DNA Methylomes and Epigenetic Age Acceleration Associations with Poor Metabolic Control in T1D

 ,
,  , ,
, ,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Human Subjects
2.2. External Databases
2.3. DNA Extraction and Illumina Infinium MethylationEPICBeadChip
2.4. Methylation Data Preprocessing and Analyses
2.5. Bisulfite Pyrosequencing
3. Results
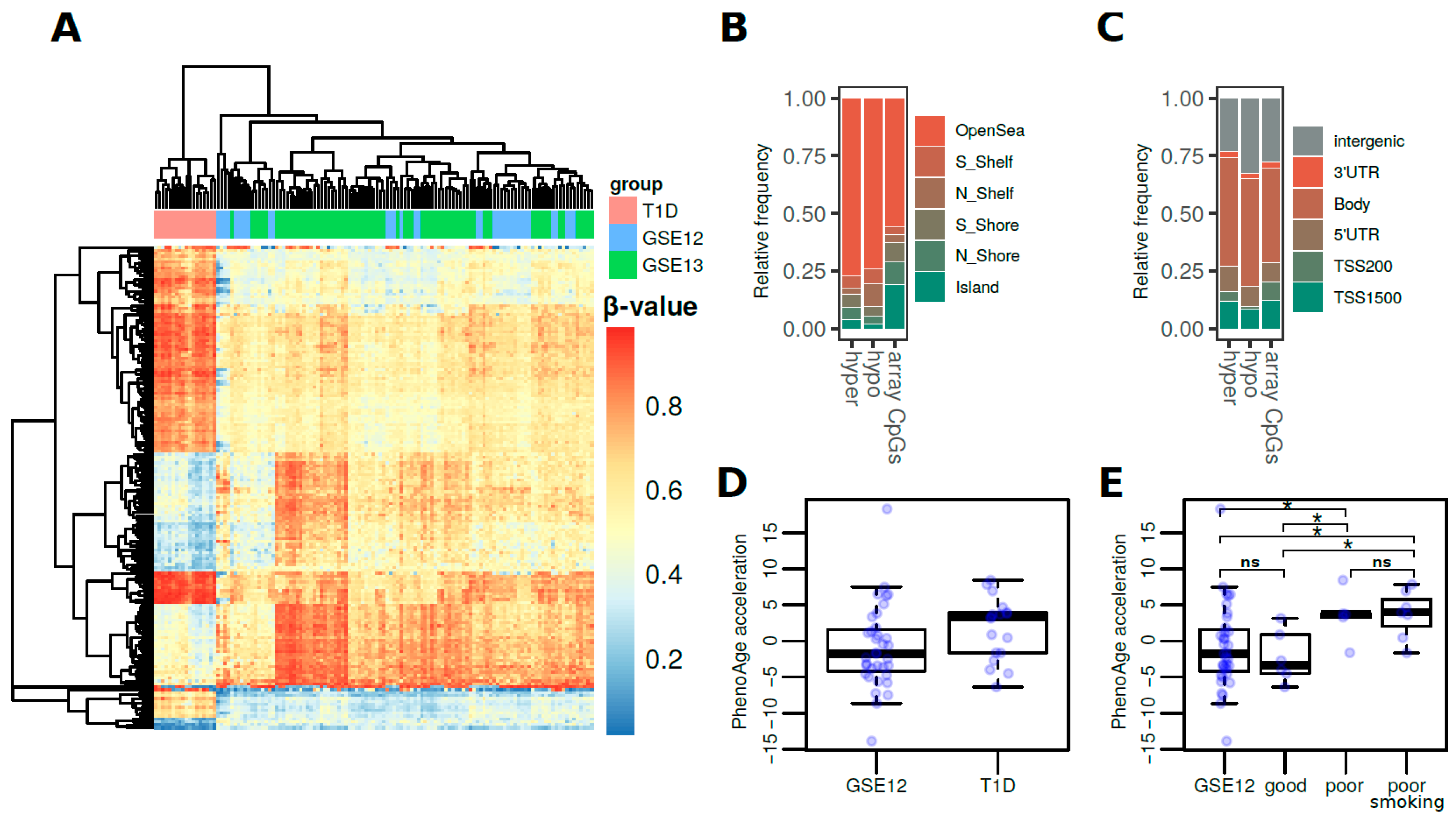
3.1. DNA Methylation Differences Associated with T1D in Whole Blood
3.2. DNA Methylation Age Is Accelerated in T1D and Is Related to Glycemic Control
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DMPs | Differentially methylated probes |
| DNAm | DNA methylation |
| HbA1c | Glycated hemoglobin |
| SVA | Surrogate variable analysis |
| T1D | Type 1 diabetes |
References
- Katsarou, A.; Gudbjornsdottir, S.; Rawshani, A.; Dabelea, D.; Bonifacio, E.; Anderson, B.J.; Jacobsen, L.M.; Schatz, D.A.; Lernmark, A. Type 1 diabetes mellitus. Nat. Rev. Dis. Primers 2017, 3, 17016. [Google Scholar] [CrossRef] [PubMed]
- Pociot, F.; Lernmark, A. Genetic risk factors for type 1 diabetes. Lancet 2016, 387, 2331–2339. [Google Scholar] [CrossRef]
- Misra, A.; Bloomgarden, Z. Metabolic memory Evolving concepts. J. Diabetes 2018, 10, 186–187. [Google Scholar] [CrossRef] [PubMed]
- Pirola, L.; Balcerczyk, A.; Okabe, J.; El-Osta, A. Epigenetic phenomena linked to diabetic complications. Nat. Rev. Endocrinol. 2010, 6, 665–675. [Google Scholar] [CrossRef]
- Longo, M.; Bellastella, G.; Maiorino, M.I.; Meier, J.J.; Esposito, K.; Giugliano, D. Diabetes and Aging From Treatment Goals to Pharmacologic Therapy. Front. Endocrinol. 2019, 10, 45. [Google Scholar] [CrossRef]
- Berezin, A. Metabolic memory phenomenon in diabetes mellitus Achieving and perspectives. Diabetes Metab. Syndr. 2016, 10 (Suppl. 1), S176–S183. [Google Scholar] [CrossRef]
- Testa, R.; Bonfigli, A.R.; Prattichizzo, F.; La Sala, L.; De Nigris, V.; Ceriello, A. The “Metabolic Memory” Theory and the Early Treatment of Hyperglycemia in Prevention of Diabetic Complications. Nutrients 2017, 9, 437. [Google Scholar] [CrossRef]
- Feil, R.; Fraga, M.F. Epigenetics and the environment emerging patterns and implications. Nat. Rev. Genet. 2012, 13, 97–109. [Google Scholar] [CrossRef]
- Jerram, S.T.; Dang, M.N.; Leslie, R.D. The Role of Epigenetics in Type 1 Diabetes. Curr. Diab. Rep. 2017, 17, 89. [Google Scholar] [CrossRef]
- Kindt, A.S.D.; Fuerst, R.W.; Knoop, J.; Laimighofer, M.; Telieps, T.; Hippich, M.; Woerheide, M.A.; Wahl, S.; Wilson, R.; Sedlmeier, E.M.; et al. Allele-specific methylation of type 1 diabetes susceptibility genes. J. Autoimmun. 2018, 89, 63–74. [Google Scholar] [CrossRef]
