Identification of Key Phospholipids That Bind and Activate Atypical PKCs

 ,
, 

 , , , ,
, , , , 
 ,
, 
Abstract
1. Introduction
2. Results
2.1. PKCζ Selectively Binds to PA and PS
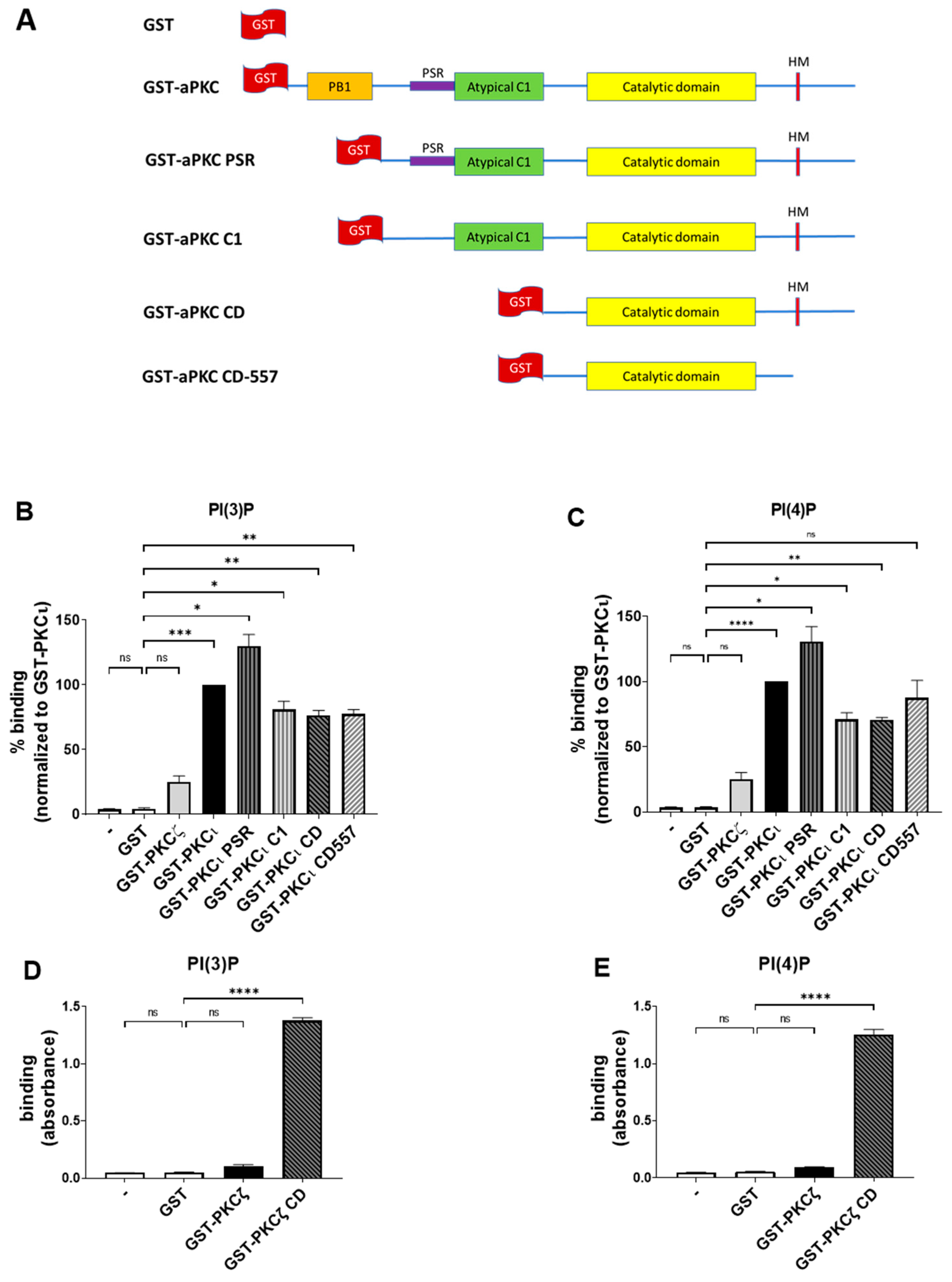
2.2. PKCι Binds to Phosphatidylinositol Monophosphates, along with PA and PS
2.3. PKCι Binds to PI(3)P and PI(4)P through the Catalytic Domain
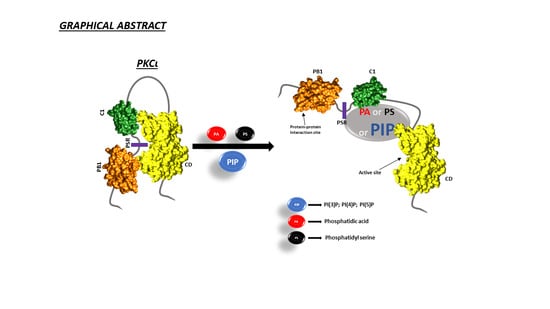
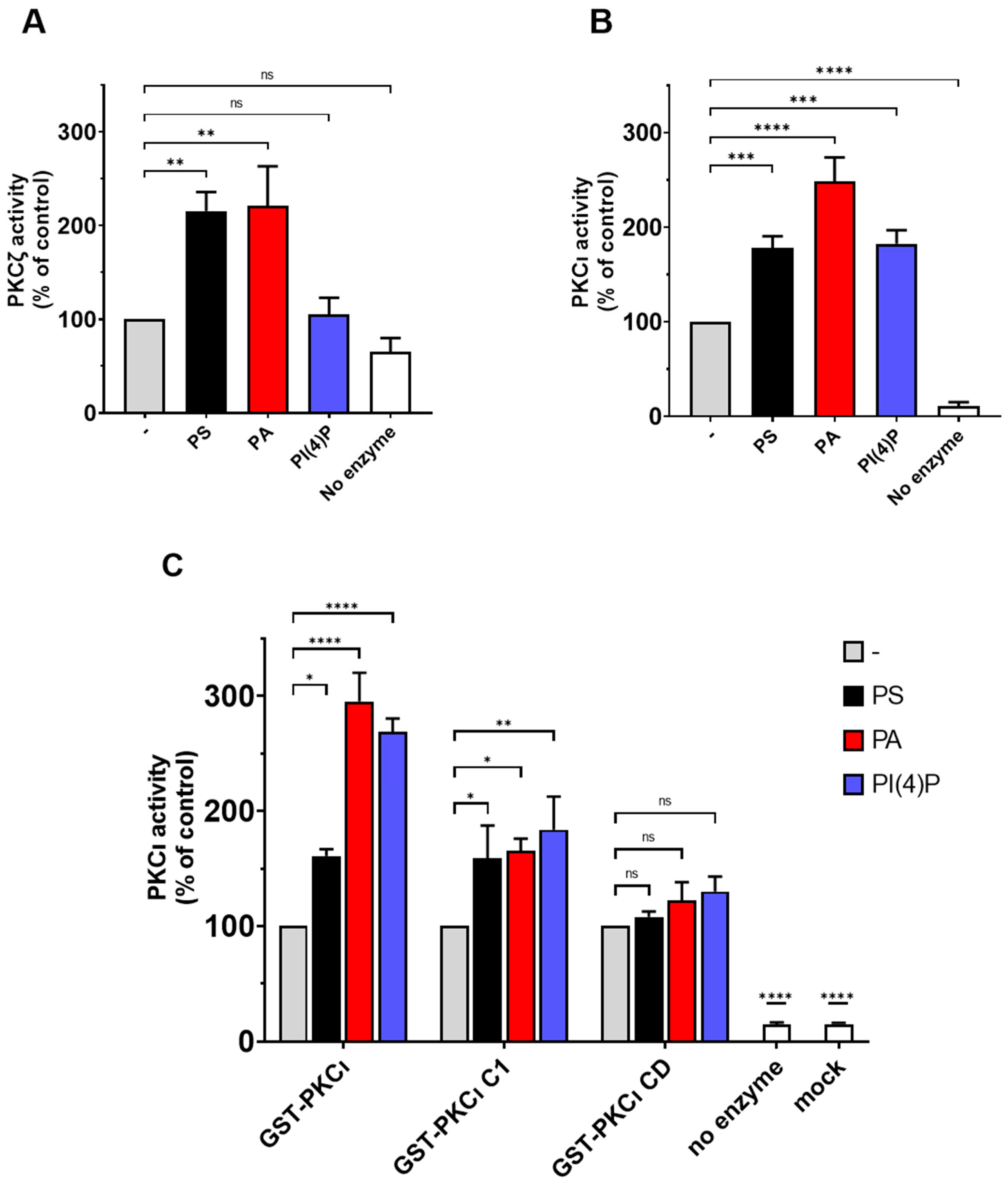
2.4. PS and PA Activates Both aPKCs, While PI(4)P Activates PKCι Selectively
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Constructs
4.3. Protein Purification
4.4. Lipid Overlay Assay
4.5. Cova PIP ELISA Assay
4.6. aPKC Activity Assay
4.7. Data Processing and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| LPA | lysophosphatidic acid |
| LPC | lysophosphocoline |
| PI | phosphatidylinositol |
| PI(3)P | phosphatidylinositol 3-phosphate |
| PI(4)P | phosphatidylinositol 4-phosphate |
| PI(5)P | phosphatidylinositol 5-phosphate |
| PI(3,4)P2 | phosphatidylinositol 3,4-bisphosphate |
| PI(3,5)P2 | phosphatidylinositol 3,5-bisphosphate |
| PI(4,5)P2 | phosphatidylinositol 4,5-bisphosphate |
| PI(3,4,5)P3 | phosphatidylinositol 3,4,5-trisphosphate |
| DAG | diacylglycerol |
| PA | phosphatidic acid |
| PS | phosphatidylserine |
| PE | phosphatidylethanolamine |
| PC | phosphatidylcholine |
| PG | phosphatidylglycerol |
| S | sphingosine |
| S1P | sphingosine-1-phosphate |
| SM | sphingomyelin |
| SPC | sphingosylphosphorycholine |
| PHS | phytosphingosine |
| CE | ceramide |
| SM4 | sulfatide |
| GM1 | monosialoganglioside |
| TAG | triacylglycerol |
| CL | cardiolipin |
| CH | Cholesterol |
References
- Hong, Y. aPKC: The Kinase that Phosphorylates Cell Polarity. F1000Research 2018, 7, 903. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Liu, M. Atypical protein kinase C in cell motility. Cell. Mol. Life Sci. 2012, 70, 3057–3066. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, M. The Par3/Par6/aPKC complex and epithelial cell polarity. Exp. Cell Res. 2013, 319, 1357–1364. [Google Scholar] [CrossRef]
- Vorhagen, S.; Niessen, C.M. Mammalian aPKC/Par polarity complex mediated regulation of epithelial division orientation and cell fate. Exp. Cell Res. 2014, 328, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Drummond, M.L.; Prehoda, K.E. Molecular Control of Atypical Protein Kinase C: Tipping the Balance between Self-Renewal and Differentiation. J. Mol. Biol. 2016, 428, 1455–1464. [Google Scholar] [CrossRef]
