HDL-Mediated Cholesterol Efflux and Plasma Loading Capacities Are Altered in Subjects with Metabolically- but Not Genetically Driven Non-Alcoholic Fatty Liver Disease (NAFLD)

 , , ,
, , ,  , , ,
, , ,  ,
,  ,
,
Abstract
1. Introduction
2. Material and Methods
2.1. Study Subjects
2.2. Measurement of HDL-Mediated Cholesterol Efflux Capacity (CEC)
2.3. Measurement of Plasma-Mediated Macrophage Cholesterol Loading Capacity (CLC)
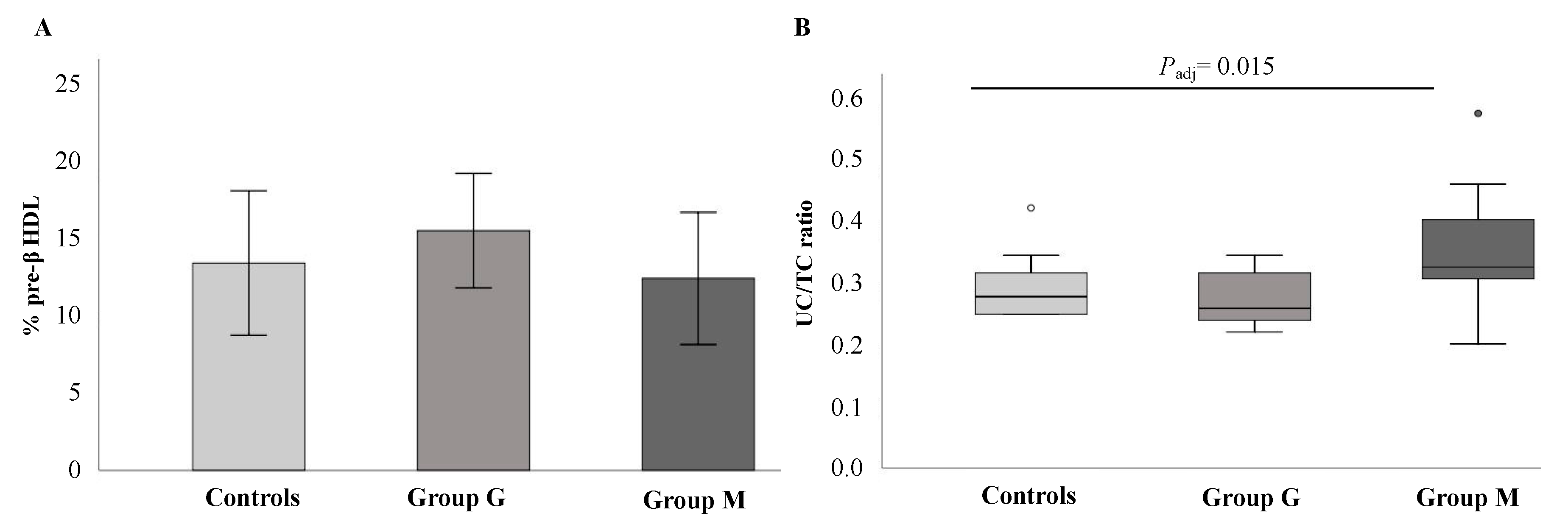
2.4. Analysis of HDL Subpopulations and Measurement of Plasma Cholesterol Esterification
2.5. Statistical Analysis
3. Results
3.1. Anthropometric and Metabolic Profiles of Study Groups
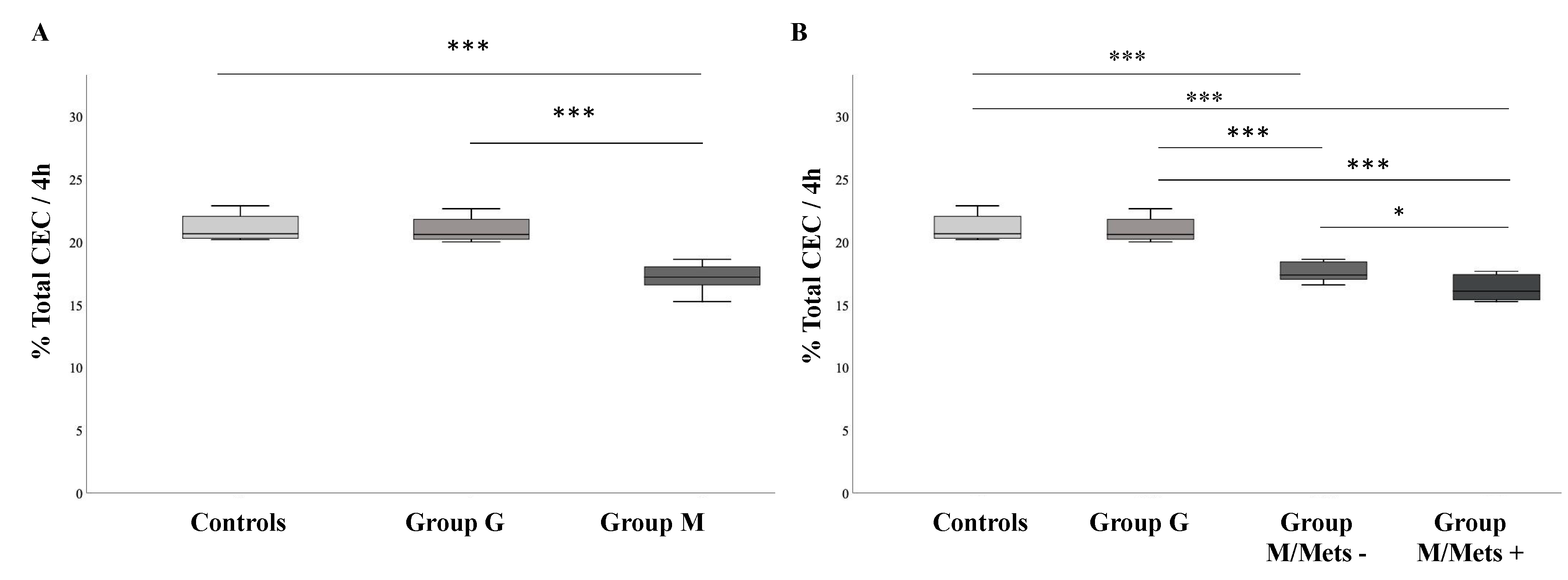
3.2. Comparison between Groups of HDL-Mediated Cholesterol Efflux Capacity (CEC)
3.3. Comparison between Groups of Plasma-Mediated Macrophage Cholesterol Loading Capacity (CLC)
3.4. Relationship of CEC and CLC with Clinical Parameters
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62, S47–S64. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M. Non-alcoholic fatty liver disease—A global public health perspective. J. Hepatol. 2019, 70, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Sookoian, S.; Chonchol, M.; Loria, P.; Targher, G. Cardiovascular and systemic risk in nonalcoholic fatty liver disease-atherosclerosis as a major player in the natural course of NAFLD. Curr. Pharm. Des. 2013, 19, 5177–5192. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Day, C.P. The Genetics of Nonalcoholic Fatty Liver Disease: Spotlight on PNPLA3 and TM6SF2. Semin. Liver. Dis. 2015, 35, 270–290. [Google Scholar] [PubMed]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [PubMed]
- Marjot, T.; Moolla, A.; Cobbold, J.F.; Hodson, L.; Tomlinson, J.W. Nonalcoholic fatty disease in adults: Current concepts in etiology, outcomes, and management. Endocr. Rev. 2020, 41, 66–117. [Google Scholar] [CrossRef]
- Targher, G.; Byrne, C.D.; Lonardo, A.; Zoppini, G.; Barbui, C. Non-alcoholic atty liver disease and risk of incident cardiovascular disease: A metaanalysis. J. Hepatol. 2016, 65, 589–600. [Google Scholar] [CrossRef]
- Baratta, F.; Pastori, D.; Angelico, F.; Balla, A.; Paganini, A.M.; Cocomello, N.; Ferro, D.; Violi, F.; Sanyal, A.J.; Del Ben, M. Nonalcoholic Fatty Liver Disease and Fibrosis Associated with Increased Risk of Cardiovascular Events in a Prospective Study. Clin. Gastroenterol. Hepatol. 2020, 18, 2324–2331.e4. [Google Scholar] [CrossRef]
- Lonardo, A.; Sookoian, S.; Pirola, C.J.; Targher, G. Non-alcoholic fatty liver disease and risk of cardiovascular disease. Metabolism 2016, 65, 1136–1150. [Google Scholar] [CrossRef]
