Isocitrate Dehydrogenase Mutations in Glioma: Genetics, Biochemistry, and Clinical Indications



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Genetics of Glioma
2.1. The Prevalence of IDH Mutation in Glioma (Lower-Grade Glioma and GBM)
2.2. Hypermethylation Phenotype in a Subset of IDH-Mutated Gliomas
3. Biochemistry of IDH
3.1. Wild-Type IDH
3.2. IDH Neomorphic Activity and D-2-HG
3.3. Distinctive Biological Patterns of IDH-Mutated Glioma
3.4. Histones and DNA Demethylases
3.5. DNA Repair Enzymes
3.6. Metabolic Enzymes
3.7. Anti-Oxidative Pathways
4. Clinical Indications Involving the Discovery of IDH-Mutated Glioma
4.1. Clinical Classification of Gliomas
4.2. Radiology—D-2-HG Imaging
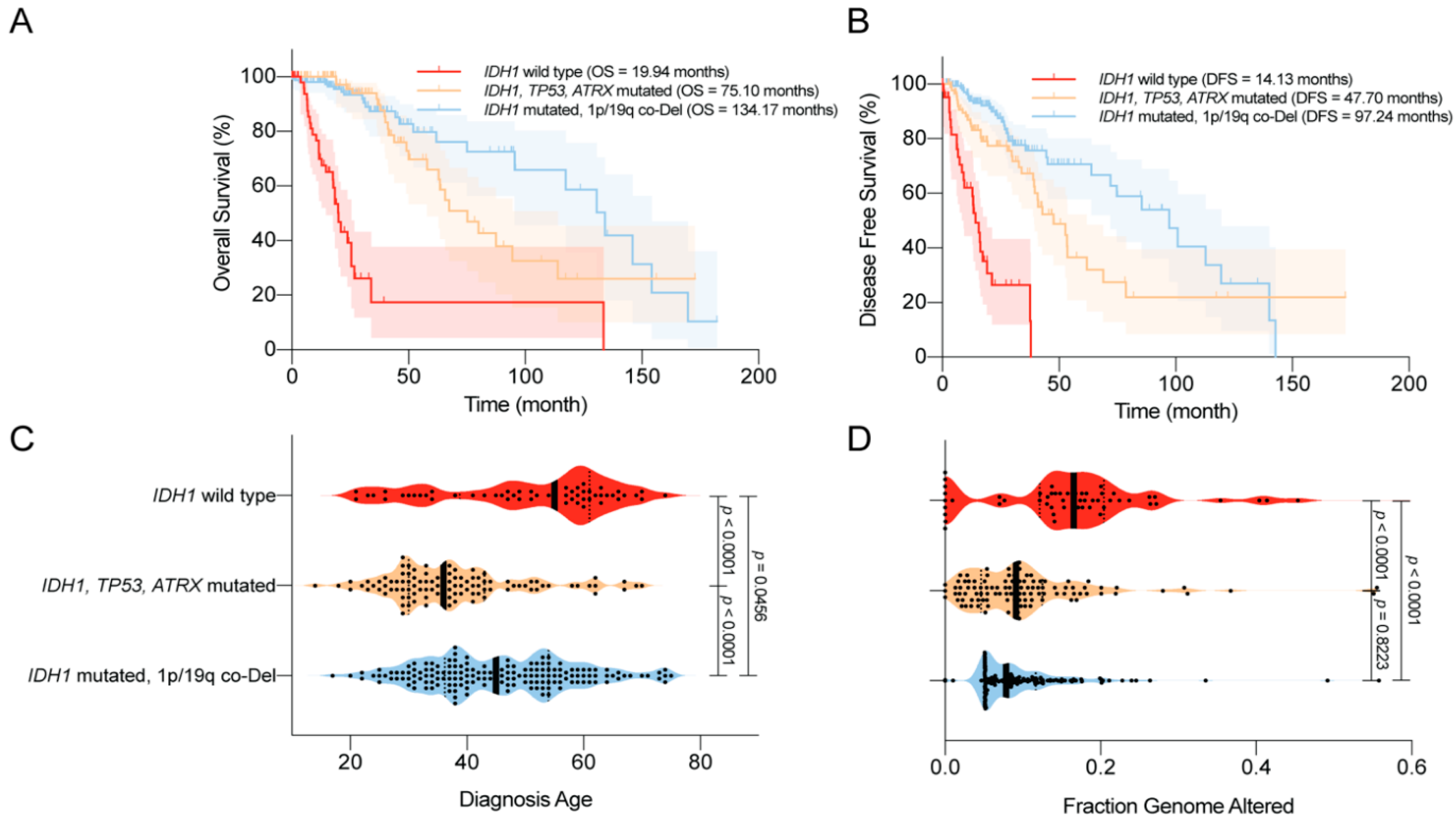
4.3. Disease Outcomes—Prolonged Survival
4.4. Complications—Epilepsy and Secondary GBM
4.5. Sensitivity to Radiotherapy and Chemotherapy
5. Novel Molecular Targeting for IDH-Mutated Glioma
5.1. IDH Mutant Inhibitors
5.2. Targeting Hypermethylation Phenotype
5.3. Targeting DNA Repair Pathways
5.4. Targeting Anti-Oxidative Pathways
5.5. Targeting Metabolic Reprogramming
5.6. Immunotherapies
6. Conclusions—Current Knowledge and Opportunities for the Future
Author Contributions
Funding
Conflicts of Interest
References
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.-M.; Gallia, G.L.; et al. An Integrated Genomic Analysis of Human Glioblastoma Multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1andIDH2Mutations in Gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Paschka, P.; Schlenk, R.F.; Gaidzik, V.I.; Habdank, M.; Krönke, J.; Bullinger, L.; Späth, D.; Kayser, S.; Zucknick, M.; Götze, K.; et al. IDH1 and IDH2 Mutations Are Frequent Genetic Alterations in Acute Myeloid Leukemia and Confer Adverse Prognosis in Cytogenetically Normal Acute Myeloid Leukemia With NPM1 Mutation Without FLT3 Internal Tandem Duplication. J. Clin. Oncol. 2010, 28, 3636–3643. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R.; Ding, L.; Dooling, D.J.; Larson, D.E.; McLellan, M.D.; Chen, K.; Koboldt, D.C.; Fulton, R.S.; Delehaunty, K.D.; McGrath, S.; et al. Recurring Mutations Found by Sequencing an Acute Myeloid Leukemia Genome. N. Engl. J. Med. 2009, 361, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Borger, D.R.; Tanabe, K.K.; Fan, K.C.; Lopez, H.U.; Fantin, V.R.; Straley, K.S.; Schenkein, D.P.; Hezel, A.F.; Ancukiewicz, M.; Liebman, H.M.; et al. Frequent Mutation of Isocitrate Dehydrogenase (IDH)1 and IDH2 in Cholangiocarcinoma Identified Through Broad-Based Tumor Genotyping. Oncologist 2011, 17, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Travins, J.; DelaBarre, B.; Lacronique, V.; Schalm, S.; Hansen, E.; Straley, K.; Kernytsky, A.; Liu, W.; Gliser, C.; et al. Targeted Inhibition of Mutant IDH2 in Leukemia Cells Induces Cellular Differentiation. Science 2013, 340, 622–626. [Google Scholar] [CrossRef]
- Amary, M.F.; Bácsi, K.; Maggiani, F.; Damato, S.; Halai, D.; Berisha, F.; Pollock, R.; O’Donnell, P.; Grigoriadis, A.; Diss, T.; et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J. Pathol. 2011, 224, 334–343. [Google Scholar] [CrossRef]
- Lopez, G.Y.; Reitman, Z.J.; Solomon, D.; Waldman, T.; Bigner, D.D.; McLendon, R.E.; A Rosenberg, S.; Samuels, Y.; Yan, H. IDH1 mutation identified in one human melanoma metastasis, but not correlated with metastases to the brain. Biochem. Biophys. Res. Commun. 2010, 398, 585–587. [Google Scholar] [CrossRef]
- Shibata, T.; Kokubu, A.; Miyamoto, M.; Sasajima, Y.; Yamazaki, N. Mutant IDH1 Confers an in Vivo Growth in a Melanoma Cell Line with BRAF Mutation. Am. J. Pathol. 2011, 178, 1395–1402. [Google Scholar] [CrossRef]
- Yan, H.; Bigner, D.D.; Velculescu, V.E.; Parsons, D.W. Mutant metabolic enzymes are at the origin of gliomas. Cancer Res. 2009, 69, 9157–9159. [Google Scholar] [CrossRef]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The Common Feature of Leukemia-Associated IDH1 and IDH2 Mutations Is a Neomorphic Enzyme Activity Converting α-Ketoglutarate to 2-Hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; Jin, S.; Su, S.M. IDH mutations in glioma and acute myeloid leukemia. Trends Mol. Med. 2010, 16, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Prensner, J.; Chinnaiyan, A.M. Metabolism unhinged: IDH mutations in cancer. Nat. Med. 2011, 17, 291–293. [Google Scholar] [CrossRef] [PubMed]
- Losman, J.-A.; Kaelin, W.G. What a difference a hydroxyl makes: Mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013, 27, 836–852. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.-H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.-T.; et al. Oncometabolite 2-Hydroxyglutarate Is a Competitive Inhibitor of α-Ketoglutarate-Dependent Dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef]
