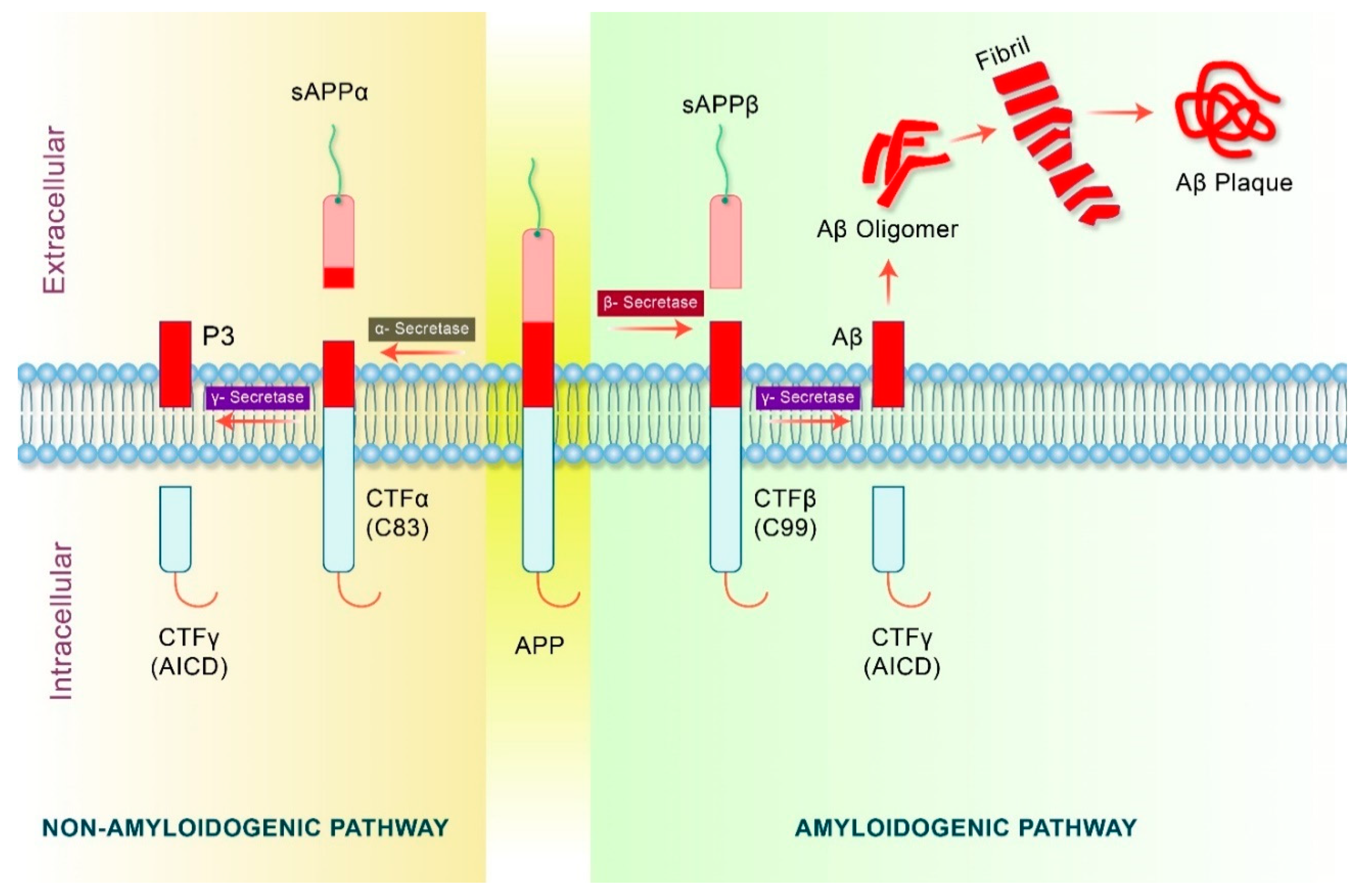
4. Proteolytic Processing of APP in Alzheimer’s Disease
5. Amyloid Precursor Protein (APP) Processing in Autophagy Pathway
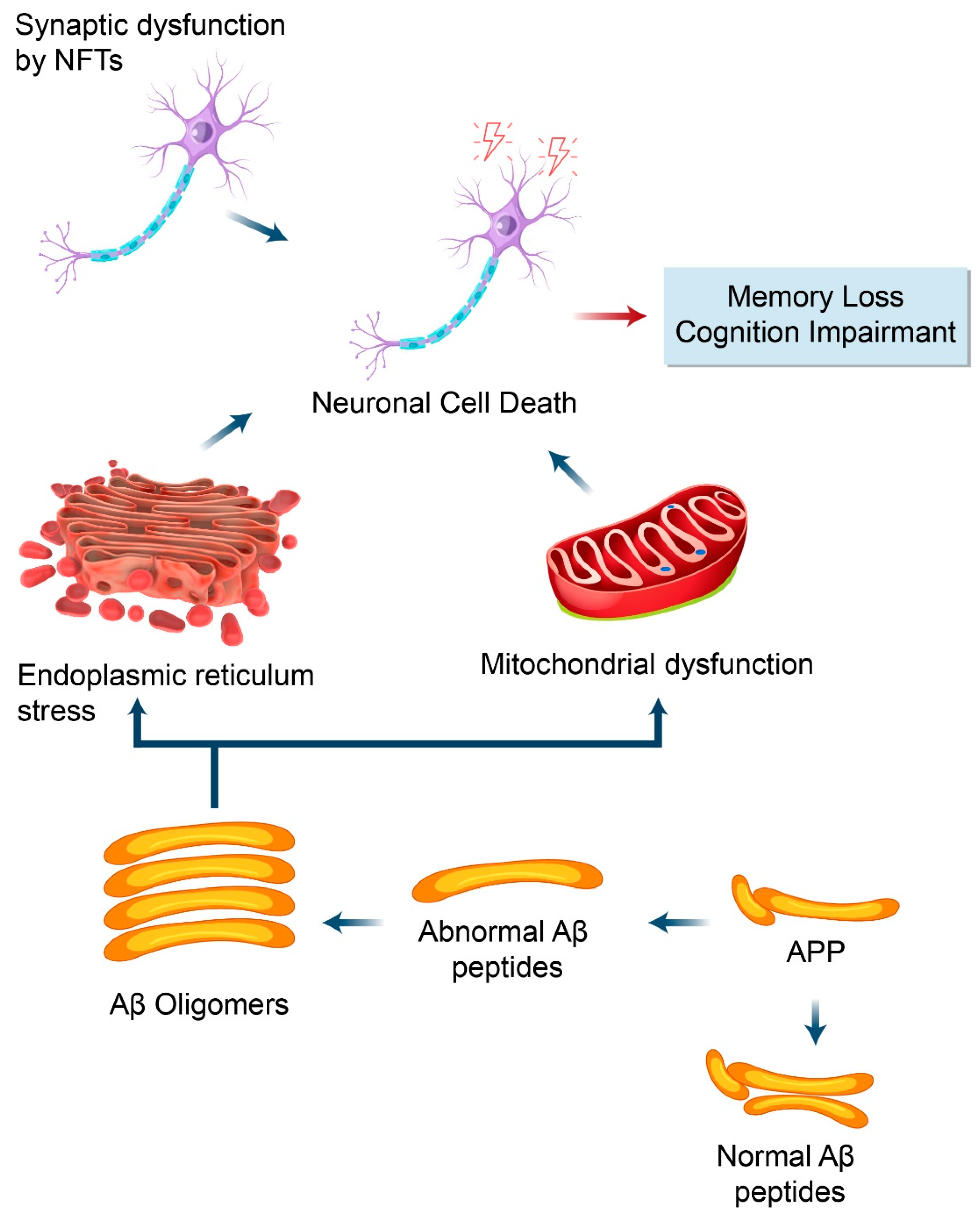
5.1. Autophagy and Aβ Processing
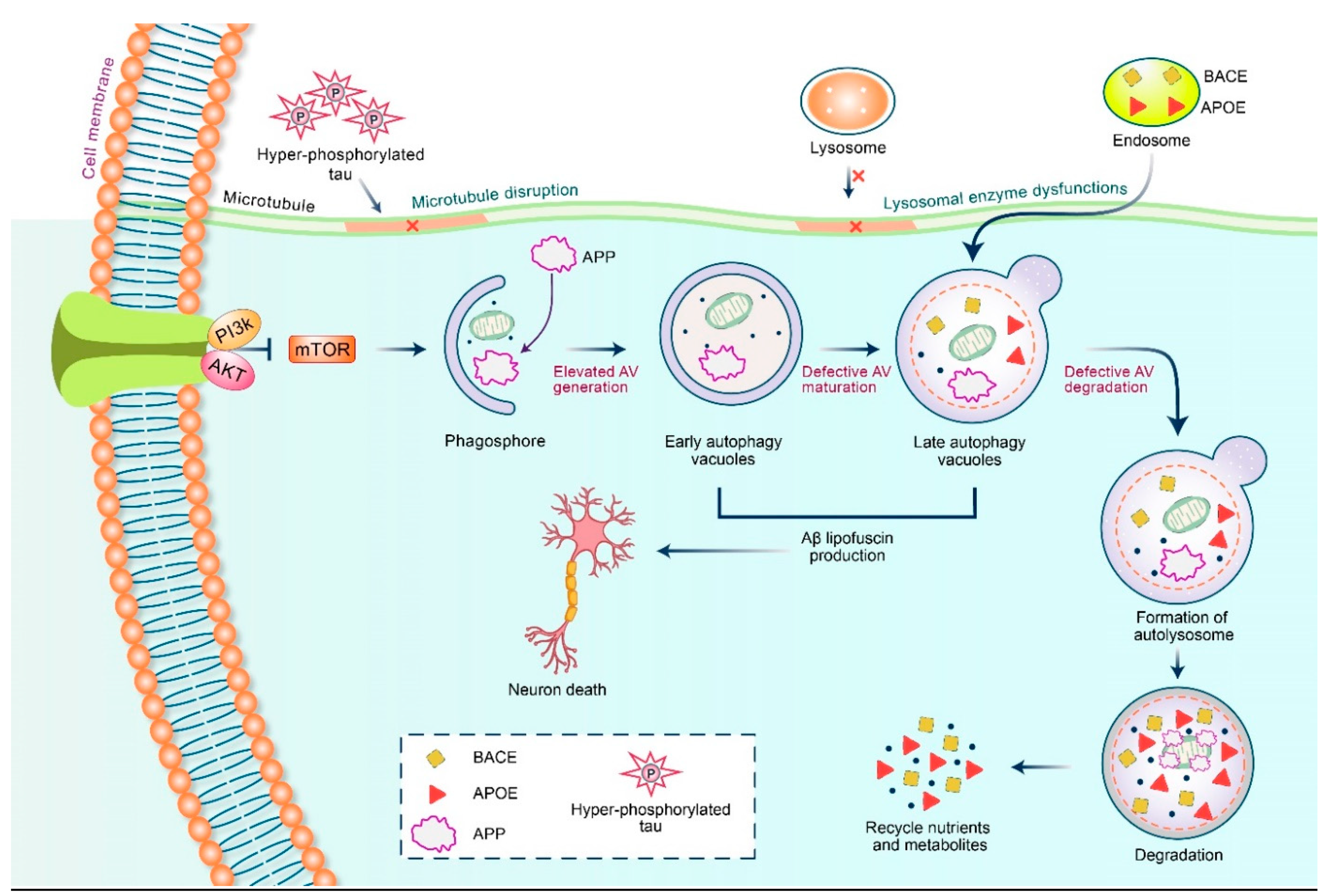
5.2. Dysfunctional Autophagy and Aβ Processing
6. Therapeutic Action of APP Triggered by Autophagy
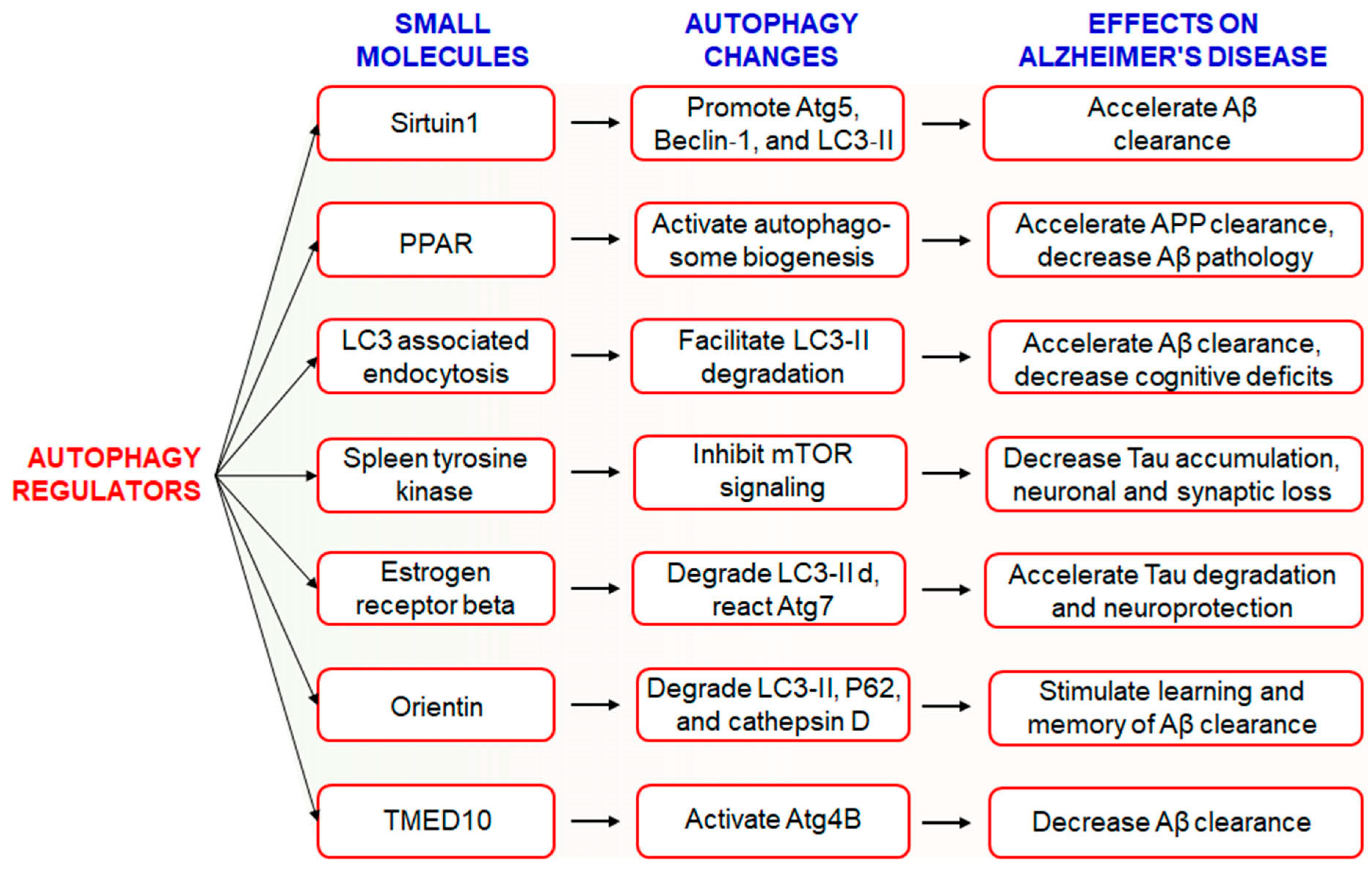
6.1. Use of Small Molecules to Modulate Autophagy in AD
6.2. Use of Natural Compounds to Modulate Autophagy in AD
6.3. Use of FDA-Approved Drugs to Modulate Autophagy in AD
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Uddin, M.S.; Al Mamun, A.; Rahman, M.A.; Behl, T.; Perveen, A.; Hafeez, A.; Bin-Jumah, M.N.; Abdel-Daim, M.M.; Ashraf, G.M. Emerging proof of protein misfolding and interaction in multifactorial alzheimer’s disease. Curr. Top. Med. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ballard, C.; Gauthier, S.; Corbett, A.; Brayne, C.; Aarsland, D.; Jones, E. Alzheimer’s disease. Lancet 2011, 377, 1019–1031. [Google Scholar] [CrossRef]
- Ghai, R.; Nagarajan, K.; Arora, M.; Grover, P.; Ali, N.; Kapoor, G. Current strategies and novel drug approaches for alzheimer disease. CNS Neurol Disord. Drug Targets 2020. [Google Scholar] [CrossRef]
- Bekris, L.M.; Yu, C.E.; Bird, T.D.; Tsuang, D.W. Genetics of alzheimer disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 213–227. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and alzheimer’s disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Thompson, R.; Zhang, H.; Xu, H. APP processing in alzheimer’s disease. Mol. Brain 2011, 4, 3. [Google Scholar] [CrossRef]
- De-Paula, V.J.; Radanovic, M.; Diniz, B.S.; Forlenza, O.V. Alzheimer’s disease. Subcell Biochem. 2012, 65, 329–352. [Google Scholar]
- Greenfield, J.P.; Tsai, J.; Gouras, G.K.; Hai, B.; Thinakaran, G.; Checler, F.; Sisodia, S.S.; Greengard, P.; Xu, H. Endoplasmic reticulum and trans-golgi network generate distinct populations of alzheimer beta-amyloid peptides. Proc. Natl. Acad. Sci. USA 1999, 96, 742–747. [Google Scholar] [CrossRef]
- Manczak, M.; Kandimalla, R.; Yin, X.; Reddy, P.H. Hippocampal mutant app and amyloid beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 1332–1342. [Google Scholar] [CrossRef]
- Sun, B.L.; Li, W.W.; Zhu, C.; Jin, W.S.; Zeng, F.; Liu, Y.H.; Bu, X.L.; Zhu, J.; Yao, X.Q.; Wang, Y.J. Clinical research on alzheimer’s disease: Progress and perspectives. Neurosci. Bull. 2018, 34, 1111–1118. [Google Scholar] [CrossRef]
- Van Giau, V.; Pyun, J.M.; Suh, J.; Bagyinszky, E.; An, S.S.A.; Kim, S.Y. A pathogenic psen1 trp165cys mutation associated with early-onset alzheimer’s disease. BMC Neurol. 2019, 19, 188. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Browne, A.; Kim, D.Y.; Tanzi, R.E. Familial alzheimer’s disease mutations in presenilin 1 do not alter levels of the secreted amyloid-protein precursor generated by beta-secretase cleavage. Curr. Alzheimer Res. 2010, 7, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, R.P.; Tepper, K.; Ronicke, R.; Soom, M.; Westermann, M.; Reymann, K.; Kaether, C.; Fandrich, M. Mechanism of amyloid plaque formation suggests an intracellular basis of abeta pathogenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 1942–1947. [Google Scholar] [CrossRef] [PubMed]
- Ercan-Herbst, E.; Ehrig, J.; Schondorf, D.C.; Behrendt, A.; Klaus, B.; Gomez Ramos, B.; Prat Oriol, N.; Weber, C.; Ehrnhoefer, D.E. A post-translational modification signature defines changes in soluble tau correlating with oligomerization in early stage alzheimer’s disease brain. Acta Neuropathol. Commun. 2019, 7, 192. [Google Scholar] [CrossRef]
- Mietelska-Porowska, A.; Wasik, U.; Goras, M.; Filipek, A.; Niewiadomska, G. Tau protein modifications and interactions: Their role in function and dysfunction. Int. J. Mol. Sci. 2014, 15, 4671–4713. [Google Scholar] [CrossRef]
- Rajmohan, R.; Reddy, P.H. Amyloid-beta and phosphorylated tau accumulations cause abnormalities at synapses of alzheimer’s disease neurons. J. Alzheimers Dis. 2017, 57, 975–999. [Google Scholar] [CrossRef]
- Rahman, M.A.; Rhim, H. Therapeutic implication of autophagy in neurodegenerative diseases. BMB Rep. 2017, 50, 345–354. [Google Scholar] [CrossRef]
- Schmukler, E.; Pinkas-Kramarski, R. Autophagy induction in the treatment of alzheimer’s disease. Drug Dev. Res. 2019. [Google Scholar] [CrossRef]
- Rasheduzzaman, M.; Yin, H.; Park, S.Y. Cardiac glycoside sensitized hepatocellular carcinoma cells to trail via ros generation, p38mapk, mitochondrial transition, and autophagy mediation. Mol. Carcinog. 2019, 58, 2040–2051. [Google Scholar] [CrossRef]