- Reina-Campos, M.; Diaz-Meco, M.T.; Moscat, J. The Dual Roles of the Atypical Protein Kinase Cs in Cancer. Cancer Cell 2019, 36, 218–235. [Google Scholar] [CrossRef]
- Du, G.-S.; Qiu, Y.; Wang, W.-S.; Peng, K.; Zhang, Z.-C.; Li, X.-S.; Xiao, W.; Yang, H. Knockdown on aPKC-ι inhibits epithelial-mesenchymal transition, migration and invasion of colorectal cancer cells through Rac1-JNK pathway. Exp. Mol. Pathol. 2019, 107, 57–67. [Google Scholar] [CrossRef]
- Qian, Y.; Yao, W.; Yang, T.; Yang, Y.; Liu, Y.; Shen, Q.; Zhang, J.; Qi, W.; Wang, J. aPKC-ι/P-Sp1/Snail signaling induces epithelial-mesenchymal transition and immunosuppression in cholangiocarcinoma. Hepatology 2017, 66, 1165–1182. [Google Scholar] [CrossRef]
- Paul, A.; Gunewardena, S.; Stecklein, S.R.; Saha, B.; Parelkar, N.; Danley, M.; Rajendran, G.; Home, P.; Ray, S.; Jokar, I.; et al. PKCλ/ι signaling promotes triple-negative breast cancer growth and metastasis. Cell Death Differ. 2014, 21, 1469–1481. [Google Scholar] [CrossRef]
- Cohen, E.E.W.; Lingen, M.W.; Zhu, B.; Zhu, H.; Straza, M.W.; Pierce, C.; Martin, L.E.; Rosner, M.R. Protein Kinase Cζ Mediates Epidermal Growth Factor–Induced Growth of Head and Neck Tumor Cells by Regulating Mitogen-Activated Protein Kinase. Cancer Res. 2006, 66, 6296–6303. [Google Scholar] [CrossRef]
- Lopez-Garcia, L.A.; Schulze, J.O.; Fröhner, W.; Zhang, H.; Süss, E.; Weber, N.; Navratil, J.; Amon, S.; Hindie, V.; Zeuzem, S.; et al. Allosteric Regulation of Protein Kinase PKCζ by the N-Terminal C1 Domain and Small Compounds to the PIF-Pocket. Chem. Biol. 2011, 18, 1463–1473. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Neimanis, S.; Lopez-Garcia, L.A.; Arencibia, J.M.; Amon, S.; Stroba, A.; Zeuzem, S.; Proschak, E.; Stark, H.; Bauer, A.F.; et al. Molecular Mechanism of Regulation of the Atypical Protein Kinase C by N-terminal Domains and an Allosteric Small Compound. Chem. Biol. 2014, 21, 754–765. [Google Scholar] [CrossRef] [PubMed]
- Corbalán-García, S.; Gómez-Fernández, J.C. Protein kinase C regulatory domains: The art of decoding many different signals in membranes. Biochim. Biophys. ActaMol. Cell Biol. Lipids 2006, 1761, 633–654. [Google Scholar] [CrossRef] [PubMed]
- Chianale, F.; Rainero, E.; Cianflone, C.; Bettio, V.; Pighini, A.; Porporato, P.E.; Filigheddu, N.; Serini, G.; Sinigaglia, F.; Baldanzi, G.; et al. Diacylglycerol kinase mediates HGF-induced Rac activation and membrane ruffling by regulating atypical PKC and RhoGDI. Proc. Natl. Acad. Sci. USA 2010, 107, 4182–4187. [Google Scholar] [CrossRef] [PubMed]