- Di Costanzo, A.; D’Erasmo, L.; Polimeni, L.; Baratta, F.; Coletta, P.; Di Martino, M.; Loffredo, L.; Perri, L.; Ceci, F.; Montali, A.; et al. Non-alcoholic fatty liver disease and subclinical atherosclerosis: A comparison of metabolically- versus genetically-driven excess fat hepatic storage. Atherosclerosis 2017, 257, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Castaldo, L.; Laguzzi, F.; Strawbridge, R.J.; Baldassarre, D.; Veglia, F.; Vigo, L.; Tremoli, E.; de Faire, U.; Eriksson, P.; Smit, A.J.; et al. Genetic Variants Associated with Non-Alcoholic Fatty Liver Disease Do Not Associate with Measures of Sub-Clinical Atherosclerosis: Results from the IMPROVE Study. Genes 2020, 11, 1243. [Google Scholar] [CrossRef] [PubMed]
- Lauridsen, B.K.; Stender, S.; Kristensen, T.S.; Kofoed, K.F.; Køber, L.; Nordestgaard, B.G.; Tybjærg-Hansen, A. Liver fat content, non-alcoholic fatty liver disease, and ischaemic heart disease: Mendelian randomization and meta-analysis of 279,013 individuals. Eur. Heart J. 2018, 39, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Camont, L.; Chapman, M.J.; Kontush, A. Biological activities of HDL subpopulations and their relevance to cardiovascular disease. Trends Mol. Med. 2011, 17, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Khera, A.V.; Cuchel, M.; de la Llera-Moya, M.; Rodrigues, A.; Burke, M.F.; Jafri, K.; French, B.C.; Phillips, J.A.; Mucksavage, M.L.; Wilensky, R.L.; et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N. Engl. J. Med. 2011, 364, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, A.; Khera, A.; Berry, J.D.; Givens, E.G.; Ayers, C.R.; Wedin, K.E.; Neeland, I.J.; Yuhanna, I.S.; Rader, D.R.; de Lemos, J.A.; et al. HDL cholesterol efflux capacity and incident cardiovascular events. N. Engl. J. Med. 2014, 371, 2383–2393. [Google Scholar] [CrossRef]
- Favari, E.; Thomas, M.J.; Sorci-Thomas, M.G. High-Density Lipoprotein Functionality as a New Pharmacological Target on Cardiovascular Disease: Unifying Mechanism That Explains High-Density Lipoprotein Protection Toward the Progression of Atherosclerosis. J. Cardiovasc. Pharmacol. 2018, 71, 325–331. [Google Scholar] [CrossRef]
- Phillips, M.C. Molecular mechanisms of cellular cholesterol efflux. J. Biol. Chem. 2014, 289, 24020–24029. [Google Scholar] [CrossRef]
- Adorni, M.P.; Zimetti, F.; Puntoni, M.; Bigazzi, F.; Sbrana, F.; Minichilli, F.; Bernini, F.; Ronda, N.; Favari, E.; Sampietro, T. Cellular cholesterol efflux and cholesterol loading capacity of serum: Effects of LDL-apheresis. J. Lipid Res. 2012, 53, 984–989. [Google Scholar] [CrossRef]
- Zimetti, F.; Favari, E.; Cagliero, P.; Adorni, M.P.; Ronda, N.; Bonardi, R.; Gomaraschi, M.; Calabresi, L.; Bernini, F.; Guardamagna, O. Cholesterol trafficking-related serum lipoprotein functions in children with cholesteryl ester storage disease. Atherosclerosis 2015, 242, 443–449. [Google Scholar] [CrossRef]