- Ryter, S.W.; Bhatia, D.; Choi, M.E. Autophagy: A lysosome-dependent process with implications in cellular redox homeostasis and human disease. Antioxid. Redox. Signal 2019, 30, 138–159. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Mamun, A.A.; Labu, Z.K.; Hidalgo-Lanussa, O.; Barreto, G.E.; Ashraf, G.M. Autophagic dysfunction in alzheimer’s disease: Cellular and molecular mechanistic approaches to halt alzheimer’s pathogenesis. J. Cell Physiol. 2019, 234, 8094–8112. [Google Scholar] [CrossRef] [PubMed]
- Mputhia, Z.; Hone, E.; Tripathi, T.; Sargeant, T.; Martins, R.; Bharadwaj, P. Autophagy modulation as a treatment of amyloid diseases. Molecules 2019, 24, 3372. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Liang, Y.; Liang, F.; Shen, N.; Shinozuka, K.; Yu, J.T.; Ran, C.; Quan, Q.; Tanzi, R.E.; Zhang, C. A curcumin analog reduces levels of the alzheimer’s disease-associated amyloid-beta protein by modulating abetapp processing and autophagy. J. Alzheimers Dis. 2019, 72, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. Lc3, a mammalian homolog of yeast apg8p, is localized in autophagosome membranes after processing (vol 19, pg 5720, 2000). EMBO J. 2003, 22, 4577. [Google Scholar]
- Liang, S.; Uchiyama, R.; Roh, Y.S.; Ohashi, K.; Zhong, Z.Y.; Seki, E. Macrophage autophagy controls mitochondrial quality and irf1 expression that regulates alcohol-induced liver injury in mice. Hepatology 2016, 64, 23a. [Google Scholar]
- Sarkar, S. Regulation of autophagy by mtor-dependent and mtor-independent pathways: Autophagy dysfunction in neurodegenerative diseases and therapeutic application of autophagy enhancers. Biochem. Soc. Trans. 2013, 41, 1103–1130. [Google Scholar] [CrossRef]
- Uddin, M.S.; Rahman, M.A.; Kabir, M.T.; Behl, T.; Mathew, B.; Perveen, A.; Barreto, G.E.; Bin-Jumah, M.N.; Abdel-Daim, M.M.; Ashraf, G.M. Multifarious roles of mtor signaling in cognitive aging and cerebrovascular dysfunction of alzheimer’s disease. Iubmb. Life 2020. [Google Scholar] [CrossRef]
- Rahman, M.A.; Hwang, H.; Nah, S.Y.; Rhim, H. Gintonin stimulates autophagic flux in primary cortical astrocytes. J. Ginseng. Res. 2020, 44, 67–78. [Google Scholar] [CrossRef]
- Rahman, M.A.; Bishayee, K.; Habib, K.; Sadra, A.; Huh, S.O. 18alpha-glycyrrhetinic acid lethality for neuroblastoma cells via de-regulating the beclin-1/bcl-2 complex and inducing apoptosis. Biochem. Pharmacol. 2016, 117, 97–112. [Google Scholar] [CrossRef]
- Rahman, M.A.; Bishayee, K.; Sadra, A.; Huh, S.O. Oxyresveratrol activates parallel apoptotic and autophagic cell death pathways in neuroblastoma cells. BBA Gen. Subj. 2017, 1861, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Ordonez-Gutierrez, L.; Benito-Cuesta, I.; Abad, J.L.; Casas, J.; Fabrias, G.; Wandosell, F. Dihydroceramide desaturase 1 inhibitors reduce amyloid-beta levels in primary neurons from an alzheimer’s disease transgenic model. Pharm. Res. Dordr. 2018, 35. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Fang, Y.; Cheng, X.; Lian, Y.; Xu, H.; Zeng, Z.; Zhu, H. Trpml1 participates in the progression of alzheimer’s disease by regulating the ppargamma/ampk/mtor signalling pathway. Cell Physiol. Biochem. 2017, 43, 2446–2456. [Google Scholar] [CrossRef]
- Rasheduzzaman, M.; Moon, J.H.; Lee, J.H.; Nazim, U.M.; Park, S.Y. Telmisartan generates ros-dependent upregulation of death receptor 5 to sensitize trail in lung cancer via inhibition of autophagy flux. Int. J. Biochem. Cell Biol. 2018, 102, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Ureshino, R.P.; Rocha, K.K.; Lopes, G.S.; Bincoletto, C.; Smaili, S.S. Calcium signaling alterations, oxidative stress, and autophagy in aging. Antioxid. Redox Signal. 2014, 21, 123–137. [Google Scholar] [CrossRef]
- Di Meco, A.; Li, J.G.; Blass, B.E.; Abou-Gharbia, M.; Lauretti, E.; Pratico, D. 12/15-lipoxygenase inhibition reverses cognitive impairment, brain amyloidosis, and tau pathology by stimulating autophagy in aged triple transgenic mice. Biol. Psychiat 2017, 81, 92–100. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Zhang, H.; Ma, Q.; Zhang, Y.W.; Xu, H. Proteolytic processing of alzheimer’s beta-amyloid precursor protein. J. Neurochem. 2012, 120 (Suppl. 1), 9–21. [Google Scholar] [CrossRef]
- Liu, F.K.; Huang, J.; Zhang, L.Y.; Chen, J.D.; Zeng, Y.; Tang, Y.J.; Liu, Z.X. Advances in cerebral organoid systems and their application in disease modeling. Neuroscience 2019, 399, 28–38. [Google Scholar] [CrossRef]
- Tue, N.T.; Dat, T.Q.; Ly, L.L.; Anh, V.D.; Yoshida, H. Insights from drosophila melanogaster model of alzheimer’s disease. Front. Biosci. (Landmark Ed.) 2020, 25, 134–146. [Google Scholar] [CrossRef]
