The Genetic Evidence of Burn-Induced Cardiac Mitochondrial Metabolism Dysfunction



Abstract
1. Introduction
2. Experimental Section
2.1. Ethics Statement
2.2. Animal Model
2.3. Preparation of Permeabilized Fibers from Trout Heart
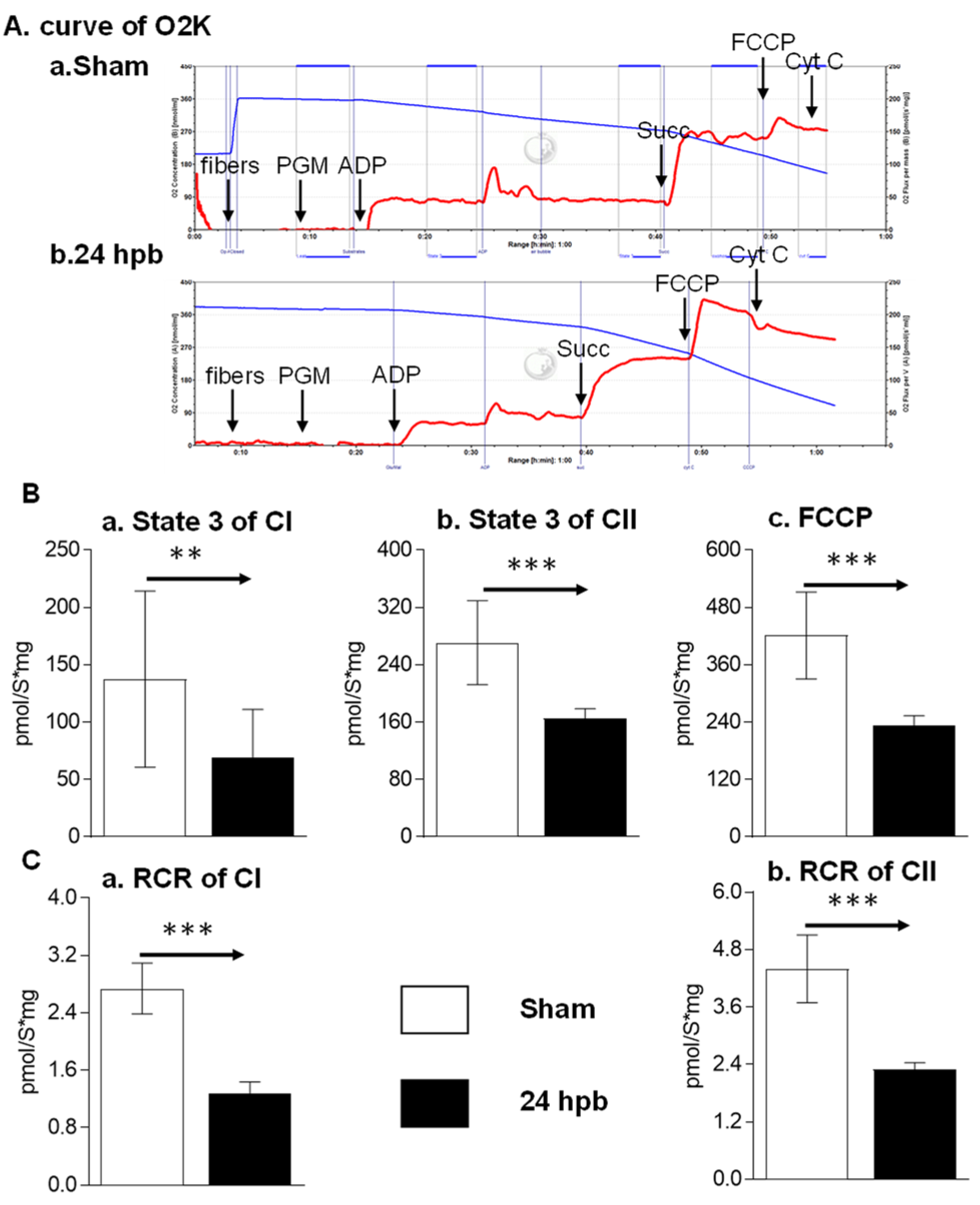
2.4. O2K Respirometer System
2.5. Isolation of Cardiac Mitochondria
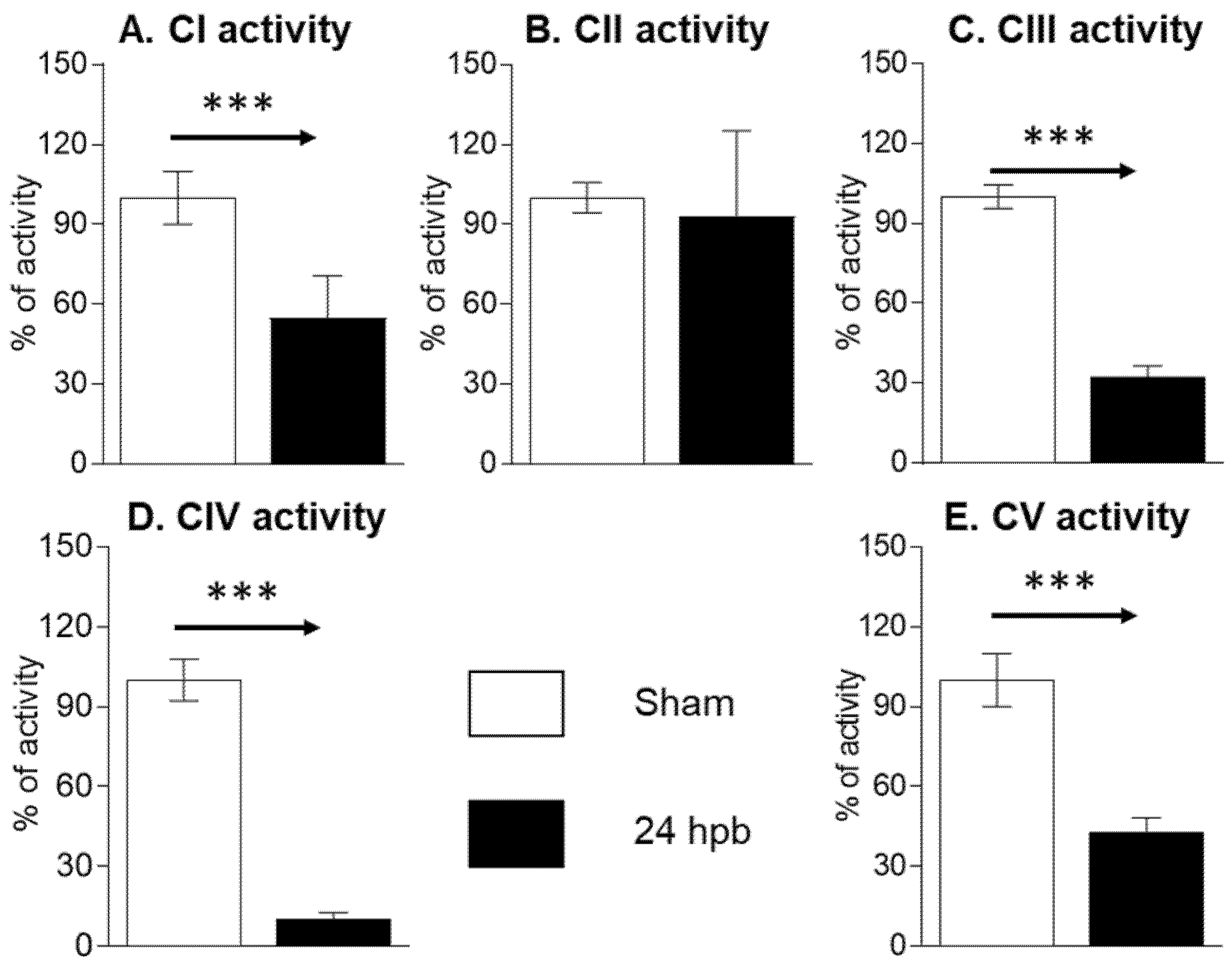
2.6. Oxidative Phosphorylation Complex Activities
2.7. Mitochondrial Copy Number
2.8. RNA Isolation
2.9. First-Strand cDNA Synthesis
2.10. Real-Time PCR Array and qPCR
2.11. Quantitative Western Blotting (WB)
2.12. Statistical Analysis
3. Results
3.1. Burn Induces Cardiac Mitochondrial Dysfunction
3.2. Burn Induces Cardiac Mitochondrial Electron Transport Chain Dysfunction
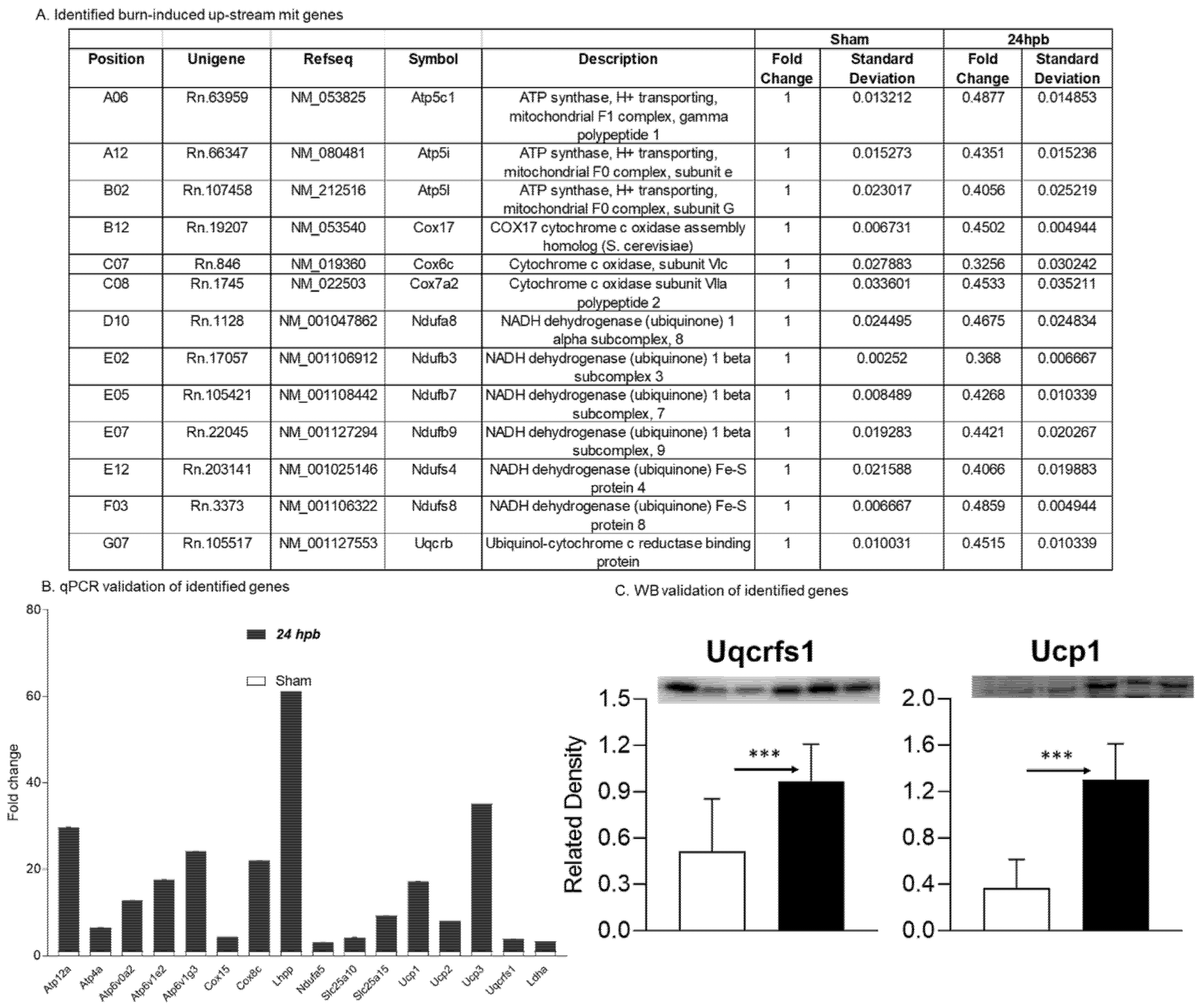
3.3. Analysis of Burn-Induced Cardiac Mitochondrial Metabolism-Related Gene Expression
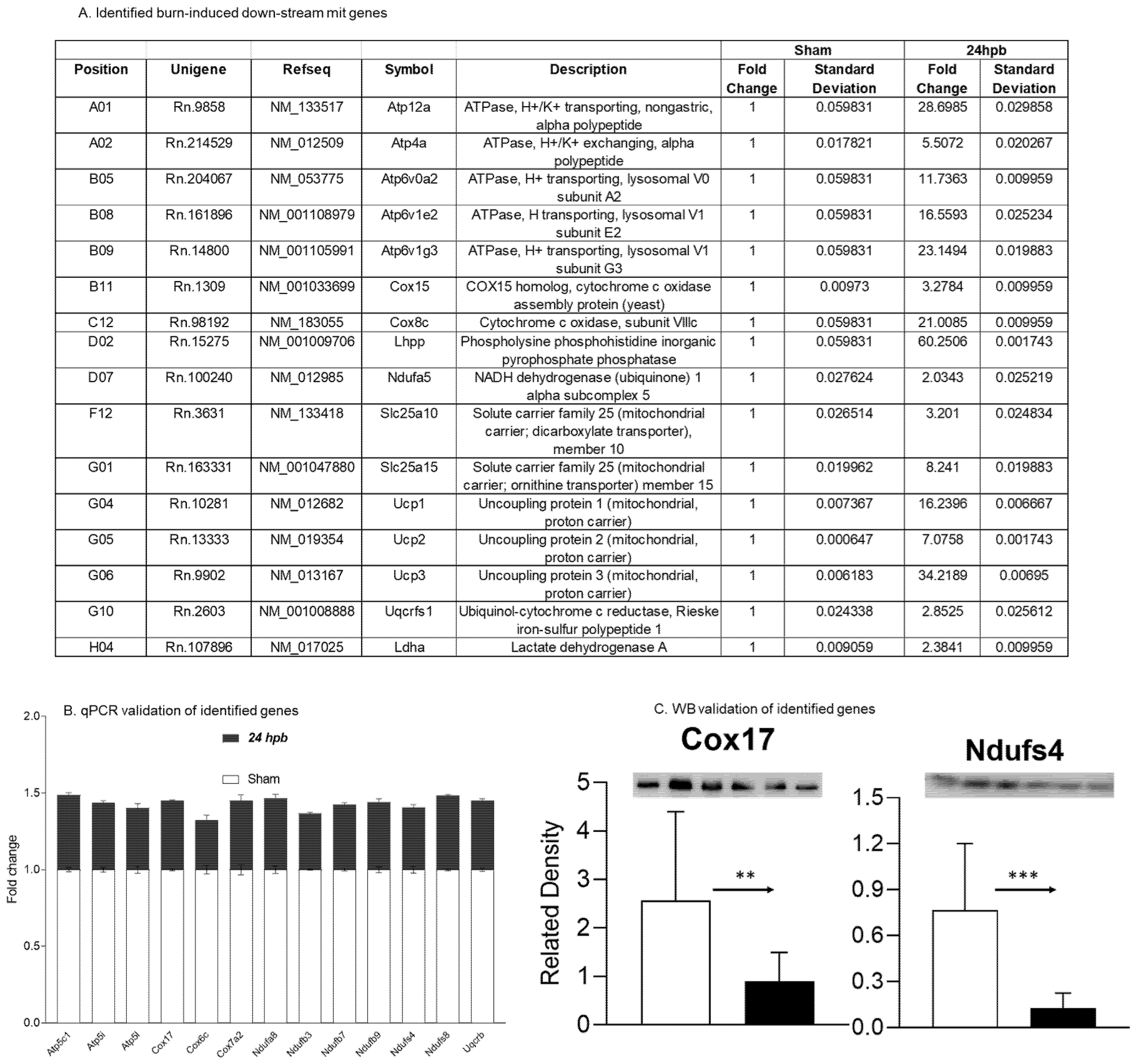
3.4. Burn-Induced Downregulated Cardiac Mitochondrial Metabolism-Related Gene Expression
3.5. Burn-Induced Upregulated Cardiac Mitochondrial Metabolism-Related Gene Expression
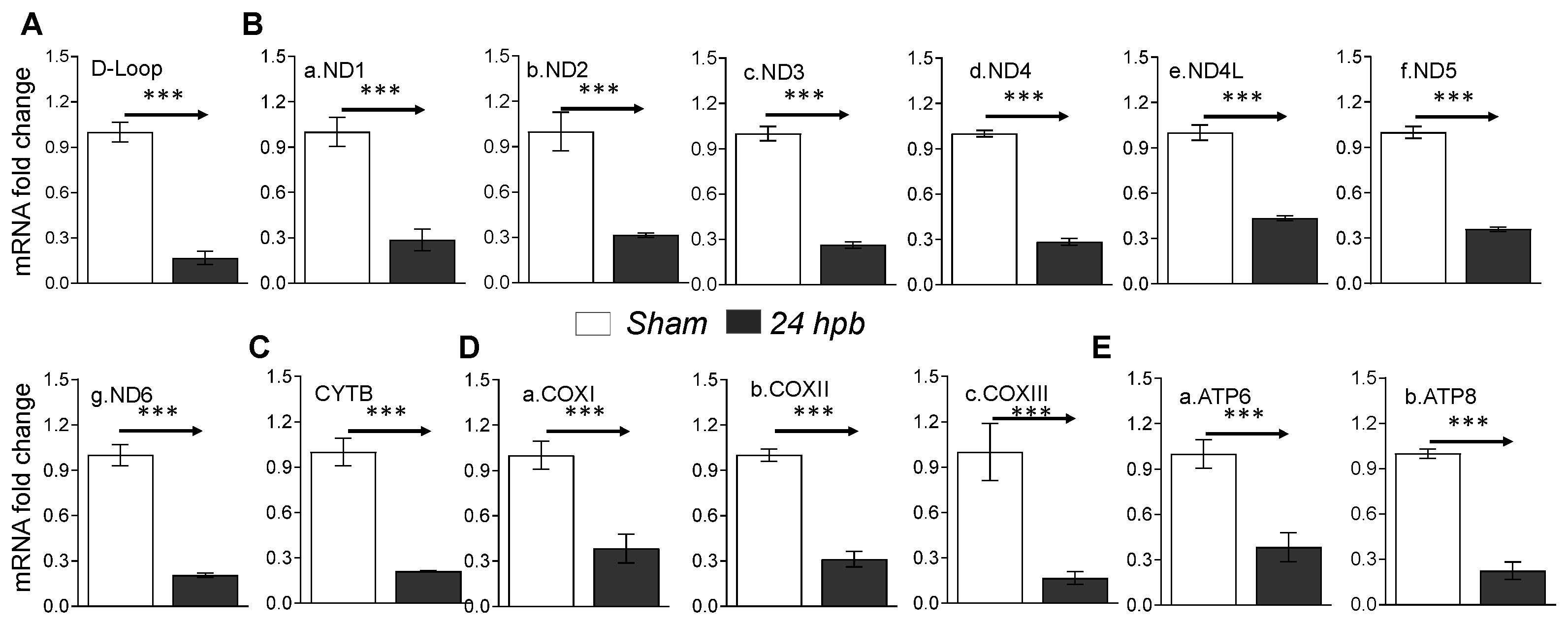
3.6. Downregulation of Cardiac Mitochondrial DNA-Encoded Genes and Proteins after Burn
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACTB | Beta-actin |
| B2M | Beta-2-microglobulin |
| CT | Cycle threshold |
| COX | Cytochrome c oxidase |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| HPRT1 | Hypoxanthine phosphoribosyl transferase 1 |
| mtDNA | Mitochondrial DNA |
| PCR | Polymerase chain reaction |
| RCR | Respiratory control ratio |
| RPLP0 | Ribosomal protein: large, P0 |
| SD | Standard deviation |