- Grundy, S.M.; Cleeman, J.I.; Daniels, S.R.; Donato, K.A.; Eckel, R.H.; Franklin, B.A.; Gordon, D.J.; Krauss, R.M.; Savage, P.J.; Smith, S.C., Jr.; et al. Diagnosis and management of the metabolic syndrome: An American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation 2005, 112, 2735–2752, Erratum in: Circulation 2005, 112, e298. [Google Scholar] [CrossRef] [PubMed]
- Di Costanzo, A.; Di Leo, E.; Noto, D.; Cefalù, A.B.; Minicocci, I.; Polito, L.; D’Erasmo, L.; Cantisani, V.; Spina, R.; Tarugi, P.; et al. Clinical and biochemical characteristics of individuals with low cholesterol syndromes: A comparison between familial hypobetalipoproteinemia and familial combined hypolipidemia. J. Clin. Lipidol. 2017, 11, 1234–1242. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa, Y.; Funahashi, T.; Kihara, S.; Shimomura, I. Adiponectin and metabolic syndrome. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Di Costanzo, A.; Pacifico, L.; Chiesa, C.; Perla, F.M.; Ceci, F.; Angeloni, A.; D’Erasmo, L.; Di Martino, M.; Arca, M. Genetic and metabolic predictors of hepatic fat content in a cohort of Italian children with obesity. Pediatr. Res. 2019, 85, 671–677. [Google Scholar] [CrossRef]
- Di Costanzo, A.; Pacifico, L.; D’Erasmo, L.; Polito, L.; Martino, M.D.; Perla, F.M.; Iezzi, L.; Chiesa, C.; Arca, M. Nonalcoholic Fatty Liver Disease (NAFLD), But not Its Susceptibility Gene Variants, Influences the Decrease of Kidney Function in Overweight/Obese Children. Int. J. Mol. Sci. 2019, 20, 4444. [Google Scholar] [CrossRef]
- Di Costanzo, A.; Belardinilli, F.; Bailetti, D.; Sponziello, M.; D’Erasmo, L.; Polimeni, L.; Baratta, F.; Pastori, D.; Ceci, F.; Montali, A.; et al. Evaluation of Polygenic Determinants of Non-Alcoholic Fatty Liver Disease (NAFLD) By a Candidate Genes Resequencing Strategy. Sci. Rep. 2018, 8, 3702. [Google Scholar] [CrossRef]
- Zimetti, F.; De Vuono, S.; Gomaraschi, M.; Adorni, M.P.; Favari, E.; Ronda, N.; Ricci, M.A.; Veglia, F.; Calabresi, L.; Lupattelli, G. Plasma cholesterol homeostasis, HDL remodeling and function during the acute phase reaction. J. Lipid Res. 2017, 58, 2051–2060. [Google Scholar] [CrossRef]
- Bortnick, A.E.; Rothblat, G.H.; Stoudt, G.; Hoppe, K.L.; Royer, L.J.; McNeish, J.; Francone, O.L. The correlation of ATP-binding cassette 1 mRNA levels with cholesterol efflux from various cell lines. J. Biol. Chem. 2000, 275, 28634–28640. [Google Scholar] [CrossRef]