- Ren, K.; Dubner, R. Activity-triggered tetrapartite neuron-glial interactions following peripheral injury. Curr. Opin. Pharmacol. 2016, 26, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Saburova, E.A.; Vasiliev, A.N.; Kravtsova, V.V.; Ryabova, E.V.; Zefirov, A.L.; Bolshakova, O.I.; Sarantseva, S.V.; Krivoi, I.I. Human app gene expression alters active zone distribution and spontaneous neurotransmitter release at the drosophila larval neuromuscular junction. Neural Plast 2017. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, D.; Suski, J.M.; Oules, B.; Debayle, D.; Gay, A.S.; Lacas-Gervais, S.; Bussiere, R.; Bauer, C.; Pinton, P.; Paterlini-Brechot, P.; et al. Localization and processing of the amyloid-beta protein precursor in mitochondria-associated membranes. J. Alzheimers Dis. 2017, 55, 1549–1570. [Google Scholar] [CrossRef] [PubMed]
- Umeda, T.; Tomiyama, T.; Sakama, N.; Tanaka, S.; Lambert, M.P.; Klein, W.L.; Mori, H. Intraneuronal amyloid beta oligomers cause cell death via endoplasmic reticulum stress, endosomal/lysosomal leakage, and mitochondrial dysfunction in vivo. J. Neurosci. Res. 2011, 89, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Muller, U.C.; Zheng, H. Physiological functions of app family proteins. Cold Spring Harb. Perspect. Med. 2012, 2, a006288. [Google Scholar] [CrossRef] [PubMed]
- Koga, K.; Liu, M.G.; Qiu, S.; Song, Q.; O’Den, G.; Chen, T.; Zhuo, M. Impaired presynaptic long-term potentiation in the anterior cingulate cortex of fmr1 knock-out mice. J. Neurosci. 2015, 35, 2033–2043. [Google Scholar] [CrossRef] [PubMed]
- McKee, A.C.; Daneshvar, D.H. The neuropathology of traumatic brain injury. Handb. Clin. Neurol 2015, 127, 45–66. [Google Scholar] [PubMed]
- Sugarman, M.A.; McKee, A.C.; Stein, T.D.; Tripodis, Y.; Besser, L.M.; Martin, B.; Palmisano, J.N.; Steinberg, E.G.; O’Connor, M.K.; Au, R.; et al. Failure to detect an association between self-reported traumatic brain injury and alzheimer’s disease neuropathology and dementia. Alzheimers Dement. 2019, 15, 686–698. [Google Scholar] [CrossRef] [PubMed]
- Marzolo, M.P.; Bu, G. Lipoprotein receptors and cholesterol in app trafficking and proteolytic processing, implications for alzheimer’s disease. Semin. Cell Dev. Biol. 2009, 20, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Kaether, C.; Thinakaran, G.; Sisodia, S. Trafficking and proteolytic processing of app. Cold Spring Harb. Perspect. Med. 2012, 2, a006270. [Google Scholar] [CrossRef]
- Chow, V.W.; Mattson, M.P.; Wong, P.C.; Gleichmann, M. An overview of app processing enzymes and products. Neuromol. Med. 2010, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.A.; Hadziselimovic, A.; Sanders, C.R. The vexing complexity of the amyloidogenic pathway. Protein Sci. 2019, 28, 1177–1193. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.A.; Rahman, M.S.; Uddin, M.J.; Mamum-Or-Rashid, A.N.M.; Pang, M.G.; Rhim, H. Emerging risk of environmental factors: Insight mechanisms of alzheimer’s diseases. Environ. Sci. Pollut. Res. 2020. [Google Scholar] [CrossRef]
- Nhan, H.S.; Chiang, K.; Koo, E.H. The multifaceted nature of amyloid precursor protein and its proteolytic fragments: Friends and foes. Acta Neuropathol. 2015, 129, 1–19. [Google Scholar] [CrossRef]
- Wilkins, H.M.; Swerdlow, R.H. Amyloid precursor protein processing and bioenergetics. Brain Res. Bull. 2017, 133, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Findlay, J.A.; Hamilton, D.L.; Sri, S.; Vargas-Caballero, M.; Ashford, M.L.J.; Smith, P.J.S. Manipulation of amyloid precursor protein processing impacts brain bioenergetics and glucose metabolism. Biophys. J. 2017, 112, 324a. [Google Scholar] [CrossRef]
- De Strooper, B.; Vassar, R.; Golde, T. The secretases: Enzymes with therapeutic potential in alzheimer disease. Nat. Rev. Neurol 2010, 6, 99–107. [Google Scholar] [CrossRef]
- Zhang, X.; Song, W. The role of app and bace1 trafficking in app processing and amyloid-beta generation. Alzheimers Res. Ther. 2013, 5, 46. [Google Scholar] [CrossRef]
- Sengupta, U.; Nilson, A.N.; Kayed, R. The role of amyloid-beta oligomers in toxicity, propagation, and immunotherapy. EBioMedicine 2016, 6, 42–49. [Google Scholar] [CrossRef]
- Bravo, R.; Parra, V.; Gatica, D.; Rodriguez, A.E.; Torrealba, N.; Paredes, F.; Wang, Z.V.; Zorzano, A.; Hill, J.A.; Jaimovich, E.; et al. Endoplasmic reticulum and the unfolded protein response: Dynamics and metabolic integration. Int. Rev. Cell Mol. Biol. 2013, 301, 215–290. [Google Scholar]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A.; Ojala, J.; Haapasalo, A.; Soininen, H.; Hiltunen, M. Impaired autophagy and app processing in alzheimer’s disease: The potential role of beclin 1 interactome. Prog. Neurobiol. 2013, 106, 33–54. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. Autophagy, amyloidogenesis and alzheimer disease. J. Cell Sci. 2007, 120, 4081–4091. [Google Scholar] [CrossRef] [PubMed]