- Chowdhury, R.; Yeoh, K.K.; Tian, Y.-M.; Hillringhaus, L.; Bagg, E.; Rose, N.R.; Leung, I.K.H.; Li, X.S.; Woon, E.C.Y.; Yang, M.; et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011, 12, 463–469. [Google Scholar] [CrossRef]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, Ş.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef]
- Turcan, Ş.; Rohle, D.; Goenka, A.; A Walsh, L.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.M.; Lu, C.; Ward, P.S.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef]
- Wang, P.; Wu, J.; Ma, S.; Zhang, L.; Yao, J.; Hoadley, K.A.; Wilkerson, M.D.; Perou, C.M.; Guan, K.-L.; Ye, D.; et al. Oncometabolite D-2-Hydroxyglutarate Inhibits ALKBH DNA Repair Enzymes and Sensitizes IDH Mutant Cells to Alkylating Agents. Cell Rep. 2015, 13, 2353–2361. [Google Scholar] [CrossRef]
- Inoue, S.; Li, W.Y.; Tseng, A.; Beerman, I.; Elia, A.J.; Bendall, S.C.; Lemonnier, F.; Kron, K.J.; Cescon, D.W.; Hao, Z.; et al. Mutant IDH1 Downregulates ATM and Alters DNA Repair and Sensitivity to DNA Damage Independent of TET2. Cancer Cell 2016, 30, 337–348. [Google Scholar] [CrossRef]
- Wang, Y.; Wild, A.T.; Turcan, S.; Wu, W.H.; Sigel, C.; Klimstra, D.S.; Ma, X.; Gong, Y.; Holland, E.C.; Huse, J.T.; et al. Targeting therapeutic vulnerabilities with PARP inhibition and radiation in IDH-mutant gliomas and cholangiocarcinomas. Sci. Adv. 2020, 6, eaaz3221. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Kwintkiewicz, J.; Liu, Y.; Tech, K.; Frady, L.N.; Su, Y.-T.; Bautista, W.; Moon, S.I.; Macdonald, J.; Ewend, M.G.; et al. Chemosensitivity of IDH1-Mutated Gliomas Due to an Impairment in PARP1-Mediated DNA Repair. Cancer Res. 2017, 77, 1709–1718. [Google Scholar] [CrossRef] [PubMed]
- Sulkowski, P.L.; Corso, C.D.; Robinson, N.D.; Scanlon, S.E.; Purshouse, K.; Bai, H.; Liu, Y.; Sundaram, R.K.; Hegan, D.C.; Fons, N.R.; et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci. Transl. Med. 2017, 9, eaal2463. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Sun, B.; Shi, W.; Zuo, H.; Cui, D.; Ni, L.; Chen, J. Decreasing GSH and increasing ROS in chemosensitivity gliomas with IDH1 mutation. Tumor Boil. 2014, 36, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Fu, X.; Liu, Y.; Yu, D.; Cai, S.J.; Yang, C. Blockade of Glutathione Metabolism in IDH1-Mutated Glioma. Mol. Cancer Ther. 2019, 19, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Gelman, S.J.; Naser, F.; Mahieu, N.G.; McKenzie, L.D.; Dunn, G.P.; Chheda, M.G.; Patti, G.J. Consumption of NADPH for 2-HG Synthesis Increases Pentose Phosphate Pathway Flux and Sensitizes Cells to Oxidative Stress. Cell Rep. 2018, 22, 512–522. [Google Scholar] [CrossRef]
- Yu, D.; Liu, Y.; Zhou, Y.; Ruiz-Rodado, V.; Larion, M.; Xu, G.; Yang, C. Triptolide suppresses IDH1-mutated malignancy via Nrf2-driven glutathione metabolism. Proc. Natl. Acad. Sci. USA 2020, 117, 9964–9972. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, Y.; Celiku, O.; Li, A.; Wu, Q.; Zhou, Y.; Yang, C. Targeting IDH1-Mutated Malignancies with NRF2 Blockade. J. Natl. Cancer Inst. 2019, 111, 1033–1041. [Google Scholar] [CrossRef]
- McBrayer, S.K.; Mayers, J.R.; DiNatale, G.J.; Shi, D.D.; Khanal, J.; Chakraborty, A.A.; Sarosiek, K.; Briggs, K.J.; Robbins, A.K.; Sewastianik, T.; et al. Transaminase Inhibition by 2-Hydroxyglutarate Impairs Glutamate Biosynthesis and Redox Homeostasis in Glioma. Cell 2018, 175, 101–116.e25. [Google Scholar] [CrossRef]
- Suzuki, H.; Aoki, K.; Chiba, K.; Sato, Y.; Shiozawa, Y.; Shiraishi, Y.; Shimamura, T.; Niida, A.; Motomura, K.; Ohka, F.; et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nat. Genet. 2015, 47, 458–468. [Google Scholar] [CrossRef]
- Philip, B.; Yu, D.X.; Silvis, M.R.; Shin, C.H.; Robinson, J.P.; Robinson, G.L.; Welker, A.E.; Angel, S.N.; Tripp, S.R.; Sonnen, J.A.; et al. Mutant IDH1 Promotes Glioma Formation In Vivo. Cell Rep. 2018, 23, 1553–1564. [Google Scholar] [CrossRef] [PubMed]
- Bardella, C.; Al-Dalahmah, O.; Krell, D.; Brazauskas, P.; Al-Qahtani, K.; Tomkova, M.; Adam, J.; Serres, S.; Lockstone, H.; Freeman-Mills, L.; et al. Expression of Idh1R132H in the Murine Subventricular Zone Stem Cell Niche Recapitulates Features of Early Gliomagenesis. Cancer Cell 2016, 30, 578–594. [Google Scholar] [CrossRef] [PubMed]
- Chung, R.; Whaley, J.; Kley, N.; Anderson, K.; Louis, D.; Menon, A.; Hettlich, C.; Freiman, R.; Hedley-Whyte, E.T.; Martuza, R.; et al. TP53 Gene Mutations and 17p Deletions in Human Astrocytomas. Genes Chromosom. Cancer 1991, 3, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, B.K.; E McLendon, R.; E Herndon, J.; Friedman, H.S.; Friedman, A.H.; Bigner, D.D.; Bigner, S.H. Alterations of the TP53 gene in human gliomas. Cancer Res. 1994, 54, 1324–1330. [Google Scholar]
- Killela, P.J.; Pirozzi, C.J.; Reitman, Z.J.; Jones, S.; Rasheed, B.A.; Lipp, E.S.; Friedman, H.; Friedman, A.H.; He, Y.; McLendon, R.E.; et al. The genetic landscape of anaplastic astrocytoma. Oncotarget 2013, 5, 1452–1457. [Google Scholar] [CrossRef]