- Rainero, E.; Cianflone, C.; Porporato, P.E.; Chianale, F.; Malacarne, V.; Bettio, V.; Ruffo, E.; Ferrara, M.; Benecchia, F.; Capello, D.; et al. The Diacylglycerol Kinase α/Atypical PKC/β1 Integrin Pathway in SDF-1α Mammary Carcinoma Invasiveness. PLoS ONE 2014, 9, e97144. [Google Scholar] [CrossRef]
- Dang, P.M.-C.; Fontayne, A.; Hakim, J.; El Benna, J.; Périanin, A. Protein Kinase C ζ Phosphorylates a Subset of Selective Sites of the NADPH Oxidase Component p47phoxand Participates in Formyl Peptide-Mediated Neutrophil Respiratory Burst. J. Immunol. 2001, 166, 1206–1213. [Google Scholar] [CrossRef]
- Akimoto, K.; Takahashi, R.; Moriya, S.; Nishioka, N.; Takayanagi, J.; Kimura, K.; Fukui, Y.; Osada, S.-I.; Mizuno, K.; Hirai, S.-I.; et al. EGF or PDGF receptors activate atypical PKClambda through phosphatidylinositol 3-kinase. EMBO J. 1996, 15, 788–798. [Google Scholar] [CrossRef]
- Wang, G.; Silva, J.; Krishnamurthy, K.; Tran, E.; Condie, B.G.; Bieberich, E. Direct Binding to Ceramide Activates Protein Kinase Cζ before the Formation of a Pro-apoptotic Complex with PAR-4 in Differentiating Stem Cells. J. Biol. Chem. 2005, 280, 26415–26424. [Google Scholar] [CrossRef] [PubMed]
- Limatola, C.; Schaap, D.; Moolenaar, W.H.; Van Blitterswijk, W.J. Phosphatidic acid activation of protein kinase C-ζ overexpressed in COS cells: Comparison with other protein kinase C isotypes and other acidic lipids. Biochem. J. 1994, 304, 1001–1008. [Google Scholar] [CrossRef]
- Pu, Y.; Peach, M.L.; Garfield, S.H.; Wincovitch, S.; Marquez, V.E.; Blumberg, P.M. Effects on Ligand Interaction and Membrane Translocation of the Positively Charged Arginine Residues Situated along the C1 Domain Binding Cleft in the Atypical Protein Kinase C Isoforms. J. Biol. Chem. 2006, 281, 33773–33788. [Google Scholar] [CrossRef]
- Chianale, F.; Cutrupi, S.; Rainero, E.; Baldanzi, G.; Porporato, P.E.; Traini, S.; Filigheddu, N.; Gnocchi, V.F.; Santoro, M.M.; Parolini, O.; et al. Diacylglycerol Kinase-α Mediates Hepatocyte Growth Factor-induced Epithelial Cell Scatter by Regulating Rac Activation and Membrane Ruffling. Mol. Biol. Cell 2007, 18, 4859–4871. [Google Scholar] [CrossRef] [PubMed]
- Müller, G.; Ayoub, M.; Storz, P.; Rennecke, J.; Fabbro, D.; Pfizenmaier, K. PKC zeta is a molecular switch in signal transduction of TNF-alpha, bifunctionally regulated by ceramide and arachidonic acid. EMBO J. 1995, 14, 1961–1969. [Google Scholar] [CrossRef]