- Krishnaswamy, S.; Verdile, G.; Groth, D.; Kanyenda, L.; Martins, R.N. The structure and function of alzheimer’s gamma secretase enzyme complex. Crit. Rev. Clin. Lab. Sci. 2009, 46, 282–301. [Google Scholar] [CrossRef] [PubMed]
- Bekris, L.M.; Galloway, N.M.; Millard, S.; Lockhart, D.; Li, G.; Galasko, D.R.; Farlow, M.R.; Clark, C.M.; Quinn, J.F.; Kaye, J.A.; et al. Amyloid precursor protein (app) processing genes and cerebrospinal fluid app cleavage product levels in alzheimer’s disease. Neurobiol. Aging 2011, 32, 556.e13–556.e23. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.L.; Vassar, R. The alzheimer’s disease beta-secretase enzyme, bace1. Mol. Neurodegener 2007, 2, 22. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Chung, T.; Vierstra, R.D. Autophagy-related11 plays a critical role in general autophagy- and senescence-induced mitophagy in arabidopsis. Plant Cell 2014, 26, 788–807. [Google Scholar] [CrossRef]
- Tian, Y.; Bustos, V.; Flajolet, M.; Greengard, P. A small-molecule enhancer of autophagy decreases levels of abeta and app-ctf via atg5-dependent autophagy pathway. FASEB J. 2011, 25, 1934–1942. [Google Scholar] [CrossRef]
- Li, C.; Gu, X.D.; Lei, M.; Wu, J.Y.; Jin, J.Z.; Shi, X.F.; Zhu, Z.Y.; Rukachaisirikul, V.; Hu, L.H.; Wen, T.Q.; et al. Thamnolia vermicularis extract improves learning ability in app/ps1 transgenic mice by ameliorating both a beta and tau pathologies. Acta Pharmacol. Sin. 2017, 38, 9–28. [Google Scholar] [CrossRef]
- Cai, Z.; Zhou, Y.; Liu, Z.; Ke, Z.; Zhao, B. Autophagy dysfunction upregulates beta-amyloid peptides via enhancing the activity of gamma-secretase complex. Neuropsychiatr. Dis. Treat. 2015, 11, 2091–2099. [Google Scholar] [CrossRef]
- Liu, J.; Li, L. Targeting autophagy for the treatment of alzheimer’s disease: Challenges and opportunities. Front. Mol. Neurosci. 2019, 12, 203. [Google Scholar] [CrossRef]
- O’Keefe, L.; Denton, D. Using drosophila models of amyloid toxicity to study autophagy in the pathogenesis of alzheimer’s disease. Biomed. Res. Int. 2018. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, P.; Saido, T.C. Dual roles for autophagy: Degradation and secretion of alzheimer’s disease abeta peptide. Bioessays 2014, 36, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Fang, F.; Guo, S.H.; Zhang, Y.; Peng, X.L.; Sun, W.M.; Wei, X.R.; He, J.S.; Hung, T. Amyloid beta-derived diffusible ligands (addls) induce abnormal autophagy associated with abeta aggregation degree. J. Mol. Neurosci. 2018, 64, 162–174. [Google Scholar] [CrossRef]
- Cordani, M.; Sanchez-Alvarez, M.; Strippoli, R.; Bazhin, A.V.; Donadelli, M. Sestrins at the interface of ros control and autophagy regulation in health and disease. Oxidative Med. Cell. Longev. 2019. [Google Scholar] [CrossRef] [PubMed]
- Derk, J.; MacLean, M.; Juranek, J.; Schmidt, A.M. The receptor for advanced glycation endproducts (rage) and mediation of inflammatory neurodegeneration. J. Alzheimers Dis. Parkinsonism 2018, 8. [Google Scholar] [CrossRef]
- Caccamo, A.; Ferreira, E.; Branca, C.; Oddo, S. P62 improves ad-like pathology by increasing autophagy. Mol. Psychiatry 2017, 22, 865–873. [Google Scholar] [CrossRef]
- Manczak, M.; Reddy, P.H. Abnormal interaction of vdac1 with amyloid beta and phosphorylated tau causes mitochondrial dysfunction in alzheimer’s disease. Hum. Mol. Genet. 2012, 21, 5131–5146. [Google Scholar] [CrossRef]
- Khalil, B.; El Fissi, N.; Aouane, A.; Cabirol-Pol, M.J.; Rival, T.; Lievens, J.C. Pink1-induced mitophagy promotes neuroprotection in huntington’s disease. Cell Death Dis. 2015, 6, e1617. [Google Scholar] [CrossRef]
- Du, F.; Yu, Q.; Yan, S.; Hu, G.; Lue, L.F.; Walker, D.G.; Wu, L.; Yan, S.F.; Tieu, K.; Yan, S.S. Pink1 signalling rescues amyloid pathology and mitochondrial dysfunction in alzheimer’s disease. Brain 2017, 140, 3233–3251. [Google Scholar] [CrossRef]
- Volpato, D.; Holzgrabe, U. Designing hybrids targeting the cholinergic system by modulating the muscarinic and nicotinic receptors: A concept to treat alzheimer’s disease. Molecules 2018, 23, 3230. [Google Scholar] [CrossRef]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the cholinergic system. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, H.; Watanabe, S.; Iwata, M.; Ueda, S.; Nobuhara, M.; Wada-Kakuda, S.; Misonou, H.; Miyasaka, T. Inhibition of microtubule assembly competent tubulin synthesis leads to accumulation of phosphorylated tau in neuronal cell bodies. Biochem. Biophys. Res. Commun. 2020, 521, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Nanduri, R.; Kalra, R.; Bhagyaraj, E.; Chacko, A.P.; Ahuja, N.; Tiwari, D.; Kumar, S.; Jain, M.; Parkesh, R.; Gupta, P. Autophagysmdb: A curated database of small molecules that modulate protein targets regulating autophagy. Autophagy 2019, 15, 1280–1295. [Google Scholar] [CrossRef] [PubMed]