| TBSA | Total body surface body area |
| UCP | Uncoupling proteins |
References
- Fozzard, H.A. Myocardial injury in burn shock. Ann. Surg. 1961. [Google Scholar] [CrossRef] [PubMed]
- Howard, T.S.; Hermann, D.G.; McQuitty, A.L.; Woodson, L.C.; Kramer, G.C.; Herndon, D.N.; Ford, P.M.; Kinsky, M.P. Burn-induced cardiac dysfunction increases length of stay in pediatric burn patients. J. Burn Care Res. 2013. [Google Scholar] [CrossRef] [PubMed]
- Papp, A.; Uusaro, A.; Parviainen, I.; Hartikainen, J.; Ruokonen, E. Myocardial function and haemodynamics in extensive burn trauma: Evaluation by clinical signs, invasive monitoring, echocardiography and cytokine concentrations. A prospective clinical study. Acta Anaesthesiol. Scand. 2003. [Google Scholar] [CrossRef] [PubMed]
- Herndon, D.N.; Tompkins, R.G. Support of the metabolic response to burn injury. Lancet 2004, 363, 1895–1902. [Google Scholar] [CrossRef]
- Guillory, A.N.; Clayton, R.P.; Herndon, D.N.; Finnerty, C.C. Cardiovascular dysfunction following burn injury: What we have learned from rat and mouse models. Int. J. Mol. Sci. 2016, 17, 53. [Google Scholar] [CrossRef]
- Bohanon, F.J.; Nunez Lopez, O.; Herndon, D.N.; Wang, X.; Bhattarai, N.; Ayadi, A.E.; Prasai, A.; Jay, J.W.; Rojas-Khalil, Y.; Toliver-Kinsky, T.E.; et al. Burn Trauma Acutely Increases the Respiratory Capacity and Function of Liver Mitochondria. Shock 2018. [Google Scholar] [CrossRef]
- Padfield, K.E.; Astrakas, L.G.; Zhang, Q.; Gopalan, S.; Dai, G.; Mindrinos, M.N.; Tompkins, R.G.; Rahme, L.G.; Tzika, A.A. Burn injury causes mitochondrial dysfunction in skeletal muscle. Proc. Natl. Acad. Sci. USA 2005. [Google Scholar] [CrossRef]