- Wang, G.; Krishnamurthy, K.; Umapathy, N.S.; Verin, A.D.; Bieberich, E. The Carboxyl-terminal Domain of Atypical Protein Kinase Cζ Binds to Ceramide and Regulates Junction Formation in Epithelial Cells. J. Biol. Chem. 2009, 284, 14469–14475. [Google Scholar] [CrossRef] [PubMed]
- Kajimoto, T.; Caliman, A.D.; Tobias, I.S.; Okada, T.; Pilo, C.A.; Van, A.-A.N.; McCammon, J.A.; Nakamura, S.-I.; Newton, A.C. Activation of atypical protein kinase C by sphingosine 1-phosphate revealed by an aPKC-specific activity reporter. Sci. Signal. 2019, 12, eaat6662. [Google Scholar] [CrossRef] [PubMed]
- Ivey, R.A.; Sajan, M.P.; Farese, R.V. Requirements for Pseudosubstrate Arginine Residues during Autoinhibition and Phosphatidylinositol 3,4,5-(PO4)3-dependent Activation of Atypical PKC. J. Biol. Chem. 2014, 289, 25021–25030. [Google Scholar] [CrossRef] [PubMed]
- Tobias, I.S.; Kaulich, M.; Kim, P.K.; Simon, N.; Jacinto, E.; Dowdy, S.F.; King, C.C.; Newton, A.C. Protein kinase Cζ exhibits constitutive phosphorylation and phosphatidylinositol-3,4,5-triphosphate-independent regulation. Biochem. J. 2016, 473, 509–523. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Lu, J.; Zhang, X.; Wu, Y.; Lettieri, K.; Hammond, G.R.; Hong, Y. A polybasic domain in aPKC mediates Par6-dependent control of membrane targeting and kinase activity. J. Cell Biol. 2020, 219. [Google Scholar] [CrossRef]
- Chou, M.M.; Hou, W.; Johnson, J.; Graham, L.K.; Lee, M.H.; Chen, C.-S.; Newton, A.C.; Schaffhausen, B.S.; Toker, A. Regulation of protein kinase C ζ by PI 3-kinase and PDK-1. Curr. Biol. 1998, 8, 1069–1078. [Google Scholar] [CrossRef]
- Li, X.; Gao, T. mTORC 2 phosphorylates protein kinase Cζ to regulate its stability and activity. EMBO Rep. 2014, 15, 191–198. [Google Scholar] [CrossRef]
- Nakanishi, H.; Brewer, K.; Exton, J.H. Activation of the zeta isozyme of protein kinase C by phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 1993, 268, 13–16. [Google Scholar] [CrossRef]
- Wang, Y.M.; Seibenhener, M.L.; Vandenplas, M.L.; Wooten, M.W. Atypical PKC zeta is activated by ceramide, resulting in coactivation of NF-kappaB/JNK kinase and cell survival. J. Neurosci. Res. 1999, 55, 293–302. [Google Scholar] [CrossRef]
- Dowler, S.; Kular, G.; Alessi, D.R. Protein Lipid Overlay Assay. Sci. Signal. 2002, 2002, pl6. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Cambronero, J.; Morris, A.; Henkels, K. PLD Protein–Protein Interactions With Signaling Molecules and Modulation by PA. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2017; Volume 583, pp. 327–357. [Google Scholar] [CrossRef]
