Attenuation of Hyperoxic Lung Injury in Newborn Thioredoxin-1-Overexpressing Mice through the Suppression of Proinflammatory Cytokine mRNA Expression

 ,
,
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Neonatal Hyperoxic Exposure and Recovery
2.3. Lung Tissue Collection
2.4. Lung Histology and Morphometry
2.5. RNA Extraction and Quantitative Real-Time PCR Analysis
2.6. Immunohistochemistry
2.7. Western Blotting
2.8. Immunoprecipitation
2.9. Statistical Analysis
3. Results
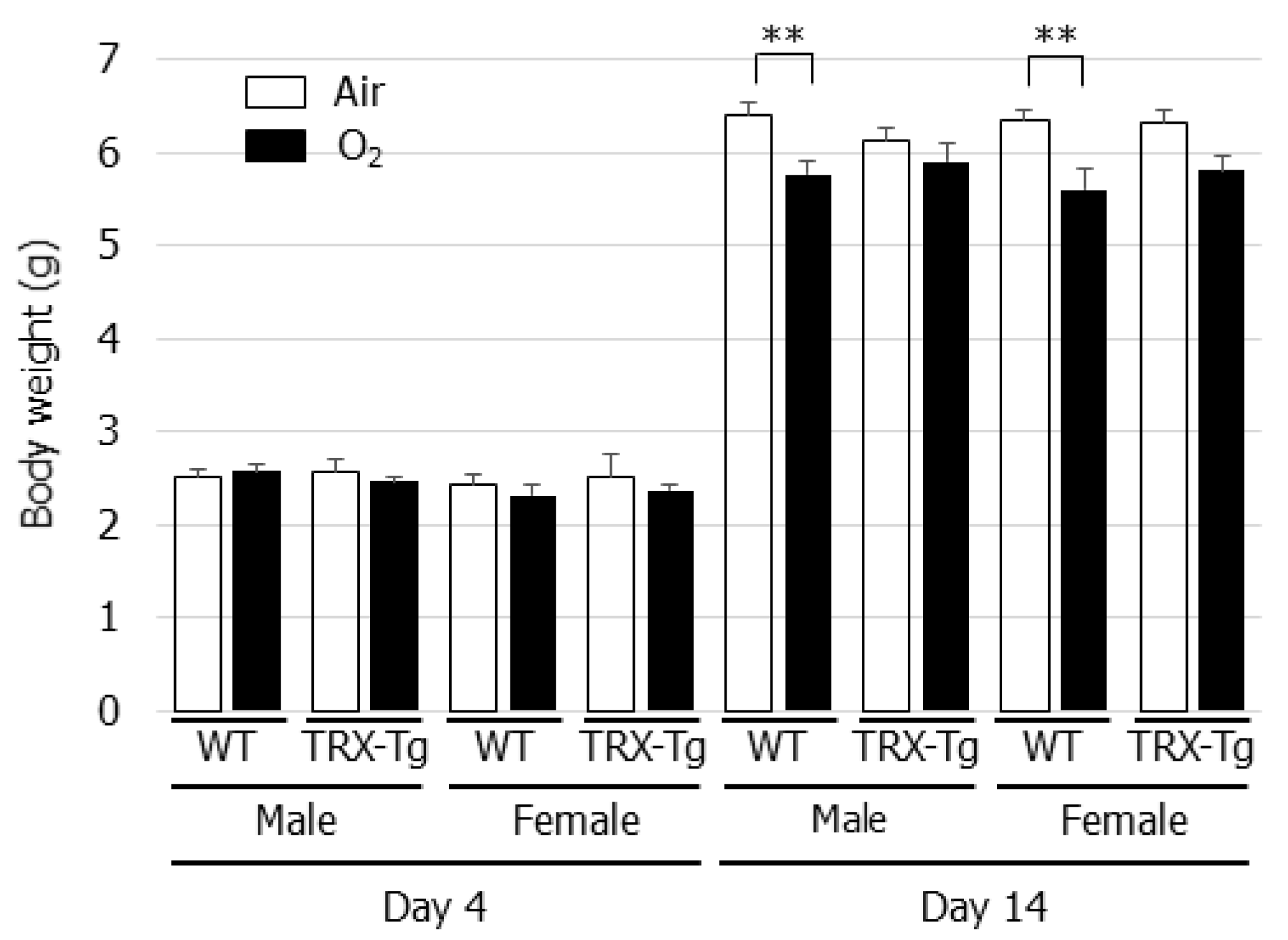
3.1. Neonatal Hyperoxia Does Not Negatively Affect Body Weight in TRX-Tg Mice
3.2. Alveolar Development Is Impaired After Neonatal Hyperoxia in Both WT and TRX-Tg Mice
3.3. Impaired Alveolar Development Is Mitigated in TRX-Tg Mouse Lungs during Recovery from Hyperoxia
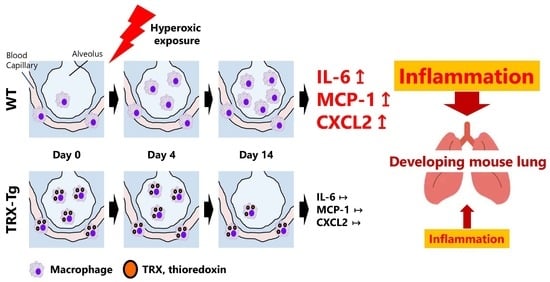
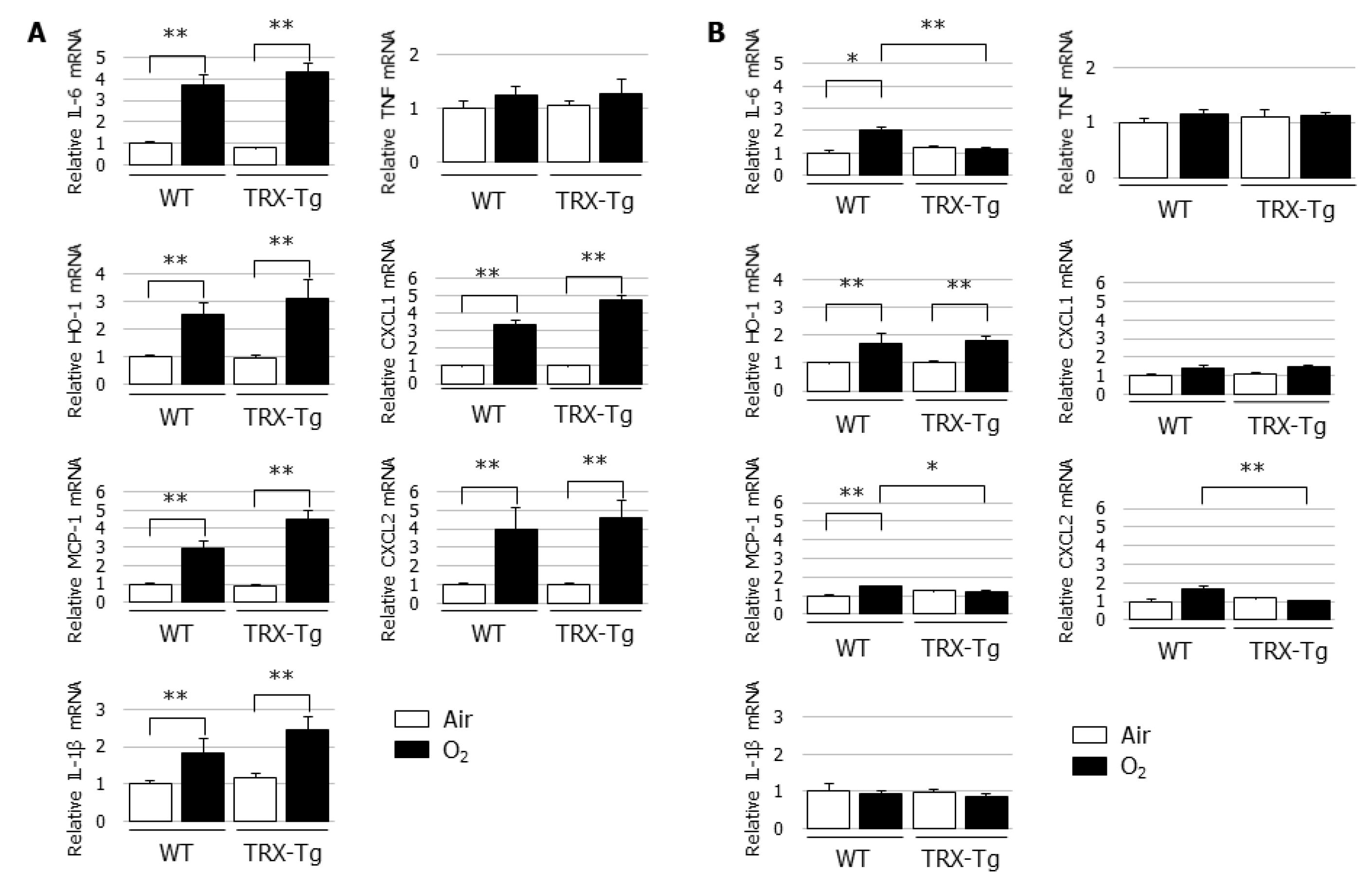
3.4. Hyperoxia-Induced Increases in Proinflammatory Cytokine and Chemokine mRNA Expression Levels Disappeared During Recovery from Hyperoxia in TRX-Tg Mouse Lungs
3.5. Macrophage Infiltration Is Reduced in the Lungs of TRX-Tg Mice after Neonatal Hyperoxia
3.6. Lung Mif mRNA and Protein Levels Are Not Different Between WT and TRX-Tg Mice after Hyperoxic Exposure
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Jobe, A.H.; Kallapur, S.G. Long term consequences of oxygen therapy in the neonatal period. Semin. Fetal Neonatal Med. 2010, 15, 230–235. [Google Scholar] [CrossRef]
- Hirata, K.; Nishihara, M.; Kimura, T.; Shiraishi, J.; Hirano, S.; Kitajima, H.; Fujimura, M. Longitudinal impairment of lung function in school-age children with extremely low birth weights. Pediatr. Pulmonol. 2017, 52, 779–786. [Google Scholar] [CrossRef]
- Jain, D.; Bancalari, E. Bronchopulmonary dysplasia: Clinical perspective. Birth Defects Res. A Clin. Mol. Teratol. 2014, 100, 134–144. [Google Scholar] [CrossRef]
- Mercier, J.C.; Hummler, H.; Durrmeyer, X.; Sanchez-Luna, M.; Carnielli, V.; Field, D.; Greenough, A.; Van Overmeire, B.; Jonsson, B.; Hallman, M.; et al. Inhaled nitric oxide for prevention of bronchopulmonary dysplasia in premature babies (EUNO): A randomised controlled trial. Lancet 2010, 376, 346–354. [Google Scholar] [CrossRef]