- Killela, P.J.; Reitman, Z.J.; Jiao, Y.; Bettegowda, C.; Agrawal, N.; Diaz, L.A.; Friedman, A.H.; Friedman, H.; Gallia, G.L.; Giovanella, B.C.; et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. USA 2013, 110, 6021–6026. [Google Scholar] [CrossRef]
- Bettegowda, C.; Agrawal, N.; Jiao, Y.; Sausen, M.; Wood, L.D.; Hruban, R.H.; Rodriguez, F.J.; Cahill, D.P.; McLendon, R.; Riggins, G.; et al. Mutations in CIC and FUBP1 Contribute to Human Oligodendroglioma. Science 2011, 333, 1453–1455. [Google Scholar] [CrossRef]
- Jiao, Y.; Killela, P.J.; Reitman, Z.J.; Rasheed, B.A.; Heaphy, C.M.; De Wilde, R.F.; Rodriguez, F.J.; Rosemberg, S.; Oba-Shinjo, S.M.; Marie, S.K.N.; et al. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget 2012, 3, 709–722. [Google Scholar] [CrossRef]
- Cairncross, J.G.; Ueki, K.; Zlatescu, M.C.; Lisle, D.K.; Finkelstein, D.M.; Hammond, R.R.; Silver, J.S.; Stark, P.C.; Macdonald, D.R.; Ino, Y.; et al. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J. Natl. Cancer Inst. 1998, 90, 1473–1479. [Google Scholar] [CrossRef]
- Labussière, M.; Idbaih, A.; Wang, X.; Marie, Y.; Boisselier, B.; Falet, C.; Paris, S.; Laffaire, J.; Carpentier, C.; Criniere, E.; et al. All the 1p19q codeleted gliomas are mutated on IDH1 or IDH2. Neurology 2010, 74, 1886–1890. [Google Scholar] [CrossRef]
- Sahm, F.; Koelsche, C.; Meyer, J.; Pusch, S.; Lindenberg, K.; Mueller, W.; Herold-Mende, C.; Von Deimling, A.; Hartmann, C. CIC and FUBP1 mutations in oligodendrogliomas, oligoastrocytomas and astrocytomas. Acta Neuropathol. 2012, 123, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Killela, P.J.; Pirozzi, C.J.; Healy, P.; Reitman, Z.J.; Lipp, E.S.; Rasheed, B.A.; Yang, R.; Diplas, B.; Wang, Z.; Greer, P.K.; et al. Mutations in IDH1, IDH2, and in the TERT promoter define clinically distinct subgroups of adult malignant gliomas. Oncotarget 2014, 5, 1515–1525. [Google Scholar] [CrossRef] [PubMed]
- Brito, C.; Azevedo, A.; Esteves, S.; Marques, A.R.; Martins, C.; Costa, I.; Mafra, M.; Marques, J.M.B.; Roque, L.; Pojo, M. Clinical insights gained by refining the 2016 WHO classification of diffuse gliomas with: EGFR amplification, TERT mutations, PTEN deletion and MGMT methylation. BMC Cancer 2019, 19, 968. [Google Scholar] [CrossRef] [PubMed]
- Noorani, I.; De La Rosa, J.; Choi, Y.H.; Strong, A.; Ponstingl, H.; Vijayabaskar, M.S.; Lee, J.; Lee, E.; Richard-Londt, A.; Friedrich, M.; et al. PiggyBac mutagenesis and exome sequencing identify genetic driver landscapes and potential therapeutic targets of EGFR-mutant gliomas. Genome Boil. 2020, 21, 1–36. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, M.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Skanderup, A.J.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data: Figure 1. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Skanderup, A.J.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG Island Methylator Phenotype that Defines a Distinct Subgroup of Glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef]
- Christensen, B.C.; Smith, A.A.; Zheng, S.; Koestler, D.C.; Houseman, E.A.; Marsit, C.J.; Wiemels, J.L.; Nelson, H.; Karagas, M.R.; Wrensch, M.R.; et al. DNA Methylation, Isocitrate Dehydrogenase Mutation, and Survival in Glioma. J. Natl. Cancer Inst. 2011, 103, 143–153. [Google Scholar] [CrossRef]
- Turcan, Ş.; Makarov, V.; Taranda, J.; Wang, Y.; Fabius, A.W.M.; Wu, W.; Zheng, Y.; El-Amine, N.; Haddock, S.; Nanjangud, G.; et al. Mutant-IDH1-dependent chromatin state reprogramming, reversibility, and persistence. Nat. Genet. 2017, 50, 62–72. [Google Scholar] [CrossRef]
- De Souza, C.F.; Sabedot, T.S.; Malta, T.M.; Stetson, L.; Morozova, O.; Sokolov, A.; Laird, P.W.; Wiznerowicz, M.; Iavarone, A.; Snyder, J.; et al. A Distinct DNA Methylation Shift in a Subset of Glioma CpG Island Methylator Phenotypes during Tumor Recurrence. Cell Rep. 2018, 23, 637–651. [Google Scholar] [CrossRef] [PubMed]
- Adler, E.; Euler, H.V.; Günther, G.; Plass, M. isoCitric dehydrogenase and glutamic acid synthesis in animal tissues. Biochem. J. 1939, 33, 1028–1045. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Liu, Y.; Cai, S.J.; Qian, M.; Ding, J.; Larion, M.; Gilbert, M.R.; Yang, C. IDH mutation in glioma: Molecular mechanisms and potential therapeutic targets. Br. J. Cancer 2020, 122, 1580–1589. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Ma, T.; Zhang, T.; Zhu, H.; Zhang, J.; Liu, Y.; Ding, J. Molecular basis for the function of the αβ heterodimer of human NAD-dependent isocitrate dehydrogenase. J. Boil. Chem. 2019, 294, 16214–16227. [Google Scholar] [CrossRef]
- Rendina, A.R.; Pietrak, B.; Smallwood, A.; Zhao, H.J.; Qi, H.; Quinn, C.; Adams, N.D.; Concha, N.; Duraiswami, C.; Thrall, S.H.; et al. Mutant IDH1 Enhances the Production of 2-Hydroxyglutarate Due to Its Kinetic Mechanism. Biochemistry 2013, 52, 4563–4577. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2018, 47, D941–D947. [Google Scholar] [CrossRef]