- O’Luanaigh, N.; Pardo, R.; Fensome, A.; Allen-Baume, V.; Jones, D.; Holt, M.R.; Cockcroft, S. Continual Production of Phosphatidic Acid by Phospholipase D Is Essential for Antigen-stimulated Membrane Ruffling in Cultured Mast Cells. Mol. Biol. Cell 2002, 13, 3730–3746. [Google Scholar] [CrossRef] [PubMed]
- Sajan, M.P.; Bandyopadhyay, G.; Kanoh, Y.; Standaert, M.L.; Quon, M.J.; Reed, B.C.; Dikic, I.; Farese, R.V. Sorbitol activates atypical protein kinase C and GLUT4 glucose transporter translocation/glucose transport through proline-rich tyrosine kinase-2, the extracellular signal-regulated kinase pathway and phospholipase D. Biochem. J. 2002, 362, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, T.; Frohman, M.A.; Matsuda, M.; Kiyokawa, E. Heterogeneity of Phosphatidic Acid Levels and Distribution at the Plasma Membrane in Living Cells as Visualized by a Förster Resonance Energy Transfer (FRET) Biosensor. J. Biol. Chem. 2010, 285, 35979–35987. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wang, Z.; Lu, M.; Yonekubo, Y.; Liang, X.; Zhang, Y.; Wu, P.; Zhou, Y.; Grinstein, S.; Hancock, J.F.; et al. Temporal Production of the Signaling Lipid Phosphatidic Acid by Phospholipase D2 Determines the Output of Extracellular Signal-Regulated Kinase Signaling in Cancer Cells. Mol. Cell. Biol. 2014, 34, 84–95. [Google Scholar] [CrossRef]
- De Matteis, M.A.; Di Campli, A.; Godi, A. The role of the phosphoinositides at the Golgi complex. Biochim. Biophys. Acta Bioenergy 2005, 1744, 396–405. [Google Scholar] [CrossRef]
- De Craene, J.-O.; Bertazzi, D.L.; Bär, S.; Friant, S. Phosphoinositides, Major Actors in Membrane Trafficking and Lipid Signaling Pathways. Int. J. Mol. Sci. 2017, 18, 634. [Google Scholar] [CrossRef]
- Phan, T.K.; Williams, S.; Bindra, G.K.; Lay, F.T.; Poon, I.K.H.; Hulett, M.D. Phosphoinositides: Multipurpose cellular lipids with emerging roles in cell death. Cell Death Differ. 2019, 26, 781–793. [Google Scholar] [CrossRef]
- Nascimbeni, A.C.; Codogno, P.; Morel, E. Phosphatidylinositol-3-phosphate in the regulation of autophagy membrane dynamics. FEBS J. 2017, 284, 1267–1278. [Google Scholar] [CrossRef]
- De Matteis, M.A.; Wilson, C.; D’Angelo, G. Phosphatidylinositol-4-phosphate: The Golgi and beyond. BioEssays 2013, 35, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Poli, A.; Zaurito, A.E.; Abdul, S.; Fiume, R.; Faenza, I.; Divecha, N. Phosphatidylinositol 5 Phosphate (PI5P): From Behind the Scenes to the Front (Nuclear) Stage. Int. J. Mol. Sci. 2019, 20, 2080. [Google Scholar] [CrossRef] [PubMed]