- Couroucli, X.I.; Placencia, J.L.; Cates, L.A.; Suresh, G.K. Should we still use vitamin A to prevent bronchopulmonary dysplasia? J. Perinatol. 2016, 36, 581–585. [Google Scholar] [CrossRef]
- Michael, Z.; Spyropoulos, F.; Ghanta, S.; Christou, H. Bronchopulmonary Dysplasia: An Update of Current Pharmacologic Therapies and New Approaches. Clin. Med. Insights Pediatr. 2018, 12, 1179556518817322. [Google Scholar] [CrossRef]
- Vogel, E.R.; Britt, R.D., Jr.; Trinidad, M.C.; Faksh, A.; Martin, R.J.; MacFarlane, P.M.; Pabelick, C.M.; Prakash, Y.S. Perinatal oxygen in the developing lung. Can. J. Physiol. Pharmacol. 2015, 93, 119–127. [Google Scholar] [CrossRef]
- Warner, B.B.; Stuart, L.A.; Papes, R.A.; Wispé, J.R. Functional and pathological effects of prolonged hyperoxia in neonatal mice. Am. J. Physiol. 1998, 275, L110–L117. [Google Scholar] [CrossRef]
- Namba, F.; Ogawa, R.; Ito, M.; Watanabe, T.; Dennery, P.A.; Tamura, M. Sex-related differences in long-term pulmonary outcomes of neonatal hyperoxia in mice. Exp. Lung Res. 2016, 42, 57–65. [Google Scholar] [CrossRef]
- Menon, R.T.; Shrestha, A.K.; Reynolds, C.L.; Barrios, R.; Shivanna, B. Long-term pulmonary and cardiovascular morbidities of neonatal hyperoxia exposure in mice. Int. J. Biochem. Cell Biol. 2018, 94, 119–124. [Google Scholar] [CrossRef]
- Holmgren, A. Thioredoxin. Annu. Rev. Biochem. 1985, 54, 237–271. [Google Scholar] [CrossRef]
- Holmgren, A. Thioredoxin and glutaredoxin systems. J. Biol. Chem. 1989, 264, 13963–13966. [Google Scholar]
- Nakamura, H.; Herzenberg, L.A.; Bai, J.; Araya, S.; Kondo, N.; Nishinaka, Y.; Herzenberg, L.A.; Yodoi, J. Circulating thioredoxin suppresses lipopolysaccharide-induced neutrophil chemotaxis. Proc. Natl. Acad. Sci. USA 2001, 98, 15143–15148. [Google Scholar] [CrossRef]
- Nakamura, H.; Tamura, S.; Watanabe, I.; Iwasaki, T.; Yodoi, J. Enhanced resistancy of thioredoxin-transgenic mice against influenza virus-induced pneumonia. Immunol. Lett. 2002, 82, 165–170. [Google Scholar] [CrossRef]