- Nakazawa, H.; Ikeda, K.; Shinozaki, S.; Kobayashi, M.; Ikegami, Y.; Fu, M.; Nakamura, T.; Yasuhara, S.; Yu, Y.M.; Martyn, J.A.J.; et al. Burn-induced muscle metabolic derangements and mitochondrial dysfunction are associated with activation of HIF-1α and mTORC1: Role of protein farnesylation. Sci. Rep. 2017. [Google Scholar] [CrossRef]
- Righi, V.; Constantinou, C.; Mintzopoulos, D.; Khan, N.; Mupparaju, S.P.; Rahme, L.G.; Swartz, H.M.; Szeto, H.H.; Tompkins, R.G.; Tzika, A.A. Mitochondria-targeted antioxidant promotes recovery of skeletal muscle mitochondrial function after burn trauma assessed by in vivo 31P nuclear magnetic resonance and electron paramagnetic resonance spectroscopy. FASEB J. 2013. [Google Scholar] [CrossRef]
- Brown, D.A.; O’Rourke, B. Cardiac mitochondria and arrhythmias. Cardiovasc. Res. 2010, 88, 241–249. [Google Scholar] [CrossRef]
- Bugger, H.; Abel, E.D. Mitochondria in the diabetic heart. Cardiovasc. Res. 2010, 88, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Rosca, M.G.; Hoppel, C.L. Mitochondrial dysfunction in heart failure. Heart Fail. Rev. 2013, 18, 607–622. [Google Scholar] [CrossRef] [PubMed]
- Marín-García, J.; Akhmedov, A.; Rybin, V.; Moe, G.W. Mitochondria and Their Role in Cardiovascular Disease; Library of Congress Control Number: 2012948132; Springer: New York, NY, USA; Heidelberg, Germany; Dordrecht, The Netherland; London, UK, 2013; ISBN1 978-1-4614-4598-2. ISBN2 978-1-4614-4599-9 (eBook). [Google Scholar] [CrossRef]
- Zang, Q.; Maass, D.L.; White, J.; Horton, J.W. Cardiac mitochondrial damage and loss of ROS defense after burn injury: The beneficial effects of antioxidant therapy. J. Appl. Physiol. 2007. [Google Scholar] [CrossRef] [PubMed]