- Kuang, H.; Tan, C.Y.; Tian, H.Z.; Liu, L.H.; Yang, M.W.; Hong, F.F.; Yang, S.L. Exploring the bi-directional relationship between autophagy and alzheimer’s disease. CNS Neurosci. Ther. 2020, 26, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.; Lee, M.R.; Huang, X.; Messina-Graham, S.; Broxmeyer, H.E. Sirt1 positively regulates autophagy and mitochondria function in embryonic stem cells under oxidative stress. Stem Cells 2014, 32, 1183–1194. [Google Scholar] [CrossRef]
- Luo, R.C.; Su, L.Y.; Li, G.Y.; Yang, J.; Liu, Q.J.; Yang, L.X.; Zhang, D.F.; Zhou, H.J.; Xu, M.; Fan, Y.; et al. Activation of ppara-mediated autophagy reduces alzheimer disease-like pathology and cognitive decline in a murine model. Autophagy 2020, 16, 52–69. [Google Scholar] [CrossRef]
- Kim, Y.S.; Silwal, P.; Kim, S.Y.; Yoshimori, T.; Jo, E.K. Autophagy-activating strategies to promote innate defense against mycobacteria. Exp. Mol. Med. 2019, 51, 151. [Google Scholar] [CrossRef]
- Heckmann, B.L.; Teubner, B.J.W.; Tummers, B.; Boada-Romero, E.; Harris, L.; Yang, M.; Guy, C.S.; Zakharenko, S.S.; Green, D.R. Lc3-associated endocytosis facilitates beta-amyloid clearance and mitigates neurodegeneration in murine alzheimer’s disease. Cell 2019, 178, 536–551. [Google Scholar] [CrossRef]
- Schweig, J.E.; Yao, H.; Coppola, K.; Jin, C.; Crawford, F.; Mullan, M.; Paris, D. Spleen tyrosine kinase (syk) blocks autophagic tau degradation in vitro and in vivo. J. Biol. Chem. 2019, 294, 13378–13395. [Google Scholar] [CrossRef]
- Wei, Y.; Zhou, J.; Wu, J.; Huang, J. Erbeta promotes abeta degradation via the modulation of autophagy. Cell Death Dis. 2019, 10, 565. [Google Scholar] [CrossRef]
- Zhong, Y.; Zheng, Q.Y.; Sun, C.Y.; Zhang, Z.; Han, K.; Jia, N. Orientin improves cognition by enhancing autophagosome clearance in an alzheimer’s mouse model. J. Mol. Neurosci. 2019, 69, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Park, S.J.; Jo, D.S.; Park, N.Y.; Kim, J.B.; Bae, J.E.; Jo, Y.K.; Hwang, J.J.; Lee, J.A.; Jo, D.G.; et al. Down-regulated tmed10 in alzheimer disease induces autophagy via atg4b activation. Autophagy 2019, 15, 1495–1505. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.A.; Saha, S.K.; Rahman, M.S.; Uddin, M.J.; Uddin, M.S.; Pang, M.G.; Rhim, H.; Cho, S.G. Molecular insights into therapeutic potential of autophagy modulation by natural products for cancer stem cells. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef]
- Rahman, M.A.; Rahman, M.R.; Zaman, T.; Uddin, M.S.; Islam, R.; Abdel-Daim, M.M.; Rhim, H. Emerging potential of naturally occurring autophagy modulators against neurodegeneration. Curr. Pharm. Des. 2020, 26, 772–779. [Google Scholar] [CrossRef] [PubMed]
- Hannan, M.A.; Dash, R.; Haque, M.N.; Mohibbullah, M.; Sohag, A.A.; Rahman, M.A.; Uddin, M.J.; Alam, M.; Moon, I. Neuroprotective potentials of marine algae and their bioactive metabolites: Pharmacological insights and therapeutic advances. Mar. Drugs 2020, 18, 347. [Google Scholar] [CrossRef] [PubMed]
- Law, B.Y.; Chan, W.K.; Xu, S.W.; Wang, J.R.; Bai, L.P.; Liu, L.; Wong, V.K. Natural small-molecule enhancers of autophagy induce autophagic cell death in apoptosis-defective cells. Sci. Rep. 2014, 4, 5510. [Google Scholar] [CrossRef]
- Li, L.S.; Lu, Y.L.; Nie, J.; Xu, Y.Y.; Zhang, W.; Yang, W.J.; Gong, Q.H.; Lu, Y.F.; Lu, Y.; Shi, J.S. Dendrobium nobile lindl alkaloid, a novel autophagy inducer, protects against axonal degeneration induced by abeta25-35 in hippocampus neurons in vitro. CNS Neurosci. Ther. 2017, 23, 329–340. [Google Scholar] [CrossRef]
- Al Rihani, S.B.; Darakjian, L.I.; Kaddoumi, A. Oleocanthal-rich extra-virgin olive oil restores the blood-brain barrier function through nlrp3 inflammasome inhibition simultaneously with autophagy induction in tgswdi mice. ACS Chem. Neurosci. 2019, 10, 3543–3554. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Y.L.; Liang, Y.B.; Chen, H.D.; Ji, X.Y.; Huang, M. Berberine mitigates cognitive decline in an alzheimer’s disease mouse model by targeting both tau hyperphosphorylation and autophagic clearance. Biomed. Pharm. 2020, 121. [Google Scholar] [CrossRef]
- Zhang, Q.C.; Bian, H.M.; Guo, L.W.; Zhu, H.X. Pharmacologic preconditioning with berberine attenuating ischemia-induced apoptosis and promoting autophagy in neuron. Am. J. Transl. Res. 2016, 8, 1197–1207. [Google Scholar]