- Favari, E.; Calabresi, L.; Adorni, M.P.; Jessup, W.; Simonelli, S.; Franceschini, G.; Bernini, F. Small discoidal pre-beta1 HDL particles are efficient acceptors of cell cholesterol via ABCA1 and ABCG1. Biochemistry 2009, 48, 11067–11074. [Google Scholar] [CrossRef]
- Favari, E.; Lee, M.; Calabresi, L.; Franceschini, G.; Zimetti, F.; Bernini, F.; Kovanen, P.T. Depletion of pre-beta-high density lipoprotein by human chymase impairs ATP-binding cassette transporter A1- but not scavenger receptor class B type I-mediated lipid efflux to high density lipoprotein. J. Biol. Chem. 2004, 279, 9930–9936. [Google Scholar] [CrossRef]
- Zanotti, I.; Favari, E.; Bernini, F. Cellular cholesterol efflux pathways: Impact on intracellular lipid trafficking and methodological considerations. Curr. Pharm. Biotechnol. 2012, 13, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Gomaraschi, M.; Ossoli, A.; Castelnuovo, S.; Simonelli, S.; Pavanello, C.; Balzarotti, G.; Arca, M.; Di Costanzo, A.; Sampietro, T.; Vaudo, G.; et al. Depletion in LpA-I:A-II particles enhances HDL-mediated endothelial protection in familial LCAT deficiency. J. Lipid Res. 2017, 58, 994–1001. [Google Scholar] [CrossRef] [PubMed]
- Fadaei, R.; Poustchi, H.; Meshkani, R.; Moradi, N.; Golmohammadi, T.; Merat, S. Impaired HDL cholesterol efflux capacity in patients with non-alcoholic fatty liver disease is associated with subclinical atherosclerosis. Sci. Rep. 2018, 8, 11691. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, E.H.; Gruppen, E.G.; Ebtehaj, S.; Bakker, S.J.L.; Tietge, U.J.F.; Dullaart, R.P.F. Cholesterol efflux capacity is impaired in subjects with an elevated Fatty Liver Index, a proxy of non-alcoholic fatty liver disease. Atherosclerosis 2018, 277, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Nestel, P.; Hoang, A.; Sviridov, D.; Straznicky, N. Cholesterol efflux from macrophages is influenced differentially by plasmas from overweight insulin-sensitive and-resistant subjects. Int. J. Obes. 2012, 36, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Lucero, D.; Sviridov, D.; Freeman, L.; López, G.I.; Fassio, E.; Remaley, A.T.; Schreier, L. Increased cholesterol efflux capacity in metabolic syndrome: Relation with qualitative alterations in HDL and LCAT. Atherosclerosis 2015, 242, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Borggreve, S.E.; De Vries, R.; Dullaart, R.P. Alterations in high-density lipoprotein metabolism and reverse cholesterol transport in insulin resistance and type 2 diabetes mellitus: Role of lipolytic enzymes, lecithin:cholesterol acyltransferase and lipid transfer proteins. Eur. J. Clin. Investig. 2003, 33, 1051–1069. [Google Scholar] [CrossRef]