- Burch, T.C.; Rhim, J.S.; Nyalwidhe, J.O. Mitochondria Biogenesis and Bioenergetics Gene Profiles in Isogenic Prostate Cells with Different Malignant Phenotypes. BioMed Res. Int. 2016. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Kobunai, T.; Yamamoto, Y.; Murono, K.; Otani, K.; Yasuda, K.; Nishikawa, T.; Kiyomatsu, T.; Kawai, K.; Hata, K.; et al. Increased copy number variation of mtDNA in an array-based digital PCR assay predicts ulcerative colitis-associated colorectal cancer. In Vivo (Brooklyn) 2017. [Google Scholar] [CrossRef]
- Li, Z.; Wang, Q.; Yu, H.; Zou, K.; Xi, Y.; Mi, W.; Ma, Y. Screening of Key Genes in Severe Burn Injury at Different Stages via Analyzing Gene Expression Data. J. Burn Care Res. 2016. [Google Scholar] [CrossRef]
- Liang, W.Y.; Tang, L.X.; Yang, Z.C.; Huang, Y.S. Calcium induced the damage of myocardial mitochondrial respiratory function in the early stage after severe burns. Burns 2002. [Google Scholar] [CrossRef]
- Pesta, D.; Gnaiger, E. High-resolution respirometry: OXPHOS protocols for human cells and permeabilized fibers from small biopsies of human muscle. Methods Mol. Biol. 2012, 810, 25–58. [Google Scholar] [CrossRef]
- Laskowski, K.R.; Russell, R.R. Uncoupling proteins in heart failure. Curr. Heart Fail. Rep. 2008, 5, 75–79. [Google Scholar] [CrossRef]
- Kozak, L.P.; Anunciado-Koza, R. UCP1: Its involvement and utility in obesity. Int. J. Obes. 2008, 32 (Suppl. 7), S32–S38. [Google Scholar] [CrossRef]
- Liu, S.-S. Generating, partitioning, targeting and functioning of superoxide in mitochondria. Biosci. Rep. 1997, 17, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Giordano, F.J. Oxygen, oxidative stress, hypoxia, and heart failure. J. Clin. Investig. 2005, 115, 500–508. [Google Scholar] [CrossRef]