- Yuan, N.N.; Cai, C.Z.; Wu, M.Y.; Su, H.X.; Li, M.; Lu, J.H. Neuroprotective effects of berberine in animal models of alzheimer’s disease: A systematic review of pre-clinical studies. BMC Complem. Altern. Med. 2019, 19. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.L.; Song, J.X.; Lu, J.H.; Yuan, Z.W.; Liu, L.F.; Durairajan, S.S.K.; Li, M. Corynoxine, a natural autophagy enhancer, promotes the clearance of alpha-synuclein via akt/mtor pathway. J. Neuroimmune Pharm. 2014, 9, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Siu, W.; Li, L.; Jin, Y.; Liang, S.; Cao, M.; Ma, M.; Wu, Z. Autophagy in alzheimer’s disease and promising modulatory effects of herbal medicine. Exp. Gerontol. 2019, 119, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.Y.; Wang, N.; Rocchi, A.; Zhang, W.R.; Vassar, R.; Zhou, Y.F.; He, C.C. Identification of natural products with neuronal and metabolic benefits through autophagy induction. Autophagy 2017, 13, 41–56. [Google Scholar] [CrossRef]
- Guo, X.D.; Lv, J.L.; Lu, J.; Fan, L.; Huang, X.; Hu, L.H.; Wang, J.Y.; Shen, X. Protopanaxadiol derivative ddpu improves behavior and cognitive deficit in ad mice involving regulation of both er stress and autophagy. Neuropharmacology 2018, 130, 77–91. [Google Scholar] [CrossRef]
- Huang, M.; Jiang, X.; Liang, Y.B.; Liu, Q.; Chen, S.Y.; Guo, Y. Berberine improves cognitive impairment by promoting autophagic clearance and inhibiting production of beta-amyloid in app/tau/ps1 mouse model of alzheimer’s disease. Exp. Gerontol. 2017, 91, 25–33. [Google Scholar] [CrossRef]
- Song, X.; Liu, B.; Cui, L.; Zhou, B.; Liu, W.; Xu, F.; Hayashi, T.; Hattori, S.; Ushiki-Kaku, Y.; Tashiro, S.I.; et al. Silibinin ameliorates anxiety/depression-like behaviors in amyloid beta-treated rats by upregulating bdnf/trkb pathway and attenuating autophagy in hippocampus. Physiol. Behav. 2017, 179, 487–493. [Google Scholar] [CrossRef]
- Meng, X.B.; Luo, Y.; Liang, T.; Wang, M.X.; Zhao, J.Y.; Sun, G.B.; Sun, X.B. Gypenoside xvii enhances lysosome biogenesis and autophagy flux and accelerates autophagic clearance of amyloid-beta through tfeb activation. J. Alzheimers Dis. 2016, 52, 1135–1150. [Google Scholar] [CrossRef]
- Liu, X.; Hao, W.L.; Qin, Y.R.; Decker, Y.; Wang, X.; Burkart, M.; Schotz, K.; Menger, M.D.; Fassbender, K.; Liu, Y. Long-term treatment with ginkgo biloba extract egb 761 improves symptoms and pathology in a transgenic mouse model of alzheimer’s disease. Brain Behav. Immun. 2015, 46, 121–131. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, Z.C.; Wang, K.F.; Chen, X.Y. A beta peptide secretion is reduced by radix polygalae-induced autophagy via activation of the ampk/mtor pathway. Mol. Med. Rep. 2015, 12, 2771–2776. [Google Scholar] [CrossRef]
- Du, B.S.; Zhang, Z.Y.; Li, N. Madecassoside prevents a beta(25-35)-induced inflammatory responses and autophagy in neuronal cells through the class iii pi3k/beclin-1/bcl-2 pathway. Int. Immunopharmacol. 2014, 20, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.M.; Tsai, S.Y.; Lin, J.A.; Wu, C.H.; Yen, G.C. Cytoprotective effects of hesperetin and hesperidin against amyloid beta-induced impairment of glucose transport through downregulation of neuronal autophagy. Mol. Nutr. Food Res. 2012, 56, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.H.; Bishayee, K.; Rahman, A.; Hong, J.S.; Lim, S.S.; Huh, S.O. Morus alba accumulates reactive oxygen species to initiate apoptosis via foxo-caspase 3-dependent pathway in neuroblastoma cells. Mol. Cells 2015, 38, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Wang, J. Wogonin increases beta-amyloid clearance and inhibits tau phosphorylation via inhibition of mammalian target of rapamycin: Potential drug to treat alzheimer’s disease. Neurol. Sci. 2015, 36, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, X.; Teng, Z.; Zhang, T.; Li, Y. Downregulation of pi3k/akt/mtor signaling pathway in curcumin-induced autophagy in app/ps1 double transgenic mice. Eur. J. Pharmacol. 2014, 740, 312–320. [Google Scholar] [CrossRef]
- Vingtdeux, V.; Giliberto, L.; Zhao, H.; Chandakkar, P.; Wu, Q.; Simon, J.E.; Janle, E.M.; Lobo, J.; Ferruzzi, M.G.; Davies, P.; et al. Amp-activated protein kinase signaling activation by resveratrol modulates amyloid-beta peptide metabolism. J. Biol. Chem. 2010, 285, 9100–9113. [Google Scholar] [CrossRef]
- Rahman, M.A.; Kim, N.H.; Kim, S.H.; Oh, S.M.; Huh, S.O. Antiproliferative and cytotoxic effects of resveratrol in mitochondria-mediated apoptosis in rat b103 neuroblastoma cells. Korean J. Physiol. Pharmacol. 2012, 16, 321–326. [Google Scholar] [CrossRef]