- Annema, W.; Dikkers, A.; de Boer, J.F.; van Greevenbroek, M.M.J.; van der Kallen, C.J.H.; Schalkwijk, C.G.; Stehouwer, C.D.A.; Dullaart, R.P.F.; Tietge, U.J.F. Impaired HDL cholesterol efflux in metabolic syndrome is unrelated to glucose tolerance status: The CODAM study. Sci. Rep. 2016, 6, 27367. [Google Scholar] [CrossRef]
- Gall, J.; Frisdal, E.; Bittar, R.; Le Goff, W.; Bruckert, E.; Lesnik, P.; Guerin, M.; Giral, P. Association of Cholesterol Efflux Capacity with Clinical Features of Metabolic Syndrome: Relevance to Atherosclerosis. J. Am. Heart Assoc. 2016, 5, e004808. [Google Scholar] [CrossRef]
- Kobayashi, A.; Takanezawa, Y.; Hirata, T.; Shimizu, Y.; Misasa, K.; Kioka, N.; Arai, H.; Ueda, K.; Matsuo, M. Efflux of sphingomyelin, cholesterol, and phosphatidylcholine by ABCG1. J. Lipid Res. 2006, 47, 1791–1802. [Google Scholar] [CrossRef]
- Meikle, P.J.; Wong, G.; Barlow, C.K.; Weir, J.M.; Greeve, M.A.; MacIntosh, G.L.; Almasy, L.; Comuzzie, A.G.; Mahaney, M.C.; Kowalczyk, A.; et al. Plasma lipid profiling shows similar associations with prediabetes and type 2 diabetes. PLoS ONE 2013, 8, e74341. [Google Scholar] [CrossRef] [PubMed]
- Sano, O.; Kobayashi, A.; Nagao, K.; Kumagai, K.; Kioka, N.; Hanada, K.; Ueda, K.; Matsuo, M. Sphingomyelin-dependence of cholesterol efflux mediated by ABCG1. J. Lipid Res. 2007, 48, 2377–2384. [Google Scholar] [CrossRef] [PubMed]
- Ronda, N.; Greco, D.; Adorni, M.P.; Zimetti, F.; Favari, E.; Hjeltnes, G.; Mikkelsen, K.; Borghi, M.O.; Favalli, E.G.; Gatti, R.; et al. Newly identified antiatherosclerotic activity of methotrexate and adalimumab: Complementary effects on lipoprotein function and macrophage cholesterol metabolism. Arthritis. Rheumatol. 2015, 67, 1155–1164. [Google Scholar] [CrossRef]
- Ebrahimi-Mamaeghani, M.; Mohammadi, S.; Arefhosseini, S.R.; Fallah, P.; Bazi, Z. Adiponectin as a potential biomarker of vascular disease. Vasc. Health Risk Manag. 2015, 11, 55–70. [Google Scholar] [PubMed]
- Eslam, M.; Sanyal, A.J.; George, J.; International Consensus Panel. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NAFLD | ||||
|---|---|---|---|---|
| Controls | GroupG | GroupM | p Value | |
| N | 10 | 10 | 19 | |
| Age, years | 55.9 ± 6.9 | 54.0 ± 8.5 | 55.6 ± 3.8 | 0.7 |
| Males, % | 80 | 78.9 | 80 | 0.9 |
| MetS, % | 0 | 0 * | 42.1 | 0.005 |
| BMI, kg/m2 | 25.5 ± 2.9 | 27.2 ± 3.6 | 30.8 ± 5.8 *** | 0.01 |
| WC, cm | 93.9 ± 9.4 | 99.0 ± 12.4 | 107.5 ± 10.1 *** | 0.006 |
| Smokers, % | 50 | 40 | 15.8 | 0.12 |
| T2DM, % | 0 | 0 | 15.8 | 0.18 |
| HFF, % | 2.06 ± 1.61 ** | 36.1 ± 22.4 | 32.0 ± 19.6 *** | <0.001 |
| Systolic BP, mmHg | 115 (103.7–120) | 120 (112.5–130) * | 130 (120–140) *** | 0.003 |
| Diastolic BP, mmHg | 70 (65–80) | 80 (72.5–84.2) | 80 (75–85) *** | 0.015 |
| Total cholesterol, mg/dL | 206.8 ± 28.5 | 206.4 ± 35.2 | 208.2 ± 27.8 | 0.9 |