- Paul, D.S.; Teschendorff, A.E.; Dang, M.A.; Lowe, R.; Hawa, M.I.; Ecker, S.; Beyan, H.; Cunningham, S.; Fouts, A.R.; Ramelius, A.; et al. Increased DNA methylation variability in type 1 diabetes across three immune effector cell types. Nat. Commun. 2016, 7, 13555. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Richardson, T.G.; McArdle, W.L.; Relton, C.L.; Gillespie, K.M.; Suderman, M.; Hemani, G. Identification of loci where DNA methylation potentially mediates genetic risk of type 1 diabetes. J. Autoimmun. 2018, 93, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Miao, F.; Chen, Z.; Genuth, S.; Paterson, A.; Zhang, L.; Wu, X.; Li, S.M.; Cleary, P.; Riggs, A.; Harlan, D.M.; et al. Evaluating the role of epigenetic histone modifications in the metabolic memory of type 1 diabetes. Diabetes 2014, 63, 1748–1762. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.L.; Bandinelli, S.; Hou, L.; Baccarelli, A.A.; Stewart, J.D.; Li, Y.; et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging 2018, 10, 573–591. [Google Scholar] [CrossRef]
- Fransquet, P.D.; Wrigglesworth, J.; Woods, R.L.; Ernst, M.E.; Ryan, J. The epigenetic clock as a predictor of disease and mortality risk a systematic review and meta-analysis. Clin. Epigenet. 2019, 11, 62. [Google Scholar] [CrossRef]
- Roshandel, D.; Chen, Z.; Canty, A.J.; Bull, S.B.; Natarajan, R.; Paterson, A.D.; Group DER. DNA methylation age calculators reveal association with diabetic neuropathy in type 1 diabetes. Clin. Epigenet. 2020, 12, 52. [Google Scholar] [CrossRef]
- Johnson, R.K.; Vanderlinden, L.A.; Dong, F.; Carry, P.M.; Seifert, J.; Waugh, K.; Shorrosh, H.; Fingerlin, T.; Frohnert, B.I.; Yang, I.V.; et al. Longitudinal DNA methylation differences precede type 1 diabetes. Sci. Rep. 2020, 10, 3721. [Google Scholar] [CrossRef]
- Zaimi, I.; Pei, D.; Koestler, D.C.; Marsit, C.J.; De Vivo, I.; Tworoger, S.S.; Shields, A.E.; Kelsey, K.T.; Michaud, D.S. Variation in DNA methylation of human blood over a 1-year period using the Illumina MethylationEPIC array. Epigenetics 2018, 13, 1056–1071. [Google Scholar] [CrossRef]
- Novakovic, B.; Lewis, S.; Halliday, J.; Kennedy, J.; Burgner, D.P.; Czajko, A.; Kim, B.; Sexton-Oates, A.; Juonala, M.; Hammarberg, K.; et al. Assisted reproductive technologies are associated with limited epigenetic variation at birth that largely resolves by adulthood. Nat. Commun. 2019, 10, 3922. [Google Scholar] [CrossRef]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef]
- Triche, T.J., Jr.; Weisenberger, D.J.; Van Den Berg, D.; Laird, P.W.; Siegmund, K.D. Low-level processing of Illumina Infinium DNA Methylation BeadArrays. Nucleic Acids Res. 2013, 41, e90. [Google Scholar] [CrossRef] [PubMed]
- Teschendorff, A.E.; Marabita, F.; Lechner, M.; Bartlett, T.; Tegner, J.; Gomez-Cabrero, D.; Beck, S. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics 2013, 29, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.A.; Lemire, M.; Choufani, S.; Butcher, D.T.; Grafodatskaya, D.; Zanke, B.W.; Gallinger, S.; Hudson, T.J.; Weksberg, R. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 2013, 8, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef]