- Givertz, M.M.; Colucci, W.S. New targets for heart-failure therapy: Endothelin, inflammatory cytokines, and oxidative stress. Lancet 1998. [Google Scholar] [CrossRef]
- Liu, Y.; Fiskum, G.; Schubert, D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J. Neurochem. 2002. [Google Scholar] [CrossRef]
- Kaminski, M.; Kiessling, M.; Suss, D.; Krammer, P.H.; Gulow, K. Novel Role for Mitochondria: Protein Kinase C -Dependent Oxidative Signaling Organelles in Activation-Induced T-Cell Death. Mol. Cell. Biol. 2007. [Google Scholar] [CrossRef]
- Zong, N.C.; Li, H.; Li, H.; Lam, M.P.Y.; Jimenez, R.C.; Kim, C.S.; Deng, N.; Kim, A.K.; Choi, J.H.; Zelaya, I.; et al. Integration of cardiac proteome biology and medicine by a specialized knowledgebase. Circ. Res. 2013. [Google Scholar] [CrossRef] [PubMed]
- Van Den Heuvel, L.; Ruitenbeek, W.; Smeets, R.; Gelman-Kohan, Z.; Elpeleg, O.; Loeffen, J.; Trijbels, F.; Mariman, E.; De Bruijn, D.; Smeitink, J. Demonstration of a new pathogenic mutation in human complex I deficiency: A 5-bp duplication in the nuclear gene encoding the 18-kD (AQDQ) subunit. Am. J. Hum. Genet. 1998. [Google Scholar] [CrossRef] [PubMed]
- Bugiani, M.; Invernizzi, F.; Alberio, S.; Briem, E.; Lamantea, E.; Carrara, F.; Moroni, I.; Farina, L.; Spada, M.; Donati, M.A.; et al. Clinical and molecular findings in children with complex I deficiency. Biochim. Biophys. Acta Bioenerget. 2004, 1659, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Sharma, L.; Lu, J.; Bai, Y. Mitochondrial Respiratory Complex I: Structure, Function and Implication in Human Diseases. Curr. Med. Chem. 2009. [Google Scholar] [CrossRef]