| HDL cholesterol, mg/dL | 64.2 ± 10.3 ** | 52.6 ± 11.8 | 45.6 ± 12.9 *** | 0.002 |
| LDL cholesterol, mg/dL | 125.1 ± 30.2 | 133.1 ± 27.9 | 126.9 ± 24.5 | 0.7 |
| Total triglycerides, mg/dL | 85.5 (59.5–102.7) | 88 (84.7–106) * | 159 (125–210) *** | 0.005 |
| ApoB, mg/dL | 97.9 ± 16.5 | 97.5 ± 16.5 | 92.0 ± 21.3 | 0.6 |
| ApoAI, mg/dL | 133 ±14.4 ** | 113.9± 14.6 | 106.6 ± 20.0 *** | 0.002 |
| Fasting glucose, mg/dL | 78.5 ± 10.0 | 82.9 ± 13.8 | 98.0 ± 21.2 *** | 0.01 |
| Fasting insulin, U/L | 5.3 (4.2–7.9) | 5.9 (4.9–10.9) * | 12.7 (8.6–18.9) *** | 0.01 |
| HOMA -IR | 0.9 (0.9–1.6) | 1.2 (0.9–2.2) * | 2.9 (1.8–4.2) *** | <0.001 |
| ADP, ng/dl | 6596.8 ± 3179.6 | 4791.5 ± 1796.4 * | 901.0 ± 413.8 *** | <0.001 |
| AST, U/L | 29.5 (19.7–36.5) | 31.5 (22.2–33.2) * | 20 (18–26) | 0.026 |
| ALT, U/L | 13 (10–22) | 16 (13.7–19.5) * | 34 (21.5–42.5) *** | <0.001 |
| y-GT, U/L | 18.5 (13.2–24.5) | 20.5 (15.2–29.2) | 25.5 (20.7–38.2) *** | 0.05 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Costanzo, A.; Ronca, A.; D’Erasmo, L.; Manfredini, M.; Baratta, F.; Pastori, D.; Di Martino, M.; Ceci, F.; Angelico, F.; Del Ben, M.; et al. HDL-Mediated Cholesterol Efflux and Plasma Loading Capacities Are Altered in Subjects with Metabolically- but Not Genetically Driven Non-Alcoholic Fatty Liver Disease (NAFLD). Biomedicines 2020, 8, 625. https://doi.org/10.3390/biomedicines8120625
Di Costanzo A, Ronca A, D’Erasmo L, Manfredini M, Baratta F, Pastori D, Di Martino M, Ceci F, Angelico F, Del Ben M, et al. HDL-Mediated Cholesterol Efflux and Plasma Loading Capacities Are Altered in Subjects with Metabolically- but Not Genetically Driven Non-Alcoholic Fatty Liver Disease (NAFLD). Biomedicines. 2020; 8(12):625. https://doi.org/10.3390/biomedicines8120625
Chicago/Turabian StyleDi Costanzo, Alessia, Annalisa Ronca, Laura D’Erasmo, Matteo Manfredini, Francesco Baratta, Daniele Pastori, Michele Di Martino, Fabrizio Ceci, Francesco Angelico, Maria Del Ben, and et al. 2020. "HDL-Mediated Cholesterol Efflux and Plasma Loading Capacities Are Altered in Subjects with Metabolically- but Not Genetically Driven Non-Alcoholic Fatty Liver Disease (NAFLD)" Biomedicines 8, no. 12: 625. https://doi.org/10.3390/biomedicines8120625
APA StyleDi Costanzo, A., Ronca, A., D’Erasmo, L., Manfredini, M., Baratta, F., Pastori, D., Di Martino, M., Ceci, F., Angelico, F., Del Ben, M., Pavanello, C., Turri, M., Calabresi, L., Favari, E., & Arca, M. (2020). HDL-Mediated Cholesterol Efflux and Plasma Loading Capacities Are Altered in Subjects with Metabolically- but Not Genetically Driven Non-Alcoholic Fatty Liver Disease (NAFLD). Biomedicines, 8(12), 625. https://doi.org/10.3390/biomedicines8120625