- Lu, A.T.; Quach, A.; Wilson, J.G.; Reiner, A.P.; Aviv, A.; Raj, K.; Hou, L.; Baccarelli, A.A.; Li, Y.; Stewart, J.D.; et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging 2019, 11, 303–327. [Google Scholar] [CrossRef]
- Liu, Z.; Kuo, P.L.; Horvath, S.; Crimmins, E.; Ferrucci, L.; Levine, M. A new aging measure captures morbidity and mortality risk across diverse subpopulations from NHANES IV A cohort study. PLoS Med. 2018, 15, e1002718. [Google Scholar] [CrossRef]
- Elboudwarej, E.; Cole, M.; Briggs, F.B.; Fouts, A.; Fain, P.R.; Quach, H.; Quach, D.; Sinclair, E.; Criswell, L.A.; Lane, J.A.; et al. Hypomethylation within gene promoter regions and type 1 diabetes in discordant monozygotic twins. J. Autoimmun. 2016, 68, 23–29. [Google Scholar] [CrossRef]


{kind=link}
| n | Age (Years) | Male/Female | T1D Duration (Years) | Age of Onset (Years) | HbA1c % (mmol/mol) | |
|---|---|---|---|---|---|---|
| T1D patients | ||||||
| Good metabolic control | 6 | 43.2 ± 10.9 | 2 / 4 | 21.8 ± 14.4 | 21.3 ± 4.4 | 6.1 ± 0.5 (44.3 ± 2.4) |
| Poor metabolic control | 12 | 48.3 ± 7.0 | 4 / 8 | 24.8 ± 9.3 | 23.5 ± 8.1 | 8.7 ± 0.6 (68.9 ± 8.8) |
| Poor metabolic control + nonsmoker | 5 | 48.0 ± 6.2 | 1 / 4 | 28.2 ± 11.5 | 19.8 ± 6.9 | 8.4 ± 0.6 (61.4 ± 5.9) |
| Poor metabolic control + smoker | 7 | 48.4 ± 8.0 | 3 / 4 | 22.3 ± 7.3 | 26.1 ± 8.4 | 8.8 ± 0.7 (74.3 ± 6.1) |
| Controls | ||||||
| GSE123914 | 35 | 58.5 ± 3.5 | 0/35 | |||
| GSE131433 | 74 | 18-35 | 23/51 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez, R.F.; Fernandez-Morera, J.L.; Romano-Garcia, J.; Menendez-Torre, E.; Delgado-Alvarez, E.; Fraga, M.F.; Fernandez, A.F. DNA Methylomes and Epigenetic Age Acceleration Associations with Poor Metabolic Control in T1D. Biomedicines 2021, 9, 13. https://doi.org/10.3390/biomedicines9010013
Pérez RF, Fernandez-Morera JL, Romano-Garcia J, Menendez-Torre E, Delgado-Alvarez E, Fraga MF, Fernandez AF. DNA Methylomes and Epigenetic Age Acceleration Associations with Poor Metabolic Control in T1D. Biomedicines. 2021; 9(1):13. https://doi.org/10.3390/biomedicines9010013
Chicago/Turabian StylePérez, Raúl F, Juan Luis Fernandez-Morera, Judit Romano-Garcia, Edelmiro Menendez-Torre, Elias Delgado-Alvarez, Mario F Fraga, and Agustin F Fernandez. 2021. "DNA Methylomes and Epigenetic Age Acceleration Associations with Poor Metabolic Control in T1D" Biomedicines 9, no. 1: 13. https://doi.org/10.3390/biomedicines9010013
APA StylePérez, R. F., Fernandez-Morera, J. L., Romano-Garcia, J., Menendez-Torre, E., Delgado-Alvarez, E., Fraga, M. F., & Fernandez, A. F. (2021). DNA Methylomes and Epigenetic Age Acceleration Associations with Poor Metabolic Control in T1D. Biomedicines, 9(1), 13. https://doi.org/10.3390/biomedicines9010013