- Ahsan, M.K.; Nakamura, H.; Tanito, M.; Yamada, K.; Utsumi, H.; Yodoi, J. Thioredoxin-1 suppresses lung injury and apoptosis induced by diesel exhaust particles (DEP) by scavenging reactive oxygen species and by inhibiting DEP-induced downregulation of Akt. Free Radic. Biol. Med. 2005, 39, 1549–1559. [Google Scholar]
- Hoshino, T.; Nakamura, H.; Okamoto, M.; Kato, S.; Araya, S.; Nomiyama, K.; Oizumi, K.; Young, H.A.; Aizawa, H.; Yodoi, J. Redox-active protein thioredoxin prevents proinflammatory cytokine- or bleomycin-induced lung injury. Am. J. Respir. Crit. Care Med. 2003, 168, 1075–1083. [Google Scholar] [CrossRef]
- Ohashi, S.; Nishio, A.; Nakamura, H.; Asada, M.; Tamaki, H.; Kawasaki, K.; Fukui, T.; Yodoi, J.; Chiba, T. Overexpression of redox-active protein thioredoxin-1 prevents development of chronic pancreatitis in mice. Antioxid Redox Signal 2006, 8, 1835–1845. [Google Scholar] [CrossRef]
- Ohashi, S.; Nishio, A.; Nakamura, H.; Kido, M.; Ueno, S.; Uza, N.; Inoue, S.; Kitamura, H.; Kiriya, K.; Asada, M.; et al. Protective roles of redox-active protein thioredoxin-1 for severe acute pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G772–G781. [Google Scholar] [CrossRef]
- Shioji, K.; Nakamura, H.; Masutani, H.; Yodoi, J. Redox regulation by thioredoxin in cardiovascular diseases. Antioxid Redox Signal 2003, 5, 795–802. [Google Scholar] [CrossRef]
- Sato, A.; Hoshino, Y.; Hara, T.; Muro, S.; Nakamura, H.; Mishima, M.; Yodoi, J. Thioredoxin-1 ameliorates cigarette smoke-induced lung inflammation and emphysema in mice. J. Pharmacol. Exp. Ther. 2008, 325, 380–388. [Google Scholar] [CrossRef]
- Kinoshita, T.; Hoshino, T.; Imaoka, H.; Ichiki, H.; Okamoto, M.; Kawayama, T.; Yodoi, J.; Kato, S.; Aizawa, H. Thioredoxin prevents the development and progression of elastase-induced emphysema. Biochem. Biophys. Res. Commun. 2007, 354, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.; Nakamura, H.; Kondo, N.; Tanito, M.; Kwon, Y.W.; Ahsan, M.K.; Matsui, H.; Narita, M.; Yodoi, J. Thioredoxin-1 attenuates indomethacin-induced gastric mucosal injury in mice. Free Radic. Res. 2007, 41, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Namba, F.; Miura, M.; Goda, T.; Nakura, Y.; Nishiumi, F.; Son, A.; Kubota, A.; Yodoi, J.; Yanagihara, I. Human