- Xu, X.; Zhao, J.; Xu, Z.; Peng, B.; Huang, Q.; Arnold, E.; Ding, J. Structures of Human Cytosolic NADP-dependent Isocitrate Dehydrogenase Reveal a Novel Self-regulatory Mechanism of Activity. J. Boil. Chem. 2004, 279, 33946–33957. [Google Scholar] [CrossRef]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2010, 465, 966. [Google Scholar] [CrossRef]
- Jin, G.; Reitman, Z.J.; Spasojevic, I.; Batinic-Haberle, I.; Yang, J.; Schmidt-Kittler, O.; Bigner, D.D.; Yan, H. 2-Hydroxyglutarate Production, but Not Dominant Negative Function, Is Conferred by Glioma-Derived NADP+-Dependent Isocitrate Dehydrogenase Mutations. PLoS ONE 2011, 6, e16812. [Google Scholar] [CrossRef]
- Laukka, T.; Myllykoski, M.; Looper, R.E.; Koivunen, P. Cancer-associated 2-oxoglutarate analogues modify histone methylation by inhibiting histone lysine demethylases. J. Mol. Boil. 2018, 430, 3081–3092. [Google Scholar] [CrossRef]
- Janke, R.; Iavarone, A.T.; Rine, J. Oncometabolite D-2-Hydroxyglutarate enhances gene silencing through inhibition of specific H3K36 histone demethylases. eLife 2017, 6, e22451. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; VasanthaKumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, W.A.; Drier, Y.; Liau, B.B.; Gillespie, S.M.; Venteicher, A.S.; Stemmer-Rachamimov, A.O.; Suvà, M.L.; Bernstein, B.E. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 2015, 529, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef]
- Polo, S.; Jackson, S.P. Dynamics of DNA damage response proteins at DNA breaks: A focus on protein modifications. Genes Dev. 2011, 25, 409–433. [Google Scholar] [CrossRef]
- Ohba, S.; Mukherjee, J.; See, W.L.; Pieper, R.O. Mutant IDH1-Driven Cellular Transformation Increases RAD51-Mediated Homologous Recombination and Temozolomide Resistance. Cancer Res. 2014, 74, 4836–4844. [Google Scholar] [CrossRef]
- Núñez, F.J.; Mendez, F.M.; Kadiyala, P.; AlGhamri, M.S.; Savelieff, M.G.; Garcia-Fabiani, M.B.; Haase, S.; Koschmann, C.; Calinescu, A.-A.; Kamran, N.; et al. IDH1-R132H acts as a tumor suppressor in glioma via epigenetic up-regulation of the DNA damage response. Sci. Transl. Med. 2019, 11, eaaq1427. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, Y.; Pang, Y.; Pacak, K.; Yang, C. Double-barreled gun: Combination of PARP inhibitor with conventional chemotherapy. Pharmacol. Ther. 2018, 188, 168–175. [Google Scholar] [CrossRef]
- Sulkowski, P.L.; Oeck, S.; Dow, J.; Economos, N.G.; Mirfakhraie, L.; Liu, Y.; Noronha, K.; Bao, X.; Li, J.; Shuch, B.M.; et al. Oncometabolites suppress DNA repair by disrupting local chromatin signalling. Nature 2020, 582, 586–591. [Google Scholar] [CrossRef]
- Grassian, A.R.; Parker, S.J.; Davidson, S.M.; Divakaruni, A.S.; Green, C.R.; Zhang, X.; Slocum, K.L.; Pu, M.; Lin, F.; Vickers, C.; et al. IDH1 mutations alter citric acid cycle metabolism and increase dependence on oxidative mitochondrial metabolism. Cancer Res. 2014, 74, 3317–3331. [Google Scholar] [CrossRef] [PubMed]
- Reitman, Z.J.; Jin, G.; Karoly, E.D.; Spasojevic, I.; Yang, J.; Kinzler, K.W.; He, Y.; Bigner, D.D.; Vogelstein, B.; Yan, H. Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations on the cellular metabolome. Proc. Natl. Acad. Sci. USA 2011, 108, 3270–3275. [Google Scholar] [CrossRef] [PubMed]
- Ohka, F.; Ito, M.; Ranjit, M.; Senga, T.; Motomura, A.; Motomura, K.; Saito, K.; Kato, K.; Kato, Y.; Wakabayashi, T.; et al. Quantitative metabolome analysis profiles activation of glutaminolysis in glioma with IDH1 mutation. Tumor Boil. 2014, 35, 5911–5920. [Google Scholar] [CrossRef] [PubMed]
- Seltzer, M.J.; Bennett, B.D.; Joshi, A.D.; Gao, P.; Thomas, A.G.; Ferraris, D.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Rabinowitz, J.D.; et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010, 70, 8981–8987. [Google Scholar] [CrossRef]
- Wen, P.Y.; Huse, J.T. 2016 World Health Organization Classification of Central Nervous System Tumors. Contin. Lifelong Learn. Neurol. 2017, 23, 1531–1547. [Google Scholar] [CrossRef]
- Riemenschneider, M.J.; Jeuken, J.W.M.; Wesseling, P.; Reifenberger, G. Molecular diagnostics of gliomas: State of the art. Acta Neuropathol. 2010, 120, 567–584. [Google Scholar] [CrossRef]
- Chen, R.; Smith-Cohn, M.; Cohen, A.L.; Colman, H. Glioma Subclassifications and Their Clinical Significance. Neurotherapeutics 2017, 14, 284–297. [Google Scholar] [CrossRef]
- Jaunmuktane, Z.; Capper, D.; Jones, D.T.W.; Schrimpf, D.; Sill, M.; Dutt, M.; Suraweera, N.; Pfister, S.M.; Von Deimling, A.; Brandner, S. Methylation array profiling of adult brain tumours: Diagnostic outcomes in a large, single centre. Acta Neuropathol. Commun. 2019, 7, 24. [Google Scholar] [CrossRef]
- Andronesi, O.C.; Rapalino, O.; Gerstner, E.; Chi, A.; Batchelor, T.T.; Cahill, D.P.; Sorensen, A.G.; Rosen, B.R. Detection of oncogenic IDH1 mutations using magnetic resonance spectroscopy of 2-hydroxyglutarate. J. Clin. Investig. 2013, 123, 3659–3663. [Google Scholar] [CrossRef]