- Chang, J.; Jung, H.J.; Jeong, S.H.; Kim, H.K.; Han, J.; Kwon, H.J. A mutation in the mitochondrial protein UQCRB promotes angiogenesis through the generation of mitochondrial reactive oxygen species. Biochem. Biophys. Res. Commun. 2014. [Google Scholar] [CrossRef]
- Jung, H.J.; Cho, M.; Kim, Y.; Han, G.; Kwon, H.J. Development of a novel class of mitochondrial ubiquinol-cytochrome c reductase binding protein (UQCRB) modulators as promising antiangiogenic leads. J. Med. Chem. 2014. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.J.; Kim, K.H.; Kim, N.D.; Han, G.; Kwon, H.J. Identification of a novel small molecule targeting UQCRB of mitochondrial complex III and its anti-angiogenic activity. Bioorg. Med. Chem. Lett. 2011. [Google Scholar] [CrossRef] [PubMed]
- Mansilla, N.; Racca, S.; Gras, D.E.; Gonzalez, D.H.; Welchen, E. The complexity of mitochondrial complex iv: An update of cytochrome c oxidase biogenesis in plants. Int. J. Mol. Sci. 2018, 19, 662. [Google Scholar] [CrossRef] [PubMed]
- Maxfield, A.B.; Heaton, D.N.; Winge, D.R. Cox17 Is Functional When Tethered to the Mitochondrial Inner Membrane. J. Biol. Chem. 2004. [Google Scholar] [CrossRef]
- Amaravadi, R.; Glerum, D.M.; Tzagoloff, A. Isolation of a cDNA encoding the human homolog of COX17, a yeast gene essential for mitochondrial copper recruitment. Hum. Genet. 1997. [Google Scholar] [CrossRef] [PubMed]
- Vitali, M.; Venturelli, E.; Galimberti, D.; Benerini Gatta, L.; Scarpini, E.; Finazzi, D. Analysis of the genes coding for subunit 10 and 15 of cytochrome c oxidase in Alzheimer’s disease. J. Neural Transm. 2009. [Google Scholar] [CrossRef] [PubMed]