- Soloff, R.S.; Katayama, C.; Lin, M.Y.; Feramisco, J.R.; Hedrick, S.M. Targeted Deletion of Protein Kinase C λ Reveals a Distribution of Functions between the Two Atypical Protein Kinase C Isoforms. J. Immunol. 2004, 173, 3250–3260. [Google Scholar] [CrossRef] [PubMed]
- Murray, N.R.; Kalari, K.R.; Fields, A.P. Protein kinase Cι expression and oncogenic signaling mechanisms in cancer. J. Cell. Physiol. 2010, 226, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Parker, P.J.; Justilien, V.; Riou, P.; Linch, M.; Fields, A.P. Atypical Protein Kinase Cι as a human oncogene and therapeutic target. Biochem. Pharmacol. 2014, 88, 1–11. [Google Scholar] [CrossRef]
- Regala, R.P.; Weems, C.; Jamieson, L.; Khoor, A.; Edell, E.S.; Lohse, C.M.; Fields, A.P. Atypical Protein Kinase Cι Is an Oncogene in Human Non–Small Cell Lung Cancer. Cancer Res. 2005, 65, 8905–8911. [Google Scholar] [CrossRef]
- Eder, A.M.; Sui, X.; Rosen, D.G.; Nolden, L.K.; Cheng, K.W.; Lahad, J.P.; Kango-Singh, M.; Lu, K.H.; Warneke, C.L.; Atkinson, E.N.; et al. Atypical PKC contributes to poor prognosis through loss of apical-basal polarity and Cyclin E overexpression in ovarian cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 12519–12524. [Google Scholar] [CrossRef]
- Du, G.; Wang, J.-M.; Lu, J.-X.; Li, Q.; Ma, C.-Q.; Du, J.-T.; Zou, S.-Q. Expression of P-aPKC-ι, E-Cadherin, and β-Catenin Related to Invasion and Metastasis in Hepatocellular Carcinoma. Ann. Surg. Oncol. 2009, 16, 1578–1586. [Google Scholar] [CrossRef]
- Kojima, Y.; Akimoto, K.; Nagashima, Y.; Ishiguro, H.; Shirai, S.; Chishima, T.; Ichikawa, Y.; Ishikawa, T.; Sasaki, T.; Kubota, Y.; et al. The overexpression and altered localization of the atypical protein kinase C λ/ι in breast cancer correlates with the pathologic type of these tumors. Hum. Pathol. 2008, 39, 824–831. [Google Scholar] [CrossRef]
- Justilien, V.; Fields, A.P. Ect2 links the PKCι–Par6α complex to Rac1 activation and cellular transformation. Oncogene 2009, 28, 3597–3607. [Google Scholar] [CrossRef]
- Regala, R.P.; Weems, C.; Jamieson, L.; Copland, J.A.; Thompson, E.A.; Fields, A.P. Atypical Protein Kinase Cι Plays a Critical Role in Human Lung Cancer Cell Growth and Tumorigenicity. J. Biol. Chem. 2005, 280, 31109–31115. [Google Scholar] [CrossRef] [PubMed]