- Andronesi, O.C.; Kim, G.S.; Gerstner, E.; Batchelor, T.; Tzika, A.A.; Fantin, V.R.; Heiden, M.G.V.; Sorensen, A.G. Detection of 2-Hydroxyglutarate in IDH-Mutated Glioma Patients by In Vivo Spectral-Editing and 2D Correlation Magnetic Resonance Spectroscopy. Sci. Transl. Med. 2012, 4, 116ra4. [Google Scholar] [CrossRef]
- Pope, W.B.; Prins, R.M.; Thomas, M.A.; Nagarajan, R.; Yen, K.E.; Bittinger, M.A.; Salamon, N.; Chou, A.P.; Yong, W.H.; Soto, H.; et al. Non-invasive detection of 2-hydroxyglutarate and other metabolites in IDH1 mutant glioma patients using magnetic resonance spectroscopy. Neuro-Oncology 2011, 107, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Natsumeda, M.; Igarashi, H.; Nomura, T.; Ogura, R.; Tsukamoto, Y.; Kobayashi, T.; Aoki, H.; Okamoto, K.; Kakita, A.; Takahashi, H.; et al. Accumulation of 2-hydroxyglutarate in gliomas correlates with survival: A study by 3.0-tesla magnetic resonance spectroscopy. Acta Neuropathol. Commun. 2014, 2, 158. [Google Scholar] [CrossRef] [PubMed]
- Lazovic, J.; Soto, H.; Piccioni, D.E.; Ru, J.R.L.; Li, S.; Mirsadraei, L.; Yong, W.; Prins, R.M.; Liau, L.M.; Ellingson, B.M.; et al. Detection of 2-hydroxyglutaric acid in vivo by proton magnetic resonance spectroscopy in U87 glioma cells overexpressing isocitrate dehydrogenase–1 mutation. Neuro-Oncology 2012, 14, 1465–1472. [Google Scholar] [CrossRef] [PubMed]
- Heo, H.; Kim, S.; Lee, H.H.; Cho, H.R.; Xu, W.J.; Lee, S.-H.; Park, C.K.; Park, S.; Choi, S.H.; Kim, H. On the Utility of Short Echo Time (TE) Single Voxel 1H–MRS in Non–Invasive Detection of 2–Hydroxyglutarate (2HG); Challenges and Potential Improvement Illustrated with Animal Models Using MRUI and LCModel. PLoS ONE 2016, 11, e0147794. [Google Scholar] [CrossRef][Green Version]
- Choi, C.; Ganji, S.K.; DeBerardinis, R.J.; Hatanpaa, K.J.; Rakheja, D.; Kovacs, Z.; Yang, X.-L.; Mashimo, T.; Raisanen, J.M.; Marin-Valencia, I.; et al. 2-hydroxyglutarate detection by magnetic resonance spectroscopy in IDH-mutated patients with gliomas. Nat. Med. 2012, 18, 624–629. [Google Scholar] [CrossRef]
- Choi, C.; Ganji, S.; Hulsey, K.; Madan, A.; Kovacs, Z.; Dimitrov, I.; Zhang, S.; Pichumani, K.; Mendelsohn, D.; Mickey, B.; et al. A comparative study of short- and long-TE ¹H MRS at 3 T for in vivo detection of 2-hydroxyglutarate in brain tumors. NMR Biomed. 2013, 26, 1242–1250. [Google Scholar] [CrossRef]
- De La Fuente, M.I.; Young, R.J.; Rubel, J.; Rosenblum, M.; Tisnado, J.; Briggs, S.; Pérez, J.A.; Cross, J.R.; Campos, C.; Straley, K.; et al. Integration of 2-hydroxyglutarate-proton magnetic resonance spectroscopy into clinical practice for disease monitoring in isocitrate dehydrogenase-mutant glioma. Neuro-Oncology 2015, 18, 283–290. [Google Scholar] [CrossRef]
- Emir, U.E.; Larkin, S.J.; De Pennington, N.; Voets, N.L.; Plaha, P.; Stacey, R.; Al-Qahtani, K.; Mccullagh, J.S.; Schofield, C.J.; Clare, S.; et al. Noninvasive Quantification of 2-Hydroxyglutarate in Human Gliomas with IDH1 and IDH2 Mutations. Cancer Res. 2015, 76, 43–49. [Google Scholar] [CrossRef]
- Sanson, M.; Marie, Y.; Paris, S.; Idbaih, A.; Laffaire, J.; Ducray, F.; El Hallani, S.; Boisselier, B.; Mokhtari, K.; Hoang-Xuan, K.; et al. Isocitrate Dehydrogenase 1 Codon 132 Mutation Is an Important Prognostic Biomarker in Gliomas. J. Clin. Oncol. 2009, 27, 4150–4154. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Nobusawa, S.; Watanabe, T.; Kleihues, P.; Ohgaki, H. IDH1 Mutations as Molecular Signature and Predictive Factor of Secondary Glioblastomas. Clin. Cancer Res. 2009, 15, 6002–6007. [Google Scholar] [CrossRef] [PubMed]
- Toedt, G.; Barbus, S.; Wolter, M.; Felsberg, J.; Tews, B.; Blond, F.; Sabel, M.; Hofmann, S.; Becker, N.; Hartmann, C.; et al. Molecular signatures classify astrocytic gliomas byIDH1mutation status. Int. J. Cancer 2010, 128, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Felsberg, J.; Hartmann, C.; Berger, H.; Steinbach, J.P.; Schramm, J.; Westphal, M.; Schackert, G.; Simon, M.; Tonn, J.C.; et al. Molecular Predictors of Progression-Free and Overall Survival in Patients With Newly Diagnosed Glioblastoma: A Prospective Translational Study of the German Glioma Network. J. Clin. Oncol. 2009, 27, 5743–5750. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.; Kharbanda, S.; Pope, W.B.; Tran, A.; Solis, O.E.; Peale, F.; Forrest, W.F.; Pujara, K.; Carrillo, J.A.; Pandita, A.; et al. Evidence for Sequenced Molecular Evolution of IDH1 Mutant Glioblastoma From a Distinct Cell of Origin. J. Clin. Oncol. 2011, 29, 4482–4490. [Google Scholar] [CrossRef]
- Balss, J.; Meyer, J.; Mueller, W.; Korshunov, A.; Hartmann, C.; Von Deimling, A. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol. 2008, 116, 597–602. [Google Scholar] [CrossRef]