- Uddin, M.S.; Mamun, A.A.; Jakaria, M.; Thangapandiyan, S.; Ahmad, J.; Rahman, M.A.; Mathew, B.; Abdel-Daim, M.M.; Aleya, L. Emerging promise of sulforaphane-mediated nrf2 signaling cascade against neurological disorders. Sci. Total Environ. 2019, 707, 135624. [Google Scholar] [CrossRef]
- Liu, J.; Su, H.; Qu, Q.M. Carnosic acid prevents beta-amyloid-induced injury in human neuroblastoma sh-sy5y cells via the induction of autophagy. Neurochem. Res. 2016, 41, 2311–2323. [Google Scholar] [CrossRef]
- Zeng, Y.Q.; Zhang, J.; Zhu, Y.G.; Zhang, J.; Shen, H.; Lu, J.P.; Pan, X.D.; Lin, N.; Dai, X.M.; Zhou, M.; et al. Tripchlorolide improves cognitive deficits by reducing amyloid beta and upregulating synapse-related proteins in a transgenic model of alzheimer’s disease. J. Neurochem. 2015, 133, 38–52. [Google Scholar] [CrossRef]
- Deng, M.Z.; Huang, L.P.; Ning, B.L.; Wang, N.B.; Zhang, Q.X.; Zhu, C.X.; Fang, Y.Q. Beta-asarone improves learning and memory and reduces acetyl cholinesterase and beta-amyloid 42 levels in app/ps1 transgenic mice by regulating beclin-1-dependent autophagy. Brain Res. 2016, 1652, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, D.J.; Shin, E.J.; Lee, B.H.; Choi, S.H.; Hwang, S.H.; Rhim, H.; Cho, I.H.; Kim, H.C.; Nah, S.Y. Effects of gintonin-enriched fraction on hippocampal cell proliferation in wild-type mice and an appswe/psen-1 double tg mouse model of alzheimer’s disease. Neurochem. Int. 2016, 101, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.; Choi, S.H.; Shim, J.Y.; Park, H.J.; Oh, M.J.; Kim, M.; Nah, S.Y. Gintonin administration is safe and potentially beneficial in cognitively impaired elderly. Alzheimer Dis. Assoc. Dis. 2018, 32, 85–87. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, N.; Rahman, M.A.; Hwang, H.; Ko, S.K.; Nah, S.Y.; Kim, H.C.; Rhim, H. Ginsenoside rk1 is a novel inhibitor of nmda receptors in cultured rat hippocampal neurons. J. Ginseng. Res. 2020, 44, 490–495. [Google Scholar] [CrossRef]
- Lin, X.; Zhang, S.J.; Huang, R.B.; Wei, L.; Tan, S.M.; Liang, C.H.; Lv, S.J.; Chen, Y.X.; Liang, S.; Tian, Y.C.; et al. Protective effect of madecassoside against cognitive impairment induced by d-galactose in mice. Pharmacol. Biochem. Behav. 2014, 124, 434–442. [Google Scholar] [CrossRef]
- Cho, Y.; Hwang, H.; Rahman, M.A.; Chung, C.; Rhim, H. Elevated o-glcnacylation induces an antidepressant-like phenotype and decreased inhibitory transmission in medial prefrontal cortex. Sci. Rep. 2020, 10, 6924. [Google Scholar] [CrossRef]
- Rahman, M.A.; Hwang, H.; Cho, Y.; Rhim, H. Modulation of o-glcnacylation regulates autophagy in cortical astrocytes. Oxid. Med. Cell. Longev. 2019, 2019, 6279313. [Google Scholar] [CrossRef]
- Gomes, B.A.Q.; Silva, J.P.B.; Romeiro, C.F.R.; dos Santos, S.M.; Rodrigues, C.A.; Goncalves, P.R.; Sakai, J.T.; Mendes, P.F.S.; Varela, E.L.P.; Monteiro, M.C. Neuroprotective mechanisms of resveratrol in alzheimer’s disease: Role of sirt1. Oxid. Med. Cell. Longev. 2018. [Google Scholar] [CrossRef]
- Rege, S.D.; Geetha, T.; Griffin, G.D.; Broderick, T.L.; Babu, J.R. Neuroprotective effects of resveratrol in alzheimer disease pathology. Front. Aging Neurosci. 2014, 6. [Google Scholar] [CrossRef]
- Song, J.; Li, N.; Xia, Y.; Gao, Z.; Zou, S.F.; Kong, L.; Yao, Y.J.; Jiao, Y.N.; Yan, Y.H.; Li, S.H.; et al. Arctigenin treatment protects against brain damage through an anti-inflammatory and anti-apoptotic mechanism after needle insertion. Front. Pharmacol. 2016, 7. [Google Scholar] [CrossRef]
- Rossi, M.; Munarriz, E.R.; Bartesaghi, S.; Milanese, M.; Dinsdale, D.; Guerra-Martin, M.A.; Bampton, E.T.; Glynn, P.; Bonanno, G.; Knight, R.A.; et al. Desmethylclomipramine induces the accumulation of autophagy markers by blocking autophagic flux. J. Cell Sci. 2009, 122, 3330–3339. [Google Scholar] [CrossRef] [PubMed]
- Donohue, E.; Tovey, A.; Vogl, A.W.; Arns, S.; Sternberg, E.; Young, R.N.; Roberge, M. Inhibition of autophagosome formation by the benzoporphyrin derivative verteporfin. J. Biol. Chem. 2011, 286, 7290–7300. [Google Scholar] [CrossRef] [PubMed]
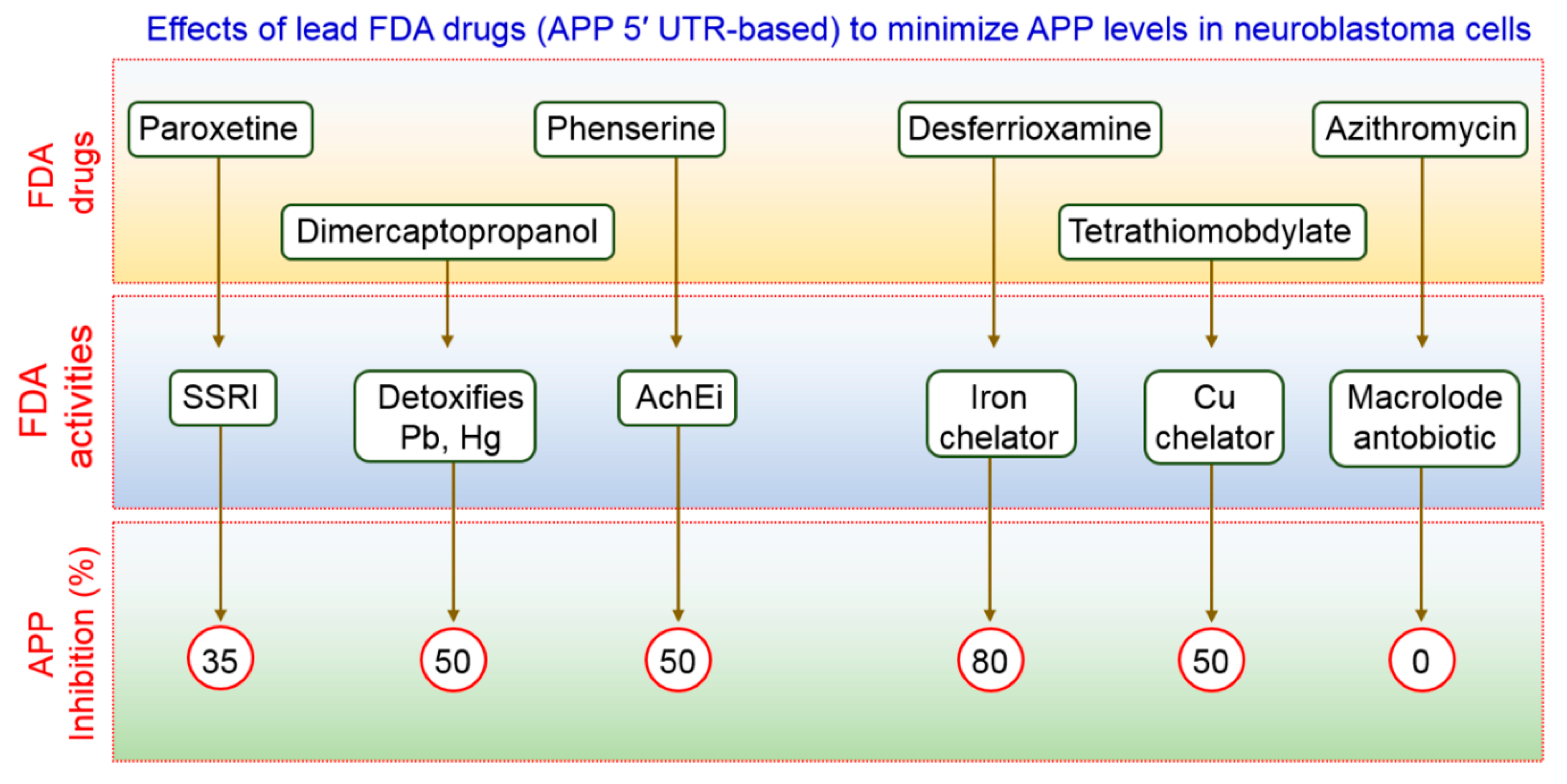
- Bandyopadhyay, S.; Ni, J.; Ruggiero, A.; Walshe, K.; Rogers, M.S.; Chattopadhyay, N.; Glicksman, M.A.; Rogers, J.T. A high-throughput drug screen targeted to the 5′untranslated region of alzheimer amyloid precursor protein mrna. J. Biomol. Screen 2006, 11, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Morse, L.J.; Payton, S.M.; Cuny, G.D.; Rogers, J.T. Fda-preapproved drugs targeted to the translational regulation and processing of the amyloid precursor protein. J. Mol. Neurosci. 2004, 24, 129–136. [Google Scholar] [CrossRef]