thioredoxin-1 attenuates the rate of lipopolysaccharide-induced preterm delivery in mice in association with its anti-inflammatory effect. Pediatr. Res. 2016, 80, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Iwasaki, Y.; Nagata, K.; Fushiki, S.; Nakamura, H.; Marunaka, Y.; Yodoi, J. Thioredoxin-1 protects against hyperoxia-induced apoptosis in cells of the alveolar walls. Pulm. Pharm. Ther. 2007, 20, 650–659. [Google Scholar] [CrossRef]
- Das, K.C. Thioredoxin-deficient mice, a novel phenotype sensitive to ambient air and hypersensitive to hyperoxia-induced lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L429–L442. [Google Scholar] [CrossRef]
- Frank, L.; Bucher, J.R.; Roberts, R.J. Oxygen toxicity in neonatal and adult animals of various species. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1978, 45, 699–704. [Google Scholar] [CrossRef]
- Takagi, Y.; Mitsui, A.; Nishiyama, A.; Nozaki, K.; Sono, H.; Gon, Y.; Hashimoto, N.; Yodoi, J. Overexpression of thioredoxin in transgenic mice attenuates focal ischemic brain damage. Proc. Natl. Acad. Sci. USA 1999, 96, 4131–4136. [Google Scholar] [CrossRef]
- Hsia, C.C.; Hyde, D.M.; Ochs, M.; Weibel, E.R. An official research policy statement of the American Thoracic Society/European Respiratory Society: Standards for quantitative assessment of lung structure. Am. J. Respir. Crit. Care Med. 2010, 181, 394–418. [Google Scholar] [CrossRef]
- Yang, G.; Hinson, M.D.; Bordner, J.E.; Lin, Q.S.; Fernando, A.P.; La, P.; Wright, C.J.; Dennery, P.A. Silencing hyperoxia-induced C/EBPα in neonatal mice improves lung architecture via enhanced proliferation of alveolar epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 301, L187–L196. [Google Scholar] [CrossRef][Green Version]
- Son, A.; Kato, N.; Horibe, T.; Matsuo, Y.; Mochizuki, M.; Mitsui, A.; Kawakami, K.; Nakamura, H.; Yodoi, J. Direct association of Thioredoxin-1 (TRX) with macrophage migration inhibitory factor (MIF): Regulatory role of TRX on MIF internalization and signaling. Antioxid Redox Signal 2009, 11, 2595–2605. [Google Scholar] [CrossRef]