- Erdogan, E.; Lamark, T.; Stallings-Mann, M.; Jamieson, L.; Pellechia, M.; Thompson, E.A.; Johansen, T.; Fields, A.P. Aurothiomalate Inhibits Transformed Growth by Targeting the PB1 Domain of Protein Kinase Cι. J. Biol. Chem. 2006, 281, 28450–28459. [Google Scholar] [CrossRef] [PubMed]
- Frederick, L.; Matthews, J.; Jamieson, L.; Justilien, V.; Thompson, E.; Radisky, D.C.; Fields, A.P. Matrix metalloproteinase-10 is a critical effector of protein kinase Cι-Par6α-mediated lung cancer. Oncogene 2008, 27, 4841–4853. [Google Scholar] [CrossRef] [PubMed]
- Justilien, V.; Jameison, L.; Der, C.J.; Rossman, K.L.; Fields, A.P. Oncogenic Activity of Ect2 Is Regulated through Protein Kinase Cι-mediated Phosphorylation. J. Biol. Chem. 2010, 286, 8149–8157. [Google Scholar] [CrossRef]
- Aranda, V.; Haire, T.; Nolan, M.E.; Calarco, J.P.; Rosenberg, A.Z.; Fawcett, J.P.; Pawson, T.; Muthuswamy, S.K. Par6–aPKC uncouples ErbB2 induced disruption of polarized epithelial organization from proliferation control. Nat. Cell Biol. 2006, 8, 1235–1245. [Google Scholar] [CrossRef]
- Hernández, A.I.; Blace, N.; Crary, J.F.; Serrano, P.A.; Leitges, M.; Libien, J.M.; Weinstein, G.; Tcherapanov, A.; Sacktor, T.C. Protein Kinase Mζ Synthesis from a Brain mRNA Encoding an Independent Protein Kinase Cζ Catalytic Domain. J. Biol. Chem. 2003, 278, 40305–40316. [Google Scholar] [CrossRef]
- Sánchez-Gómez, P.; De Cárcer, G.; Sandoval, I.V.; Moscat, J.; Diaz-Meco, M.T. Localization of Atypical Protein Kinase C Isoforms into Lysosome-Targeted Endosomes through Interaction with p62. Mol. Cell. Biol. 1998, 18, 3069–3080. [Google Scholar] [CrossRef]
- Standaert, M.L.; Bandyopadhyay, G.; Kanoh, Y.; Sajan, M.P.; Farese, R.V. Insulin and PIP3Activate PKC-ζ by Mechanisms That Are Both Dependent and Independent of Phosphorylation of Activation Loop (T410) and Autophosphorylation (T560) Sites. Biochemistry 2001, 40, 249–255. [Google Scholar] [CrossRef]
- Fox, T.E.; Houck, K.L.; Oneill, S.M.; Nagarajan, M.; Stover, T.C.; Pomianowski, P.T.; Unal, O.; Yun, J.K.; Naides, S.J.; Kester, M. Ceramide Recruits and Activates Protein Kinase C ζ (PKCζ) within Structured Membrane Microdomains. J. Biol. Chem. 2007, 282, 12450–12457. [Google Scholar] [CrossRef]
- Bourbon, N.A.; Yun, J.K.; Kester, M. Ceramide Directly Activates Protein Kinase C ζ to Regulate a Stress-activated Protein Kinase Signaling Complex. J. Biol. Chem. 2000, 275, 35617–35623. [Google Scholar] [CrossRef]