- Chaichana, K.L.; Parker, S.L.; Olivi, A.; Quinones-Hinojosa, A. Long-term seizure outcomes in adult patients undergoing primary resection of malignant brain astrocytomas. J. Neurosurg. 2009, 111, 282–292. [Google Scholar] [CrossRef]
- Chang, E.F.; Potts, M.B.; Keles, G.E.; Lamborn, K.R.; Chang, S.M.; Barbaro, N.M.; Berger, M.S. Seizure characteristics and control following resection in 332 patients with low-grade gliomas. J. Neurosurg. 2008, 108, 227–235. [Google Scholar] [CrossRef]
- Kerkhof, M.; Vecht, C.J. Seizure characteristics and prognostic factors of gliomas. Epilepsia 2013, 54, 12–17. [Google Scholar] [CrossRef]
- Van Breemen, M.S.; Wilms, E.B.; Vecht, C.J. Epilepsy in patients with brain tumours: Epidemiology, mechanisms, and management. Lancet Neurol. 2007, 6, 421–430. [Google Scholar] [CrossRef]
- Klein, M.; Engelberts, N.H.J.; Van Der Ploeg, H.M.; Trenité, D.G.A.K.-N.; Aaronson, N.K.; Taphoorn, M.J.B.; Baaijen, H.; Vandertop, W.P.; Müller, M.; Postma, T.J.; et al. Epilepsy in low-grade gliomas: The impact on cognitive function and quality of life. Ann. Neurol. 2003, 54, 514–520. [Google Scholar] [CrossRef]
- Stockhammer, F.; Misch, M.; Helms, H.-J.; Lengler, U.; Prall, F.; Von Deimling, A.; Hartmann, C. IDH1/2 mutations in WHO grade II astrocytomas associated with localization and seizure as the initial symptom. Seizure 2012, 21, 194–197. [Google Scholar] [CrossRef] [PubMed]
- Liubinas, S.V.; D’Abaco, G.M.; Moffat, B.M.; Gonzales, M.; Feleppa, F.; Nowell, C.J.; Gorelik, A.; Drummond, K.J.; O’Brien, T.J.; Kaye, A.H.; et al. IDH1 mutation is associated with seizures and protoplasmic subtype in patients with low-grade gliomas. Epilepsia 2014, 55, 1438–1443. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Qing, M.; Wang, X.; Liu, Y.; Mao, Y.; Zhou, Q.; Luo, J. An analysis of 170 glioma patients and systematic review to investigate the association between IDH-1 mutations and preoperative glioma-related epilepsy. J. Clin. Neurosci. 2016, 31, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Judkins, J.; Thomas, C.; Wu, M.; Khoury, L.; Benjamin, C.; Pacione, D.; Golfinos, J.G.; Kumthekar, P.; Ghamsari, F.; et al. Mutant IDH1 and seizures in patients with glioma. Neurology 2017, 88, 1805–1813. [Google Scholar] [CrossRef] [PubMed]
- Toledo, M.; Sarria-Estrada, S.; Quintana, M.; Maldonado, X.; Martínez-Ricarte, F.; Rodon, J.; Auger, C.; Aizpurua, M.; Salas-Puig, J.; Santamarina, E.; et al. Epileptic features and survival in glioblastomas presenting with seizures. Epilepsy Res. 2017, 130, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Moussawi, K.; Riegel, A.; Nair, S.; Kalivas, P.W. Extracellular Glutamate: Functional Compartments Operate in Different Concentration Ranges. Front. Syst. Neurosci. 2011, 5, 94. [Google Scholar] [CrossRef]
- Elkhaled, A.; Jalbert, L.; Constantin, A.; Yoshihara, H.A.I.; Phillips, J.J.; Molinaro, A.M.; Chang, S.M.; Nelson, S.J. Characterization of metabolites in infiltrating gliomas usingex vivo1H high-resolution magic angle spinning spectroscopy. NMR Biomed. 2014, 27, 578–593. [Google Scholar] [CrossRef]
- Vaarmann, A.; Kovac, S.; Holmstrom, K.; Gandhi, S.; Abramov, A.Y. Dopamine protects neurons against glutamate-induced excitotoxicity. Cell Death Dis. 2013, 4, e455. [Google Scholar] [CrossRef]
- Rohle, D.; Popovici-Muller, J.; Palaskas, N.; Turcan, Ş.; Grommes, C.; Campos, C.; Tsoi, J.; Clark, O.; Oldrini, B.; Komisopoulou, E.; et al. An Inhibitor of Mutant IDH1 Delays Growth and Promotes Differentiation of Glioma Cells. Science 2013, 340, 626–630. [Google Scholar] [CrossRef]
- Hartmann, C.; Hentschel, B.; Wick, W.; Capper, D.; Felsberg, J.; Simon, M.; Westphal, M.; Schackert, G.; Meyermann, R.; Pietsch, T.; et al. Patients with IDH1 wild type anaplastic astrocytomas exhibit worse prognosis than IDH1-mutated glioblastomas, and IDH1 mutation status accounts for the unfavorable prognostic effect of higher age: Implications for classification of gliomas. Acta Neuropathol. 2010, 120, 707–718. [Google Scholar] [CrossRef]
- Houillier, C.; Wang, X.; Kaloshi, G.; Mokhtari, K.; Guillevin, R.; Laffaire, J.; Paris, S.; Boisselier, B.; Idbaih, A.; Laigle-Donadey, F.; et al. IDH1 or IDH2 mutations predict longer survival and response to temozolomide in low-grade gliomas. Neurology 2010, 75, 1560–1566. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, C.; Hentschel, B.; Tatagiba, M.; Schramm, J.; Schnell, O.; Seidel, C.; Stein, R.; Reifenberger, G.; Pietsch, T.; Von Deimling, A.; et al. Molecular Markers in Low-Grade Gliomas: Predictive or Prognostic? Clin. Cancer Res. 2011, 17, 4588–4599. [Google Scholar] [CrossRef] [PubMed]
- Bent, M.J.V.D.; Dubbink, H.J.; Marie, Y.; Brandes, A.A.; Taphoorn, M.J.; Wesseling, P.; Frenay, M.; Tijssen, C.C.; Lacombe, D.; Idbaih, A.; et al. IDH1 and IDH2 Mutations Are Prognostic but not Predictive for Outcome in Anaplastic Oligodendroglial Tumors: A Report of the European Organization for Research and Treatment of Cancer Brain Tumor Group. Clin. Cancer Res. 2010, 16, 1597–1604. [Google Scholar] [CrossRef] [PubMed]