- Tanabe, N.; Hoshino, Y.; Marumo, S.; Kiyokawa, H.; Sato, S.; Kinose, D.; Uno, K.; Muro, S.; Hirai, T.; Yodoi, J.; et al. Thioredoxin-1 protects against neutrophilic inflammation and emphysema progression in a mouse model of chronic obstructive pulmonary disease exacerbation. PLoS ONE 2013, 8, e79016. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Matsuo, Y.; Fukunaga, A.; Ono, R.; Nishigori, C.; Yodoi, J. Thioredoxin ameliorates cutaneous inflammation by regulating the epithelial production and release of pro-inflammatory cytokines. Front. Immunol. 2013, 4, 269. [Google Scholar] [CrossRef]
- Yashiro, M.; Tsukahara, H.; Matsukawa, A.; Yamada, M.; Fujii, Y.; Nagaoka, Y.; Tsuge, M.; Yamashita, N.; Ito, T.; Yamada, M.; et al. Redox-active protein thioredoxin-1 administration ameliorates influenza A virus (H1 N1)-induced acute lung injury in mice. Crit. Care Med. 2013, 41, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Bloom, B.R.; Bennett, B. Mechanism of a reaction in vitro associated with delayed-type hypersensitivity. Science 1966, 153, 80–82. [Google Scholar] [CrossRef]
- Conroy, H.; Mawhinney, L.; Donnelly, S.C. Inflammation and cancer: Macrophage migration inhibitory factor (MIF)—The potential missing link. QJM 2010, 103, 831–836. [Google Scholar] [CrossRef]
- Kudrin, A.; Ray, D. Cunning factor: Macrophage migration inhibitory factor as a redox-regulated target. Immunol. Cell Biol. 2008, 86, 232–238. [Google Scholar] [CrossRef]
- Yamada, Y.; Nakamura, H.; Adachi, T.; Sannohe, S.; Oyamada, H.; Kayaba, H.; Yodoi, J.; Chihara, J. Elevated serum levels of thioredoxin in patients with acute exacerbation of asthma. Immunol. Lett. 2003, 86, 199–205. [Google Scholar] [CrossRef]
- Kato, A.; Odamaki, M.; Nakamura, H.; Yodoi, J.; Hishida, A. Elevation of blood thioredoxin in hemodialysis patients with hepatitis C virus infection. Kidney Int. 2003, 63, 2262–2268. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hofer, S.; Rosenhagen, C.; Nakamura, H.; Yodoi, J.; Bopp, C.; Zimmermann, J.B.; Goebel, M.; Schemmer, P.; Hoffmann, K.; Schulze-Osthoff, K.; et al. Thioredoxin in human and experimental sepsis. Crit. Care Med. 2009, 37, 2155–2159. [Google Scholar] [CrossRef]