- Payton, S.; Cahill, C.M.; Randall, J.D.; Gullans, S.R.; Rogers, J.T. Drug discovery targeted to the alzheimer’s app mrna 5′-untranslated region: The action of paroxetine and dimercaptopropanol. J. Mol. Neurosci. 2003, 20, 267–275. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Natural Products | AD Model | Activities/Effects | Molecular Mechanism | References |
|---|---|---|---|---|
| Dendrobium nobile Lindl alkaloid, DNLA | Hippocampus neurons of Aβ25-35 | Protective effects of axonal degeneration | Autophagic flux enhancement | [97] |
| Extra-Virgin Olive Oil (EVOO) | TgSwDI mice | Neuroinflammation reduction | AMPK-ULK1 pathway induction | [98] |
| Ginsenoside Rg2 | 5×FAD transgenic mice | Removal of Aβ aggregation | AMPK/ULK1-mediated autophagy induction | [104] |
| Protopanaxadiol derivative DDPU | APP/PS1 mice model | Stimulates the clearance of Aβ | Inhibition of PI3K/mTOR-mediated autophagy induction | [105] |
| Berberine | 3×Tg-AD mice | Promotes the clearance of Aβ | Activates Bcl2/Beclin1-mediated autophagy induction | [101,106] |
| Flavonoids Silibinin | Aβ1-42-induced rat model | Attenuates neuronal damage | Inhibits autophagy | [107] |
| Corynoxine B | Tg2567 mice, N2a-SwedAPP cell model | Augments APP and Aβ degradation | Pathway that induces autophagy is unknown | [103] |
| Gypenoside XVII | APP/PS1 transgenic mice | Prevents Aβ accumulation | Promotes TFEB to induce autophagy | [108] |
| Ginkgo biloba extract | TgCRND8 mice | Improves cognitive function | Induces autophagy | [109] |
| Radix polygalae extract | Cell model of CHO-APP/BACE1 | Decreases Aβ1-40 levels | Activates AMPK/mTOR and promotes autophagy | [110] |
| Madecassoside | D-galactose-induced mouse model | Autophagy inhibition | Increases Bcl-2 and decreases Beclin-1 | [111] |
| Hesperetin | N2a cell model | Increases Aβdamage | Autophagy inhibition | [112] |
| Morus alba extract | SH-SY5Y cells | Autophagy induction | mTOR-dependent autophagy pathway | [94,113] |
| Wogonin | SH-SY5Y-APP primary cortical astrocytes | Enhances Aβ removal | Activates ULK1/mTOR and induces autophagy | [114] |
| Curcumin | APP/PS1 transgenic mice | Prevents Aβ deposition | Inhibits PI3K/mTOR and induces autophagy | [115] |
| Resveratrol | N2a-APP cells, HEK293-APP cells | Decreases Aβ production and aggregation | Induces autophagy by activating AMPK/mTOR signaling | [116,117] |
| Sulforaphane | AD model | Nrf2 signaling | Induces autophagy | [118] |
| Carnosic acid | Aβ25-35-induced SHSY5Y cells | Inhibition of Aβ1-42 aggregation | Activates AMPK/mTOR and induces autophagy | [119] |
| Tripchlorolide | 5×FAD transgenicmice | Reduces cerebral Aβ deposits | Activates PI3K/mTOR pathway | [120] |
| β-asarone | APP/PS1 transgenic mice | Decreases Aβ level | Activates PI3K/mTOR and inhibits autophagy | [121] |
| Oxyresveratrol | SH-SY5Y cell model | Stimulates autophagy | Atg5/7, Beclin-1, and LC-3 induction | [31] |
| 18α-Glycyrrhetinic acid | SH-SY5Y cell model | Induction of autophagy flux | mTOR-dependent autophagy induction | [30] |
| Gintonin | Mouse cortical Astrocytes, APPswe/PSEN-1 | Autophagic flux induction, cognition improvements | Beclin-1, Atg5/7, LAMP-1 induction, elevation of hippocampal neurogenesis | [29,122,123] |
| Emodin | APP/PS1 mice | Autophagy inhibition | Activates Bcl-2/Beclin-1/PIK3C3 pathway | [103] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, M.A.; Rahman, M.S.; Rahman, M.H.; Rasheduzzaman, M.; Mamun-Or-Rashid, A.; Uddin, M.J.; Rahman, M.R.; Hwang, H.; Pang, M.-G.; Rhim, H. Modulatory Effects of Autophagy on APP Processing as a Potential Treatment Target for Alzheimer’s Disease. Biomedicines 2021, 9, 5. https://doi.org/10.3390/biomedicines9010005
Rahman MA, Rahman MS, Rahman MH, Rasheduzzaman M, Mamun-Or-Rashid A, Uddin MJ, Rahman MR, Hwang H, Pang M-G, Rhim H. Modulatory Effects of Autophagy on APP Processing as a Potential Treatment Target for Alzheimer’s Disease. Biomedicines. 2021; 9(1):5. https://doi.org/10.3390/biomedicines9010005
Chicago/Turabian StyleRahman, Md. Ataur, Md Saidur Rahman, MD. Hasanur Rahman, Mohammad Rasheduzzaman, ANM Mamun-Or-Rashid, Md Jamal Uddin, Md Rezanur Rahman, Hongik Hwang, Myung-Geol Pang, and Hyewhon Rhim. 2021. "Modulatory Effects of Autophagy on APP Processing as a Potential Treatment Target for Alzheimer’s Disease" Biomedicines 9, no. 1: 5. https://doi.org/10.3390/biomedicines9010005
APA StyleRahman, M. A., Rahman, M. S., Rahman, M. H., Rasheduzzaman, M., Mamun-Or-Rashid, A., Uddin, M. J., Rahman, M. R., Hwang, H., Pang, M.-G., & Rhim, H. (2021). Modulatory Effects of Autophagy on APP Processing as a Potential Treatment Target for Alzheimer’s Disease. Biomedicines, 9(1), 5. https://doi.org/10.3390/biomedicines9010005