- Popovici-Muller, J.; Saunders, J.O.; Salituro, F.G.; Travins, J.M.; Yan, S.; Zhao, F.; Gross, S.; Dang, L.; Yen, K.E.; Yang, H.; et al. Discovery of the First Potent Inhibitors of Mutant IDH1 That Lower Tumor 2-HG in Vivo. ACS Med. Chem. Lett. 2012, 3, 850–855. [Google Scholar] [CrossRef] [PubMed]
- Dinardo, C.; Stein, E.M.; De Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable Remissions with Ivosidenib inIDH1-Mutated Relapsed or Refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398. [Google Scholar] [CrossRef] [PubMed]
- Konteatis, Z.; Artin, E.; Nicolay, B.; Straley, K.; Padyana, A.K.; Jin, L.; Chen, Y.; Narayaraswamy, R.; Tong, S.; Wang, F.; et al. Vorasidenib (AG-881): A First-in-Class, Brain-Penetrant Dual Inhibitor of Mutant IDH1 and 2 for Treatment of Glioma. ACS Med. Chem. Lett. 2020, 11, 101–107. [Google Scholar] [CrossRef]
- Fan, B.; Mellinghoff, I.K.; Wen, P.Y.; Lowery, M.A.; Goyal, L.; Tap, W.D.; Pandya, S.S.; Manyak, E.; Jiang, L.; Liu, G.; et al. Clinical pharmacokinetics and pharmacodynamics of ivosidenib, an oral, targeted inhibitor of mutant IDH1, in patients with advanced solid tumors. Investig. New Drugs 2019, 38, 433–444. [Google Scholar] [CrossRef]
- Popovici-Muller, J.; Lemieux, R.M.; Artin, E.; Saunders, J.O.; Salituro, F.G.; Travins, J.; Cianchetta, G.; Cai, Z.; Zhou, D.; Cui, D.; et al. Discovery of AG-120 (Ivosidenib): A First-in-Class Mutant IDH1 Inhibitor for the Treatment of IDH1 Mutant Cancers. ACS Med. Chem. Lett. 2018, 9, 300–305. [Google Scholar] [CrossRef]
- Mellinghoff, I.K.; Ellingson, B.M.; Touat, M.; Maher, E.; De La Fuente, M.I.; Holdhoff, M.; Cote, G.M.; Burris, H.; Janku, F.; Young, R.J.; et al. Ivosidenib in Isocitrate Dehydrogenase 1–Mutated Advanced Glioma. J. Clin. Oncol. 2020, JCO1903327. [Google Scholar] [CrossRef]
- Molenaar, R.J.; Radivoyevitch, T.; Nagata, Y.; Khurshed, M.; Przychodzen, B.; Makishima, H.; Xu, M.; Bleeker, F.E.; Wilmink, J.W.; Carraway, H.E.; et al. IDH1/2Mutations Sensitize Acute Myeloid Leukemia to PARP Inhibition and This Is Reversed by IDH1/2-Mutant Inhibitors. Clin. Cancer Res. 2018, 24, 1705–1715. [Google Scholar] [CrossRef]
- Borodovsky, A.; Salmasi, V.; Turcan, Ş.; Fabius, A.W.M.; Baia, G.S.; Eberhart, C.G.; Weingart, J.D.; Gallia, G.L.; Baylin, S.B.; Chan, T.A.; et al. 5-azacytidine reduces methylation, promotes differentiation and induces tumor regression in a patient-derived IDH1 mutant glioma xenograft. Oncotarget 2013, 4, 1737–1747. [Google Scholar] [CrossRef] [PubMed]
- Turcan, Ş.; Fabius, A.W.M.; Borodovsky, A.; Pedraza, A.; Brennan, C.W.; Huse, J.; Viale, A.; Riggins, G.J.; Chan, T.A. Efficient induction of differentiation and growth inhibition in IDH1 mutant glioma cells by the DNMT Inhibitor Decitabine. Oncotarget 2013, 4, 1729–1736. [Google Scholar] [CrossRef] [PubMed]
- Fathi, A.T.; Abdel-Wahab, O. Mutations in Epigenetic Modifiers in Myeloid Malignancies and the Prospect of Novel Epigenetic-Targeted Therapy. Adv. Hematol. 2012, 2012, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gursoy-Yuzugullu, O.; Carman, C.; Serafim, R.B.; Myronakis, M.; Valente, V.; Price, B.D. Epigenetic therapy with inhibitors of histone methylation suppresses DNA damage signaling and increases glioma cell radiosensitivity. Oncotarget 2017, 8, 24518–24532. [Google Scholar] [CrossRef] [PubMed]
- Bent, M.J.V.D.; Mellinghoff, I.K.; Bindra, R.S.; Binja, R. Gray Areas in the Gray Matter: IDH1/2 Mutations in Glioma. Am. Soc. Clin. Oncol. Educ. Book 2020, 40, 96–103. [Google Scholar] [CrossRef]
- Esteller, M.; García-Foncillas, J.; Andion, E.; Goodman, S.N.; Hidalgo, O.F.; Vanaclocha, V.; Baylin, S.B.; Herman, J.G. Inactivation of the DNA-Repair GeneMGMTand the Clinical Response of Gliomas to Alkylating Agents. N. Engl. J. Med. 2000, 343, 1350–1354. [Google Scholar] [CrossRef]
- Hegi, M.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; De Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMTGene Silencing and Benefit from Temozolomide in Glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef]
- Cai, S.J.; Liu, Y.; Han, S.; Yang, C. Brusatol, an NRF2 inhibitor for future cancer therapeutic. Cell Biosci. 2019, 9, 45. [Google Scholar] [CrossRef]
- Emadi, A.; Jun, S.A.; Tsukamoto, T.; Fathi, A.T.; Minden, M.D.; Dang, C.V. Inhibition of glutaminase selectively suppresses the growth of primary acute myeloid leukemia cells with IDH mutations. Exp. Hematol. 2014, 42, 247–251. [Google Scholar] [CrossRef]
- Matre, P.; Velez, J.; Jácamo, R.; Qi, Y.; Su, X.; Cai, T.; Chan, S.M.; Lodi, A.; Sweeney, S.R.; Ma, H.; et al. Inhibiting glutaminase in acute myeloid leukemia: Metabolic dependency of selected AML subtypes. Oncotarget 2016, 7, 79722–79735. [Google Scholar] [CrossRef]