- Callister, M.E.; Burke-Gaffney, A.; Quinlan, G.J.; Nicholson, A.G.; Florio, R.; Nakamura, H.; Yodoi, J.; Evans, T.W. Extracellular thioredoxin levels are increased in patients with acute lung injury. Thorax 2006, 61, 521–527. [Google Scholar] [CrossRef]
- Yu, T.; Zhang, W.; Lin, Y.; Li, Q.; Xue, J.; Cai, Z.; Cheng, Y.; Shao, B. Prognostic value of serum thioredoxin levels in ischemic stroke. Neurol. Res. 2017, 39, 988–995. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Yang, L.; Zhong, L. Decreased serum levels of thioredoxin in patients with coronary artery disease plus hyperhomocysteinemia is strongly associated with the disease severity. Atherosclerosis 2010, 212, 351–355. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagano, N.; Tanaka, K.; Ozawa, J.; Watanabe, T.; Miyake, F.; Matsumura, S.; Osada, K.; Matsuoka, K.; Tamura, M.; Namba, F. Attenuation of Hyperoxic Lung Injury in Newborn Thioredoxin-1-Overexpressing Mice through the Suppression of Proinflammatory Cytokine mRNA Expression. Biomedicines 2020, 8, 66. https://doi.org/10.3390/biomedicines8030066
Nagano N, Tanaka K, Ozawa J, Watanabe T, Miyake F, Matsumura S, Osada K, Matsuoka K, Tamura M, Namba F. Attenuation of Hyperoxic Lung Injury in Newborn Thioredoxin-1-Overexpressing Mice through the Suppression of Proinflammatory Cytokine mRNA Expression. Biomedicines. 2020; 8(3):66. https://doi.org/10.3390/biomedicines8030066
Chicago/Turabian StyleNagano, Nobuhiko, Kosuke Tanaka, Junichi Ozawa, Takaaki Watanabe, Fuyu Miyake, Shun Matsumura, Kohei Osada, Kikumi Matsuoka, Masanori Tamura, and Fumihiko Namba. 2020. "Attenuation of Hyperoxic Lung Injury in Newborn Thioredoxin-1-Overexpressing Mice through the Suppression of Proinflammatory Cytokine mRNA Expression" Biomedicines 8, no. 3: 66. https://doi.org/10.3390/biomedicines8030066
APA StyleNagano, N., Tanaka, K., Ozawa, J., Watanabe, T., Miyake, F., Matsumura, S., Osada, K., Matsuoka, K., Tamura, M., & Namba, F. (2020). Attenuation of Hyperoxic Lung Injury in Newborn Thioredoxin-1-Overexpressing Mice through the Suppression of Proinflammatory Cytokine mRNA Expression. Biomedicines, 8(3), 66. https://doi.org/10.3390/biomedicines8030066