- Chalfant, C.E.; Szulc, Z.; Roddy, P.; Bielawska, A.; Hannun, Y.A. The structural requirements for ceramide activation of serine-threonine protein phosphatases. J. Lipid Res. 2003, 45, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Pastor-Flores, D.; Schulze, J.O.; Bahí, A.; Süß, E.; Casamayor, A.; Biondi, R.M. Lipid regulators of Pkh2 in Candida albicans, the protein kinase ortholog of mammalian PDK1. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2016, 1861, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Heras-Martínez, G.D.L.; Calleja, V.; Bailly, R.; Dessolin, J.; Larijani, B.; Requejo-Isidro, J. A Complex Interplay of Anionic Phospholipid Binding Regulates 3′-Phosphoinositide-Dependent-Kinase-1 Homodimer Activation. Sci. Rep. 2019, 9, 1–18. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PKCZ | PKCι | |
|---|---|---|
| PI | − | − |
| PI(3)P | − | ++ |
| PI(4)P | − | ++ |
| PI(5)P | − | ++ |
| PI(3,4)P2 | − | − |
| PI(3,5)P2 | − | − |
| PI(4,5)P2 | − | − |
| PI(3,4,5)P3 | − | − |
| PA | ++ | ++ |
| LPA | − | − |
| PC | − | − |
| LPC | − | − |
| PS | ++ | ++ |
| PE | − | − |
| PG | + | + |
| DAG | − | − |
| S | − | − |
| S1P | − | − |
| SPC | − | − |
| PHS | − | − |
| CE | − | − |
| SM | − | − |
| SM4 | − | + |
| GM1 | − | − |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Velnati, S.; Centonze, S.; Girivetto, F.; Capello, D.; Biondi, R.M.; Bertoni, A.; Cantello, R.; Ragnoli, B.; Malerba, M.; Graziani, A.; et al. Identification of Key Phospholipids That Bind and Activate Atypical PKCs. Biomedicines 2021, 9, 45. https://doi.org/10.3390/biomedicines9010045
Velnati S, Centonze S, Girivetto F, Capello D, Biondi RM, Bertoni A, Cantello R, Ragnoli B, Malerba M, Graziani A, et al. Identification of Key Phospholipids That Bind and Activate Atypical PKCs. Biomedicines. 2021; 9(1):45. https://doi.org/10.3390/biomedicines9010045
Chicago/Turabian StyleVelnati, Suresh, Sara Centonze, Federico Girivetto, Daniela Capello, Ricardo M. Biondi, Alessandra Bertoni, Roberto Cantello, Beatrice Ragnoli, Mario Malerba, Andrea Graziani, and et al. 2021. "Identification of Key Phospholipids That Bind and Activate Atypical PKCs" Biomedicines 9, no. 1: 45. https://doi.org/10.3390/biomedicines9010045
APA StyleVelnati, S., Centonze, S., Girivetto, F., Capello, D., Biondi, R. M., Bertoni, A., Cantello, R., Ragnoli, B., Malerba, M., Graziani, A., & Baldanzi, G. (2021). Identification of Key Phospholipids That Bind and Activate Atypical PKCs. Biomedicines, 9(1), 45. https://doi.org/10.3390/biomedicines9010045