- Tateishi, K.; Wakimoto, H.; Iafrate, A.J.; Tanaka, S.; Loebel, F.; Lelic, N.; Wiederschain, D.; Bedel, O.; Deng, G.; Zhang, B.; et al. Extreme Vulnerability of IDH1 Mutant Cancers to NAD+ Depletion. Cancer Cell 2015, 28, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Tateishi, K.; Higuchi, F.; Miller, J.J.; Koerner, M.V.; Lelic, N.; Shankar, G.M.; Tanaka, S.; Fisher, D.E.; Batchelor, T.T.; Iafrate, A.J.; et al. The Alkylating Chemotherapeutic Temozolomide Induces Metabolic Stress in IDH1-Mutant Cancers and Potentiates NAD+ Depletion-Mediated Cytotoxicity. Cancer Res. 2017, 77, 4102–4115. [Google Scholar] [CrossRef] [PubMed]
- Bunse, L.; Pusch, S.; Bunse, T.; Sahm, F.; Sanghvi, K.; Friedrich, M.; AlAnsary, D.; Sonner, J.K.; Green, E.; Deumelandt, K.; et al. Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat. Med. 2018, 24, 1192–1203. [Google Scholar] [CrossRef] [PubMed]
- Kohanbash, G.; Carrera, D.A.; Shrivastav, S.; Ahn, B.J.; Jahan, N.; Mazor, T.; Chheda, Z.S.; Downey, K.M.; Watchmaker, P.B.; Beppler, C.; et al. Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. J. Clin. Investig. 2017, 127, 1425–1437. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.; Bunse, L.; Pusch, S.; Sahm, F.; Wiestler, B.; Quandt, J.; Menn, O.; Osswald, M.; Oezen, I.; Ott, M.; et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature 2014, 512, 324–327. [Google Scholar] [CrossRef] [PubMed]
- Curry, W.T.; Lim, M. Immunomodulation: Checkpoint blockade etc.: Figure 1. Neuro-Oncology 2015, 17, vii26–vii31. [Google Scholar] [CrossRef]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; A Goldlust, S.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef]
- Weller, M.; Roth, P.; Preusser, M.; Wick, W.; Reardon, D.A.; Platten, M.; Sampson, J.H. Vaccine-based immunotherapeutic approaches to gliomas and beyond. Nat. Rev. Neurol. 2017, 13, 363–374. [Google Scholar] [CrossRef]
- Fecci, P.E.; Sampson, J.H. The current state of immunotherapy for gliomas: An eye toward the future. J. Neurosurg. 2019, 131, 657–666. [Google Scholar] [CrossRef]
- Elhammali, A.; Ippolito, J.E.; Collins, L.; Crowley, J.; Marasa, J.; Piwnica-Worms, D.R. A high-throughput fluorimetric assay for 2-hydroxyglutarate identifies Zaprinast as a glutaminase inhibitor. Cancer Discov. 2014, 4, 828–839. [Google Scholar] [CrossRef]
- Sulkowski, P.L.; Sundaram, R.K.; Oeck, S.; Corso, C.D.; Liu, Y.; Noorbakhsh, S.; Niger, M.; Boeke, M.; Ueno, D.; Kalathil, A.N.; et al. Krebs-cycle-deficient hereditary cancer syndromes are defined by defects in homologous-recombination DNA repair. Nat. Genet. 2018, 50, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, T.-C.A.; Mukherjee, J.; Viswanath, P.; Ohba, S.; Ronen, S.M.; Bjerkvig, R.; Pieper, R.O. Rapid Conversion of Mutant IDH1 from Driver to Passenger in a Model of Human Gliomagenesis. Mol. Cancer Res. 2016, 14, 976–983. [Google Scholar] [CrossRef]
- Molenaar, R.J.; Botman, D.; Smits, M.A.; Hira, V.V.; Van Lith, S.A.; Stap, J.; Henneman, P.; Khurshed, M.; Lenting, K.; Mul, A.N.; et al. Radioprotection of IDH1-Mutated Cancer Cells by the IDH1-Mutant Inhibitor AGI-5198. Cancer Res. 2015, 75, 4790–4802. [Google Scholar] [CrossRef] [PubMed]
- Pusch, S.; Krausert, S.; Fischer, V.; Balss, J.; Ott, M.; Schrimpf, D.; Capper, D.; Sahm, F.; Eisel, J.; Beck, A.-C.; et al. Pan-mutant IDH1 inhibitor BAY 1436032 for effective treatment of IDH1 mutant astrocytoma in vivo. Acta Neuropathol. 2017, 133, 629–644. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, A.; Herbst, L.; Pusch, S.; Klett, L.; Goparaju, R.; Stichel, D.; Kaulfuss, S.; Panknin, O.; Zimmermann, K.; Toschi, L.; et al. Pan-mutant-IDH1 inhibitor BAY1436032 is highly effective against human IDH1 mutant acute myeloid leukemia in vivo. Leukemia 2017, 31, 2020–2028. [Google Scholar] [CrossRef]
- Noorani, I. Genetically Engineered Mouse Models of Gliomas: Technological Developments for Translational Discoveries. Cancers 2019, 11, 1335. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Lang, F.; Chou, F.-J.; Zaghloul, K.A.; Yang, C. Isocitrate Dehydrogenase Mutations in Glioma: Genetics, Biochemistry, and Clinical Indications. Biomedicines 2020, 8, 294. https://doi.org/10.3390/biomedicines8090294
Liu Y, Lang F, Chou F-J, Zaghloul KA, Yang C. Isocitrate Dehydrogenase Mutations in Glioma: Genetics, Biochemistry, and Clinical Indications. Biomedicines. 2020; 8(9):294. https://doi.org/10.3390/biomedicines8090294
Chicago/Turabian StyleLiu, Yang, Fengchao Lang, Fu-Ju Chou, Kareem A. Zaghloul, and Chunzhang Yang. 2020. "Isocitrate Dehydrogenase Mutations in Glioma: Genetics, Biochemistry, and Clinical Indications" Biomedicines 8, no. 9: 294. https://doi.org/10.3390/biomedicines8090294
APA StyleLiu, Y., Lang, F., Chou, F.-J., Zaghloul, K. A., & Yang, C. (2020). Isocitrate Dehydrogenase Mutations in Glioma: Genetics, Biochemistry, and Clinical Indications. Biomedicines, 8(9), 294. https://doi.org/10.3390/biomedicines8090294