- Capaldi, R.A.; Aggeler, R.; Turina, P.; Wilkens, S. Coupling between catalytic sites and the proton channel in F1F0-type ATPases. Trends Biochem. Sci. 1994, 19, 284–289. [Google Scholar] [CrossRef]
- Nijtmans, L.G.J.; Klement, P.; Houštěk, J.; van den Bogert, C. Assembly of mitochondrial ATP synthase in cultured human cells: Implications for mitochondrial diseases. BBA Mol. Basis Dis. 1995. [Google Scholar] [CrossRef]
- Zeviani, M.; Di Donato, S. Mitochondrial disorders. Brain 2004, 127, 2153–2172. [Google Scholar] [CrossRef]
- Scudieri, P.; Musante, I.; Caci, E.; Venturini, A.; Morelli, P.; Walter, C.; Tosi, D.; Palleschi, A.; Martin-Vasallo, P.; Sermet-Gaudelus, I.; et al. Increased expression of ATP12A proton pump in cystic fibrosis airways. JCI Insight 2018. [Google Scholar] [CrossRef]
- Crambert, G. H-K-ATpase type 2: Relevance for renal physiology and beyond. Am. J. Physiol. Ren. Physiol. 2014, 306, F693–F700. [Google Scholar] [CrossRef] [PubMed]
- Knez, J.; Salvi, E.; Tikhonoff, V.; Stolarz-Skrzypek, K.; Ryabikov, A.; Thijs, L.; Braga, D.; Kloch-Badelek, M.; Malyutina, S.; Casiglia, E.; et al. Left ventricular diastolic function associated with common genetic variation in ATP12A in a general population. BMC Med. Genet. 2014. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, K.; Ashenagar, M.S.; Tabuchi, M.; Higashino, H. Whole rat DNA array survey for candidate genes related to hypertension in kidneys from three spontaneously hypertensive rat substrains at two stages of age and with hypotensive induction caused by hydralazine hydrochloride. Exp. Ther. Med. 2011. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.J.; Cummins, C.B.; Szczesny, B.; Radhakrishnan, R.S. Cardiac Dysfunction after Burn Injury: Role of the AMPK-SIRT1-PGCalpha-NFE2L2-ARE Pathway. J. Am. Coll. Surg. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.J.; Cummins, C.B.; Radhakrishnan, R.S. Burn-Induced Cardiac Mitochondrial Dysfunction via Interruption of the PDE5A-cGMP-PKG Pathway. Int. J. Mol. Sci. 2020, 21, 2350. [Google Scholar] [CrossRef]
- Wen, J.J.; Cummins, C.; Radhakrishnan, R.S. Sildenafil Recovers Burn-Induced Cardiomyopathy. Cells 2020, 9, 1393. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | 5′-Forward-3′ | 5′-Reverse-3′ | Amplicon Size (bp) | Accession # |
|---|---|---|---|---|
| Atp5f1c | AGATGCATCGGTCATTGCCT | AGCCACGACGGTACTGAAAG | 142 | NM_053825.2 |
| Atp5me | GTCACGGACAAAATGGTGCC | GTATGCCATGCCGAGGATCA | 87 | NM_080481.1 |
| Atp5mg | GGCCAAGTTCATCCGTAACC | GAACCAGCTCAACCCTAGCA | 116 | NM_212516.2 |
| Cox17 | TGTGGACATCTCATCGAGGC | ATTCACAAAGTGGGCCACCA | 82 | NM_053540.2 |
| Cox6c | CCACAGATGCGTGGTCTTCT | TCCTAGGGCCACAACGAATG | 72 | NM_019360.2 |
| Cox 7a2 | GAGTTCCGTTTCCGGTCTGG | CCTTCGTGAAGTGGTGCTGA | 143 | NM_022503.2 |
| Ndufa8 | GCCTGCCGTAAGCTAAGCAA | AGGGACCTGAGAGGTAACCG | 105 | NM_001047862.2 |
| Ndufb3 | CGCTAGTCCCGGAACGTTTA | TGACAGCAATGTGACCTCCC | 132 | NM_001106912.1 |
| Ndufb7 | AAGGTAGGGCAGAGTAGCCA | CTCCGGAAGACCGTAGTTCG | 118 | NM_001108442.1 |
| Ndufb9 | GAGCTGGGATCGGGAGGTTA | AAGTCGCCTTCCTTTCTGGC | 96 | NM_001127294.2 |
| Ndufs4 | TCCTGCTCGCAATAACATGC | CATGTTGGAGAGGGGATCGG | 130 | NM_001025146.1 |
| Ndufs8 | AGCCGCTGCACTTCAAGAT | ACTGCACTGCTATGAAGGCT | 112 | NM_001106322.2 |
| Nqcrb | GGGATTAGGCAACCAGCACT | TCTGAAAGGCTGTGTCTCGG | 116 | NM_001127553.2 |
| Gene | 5′-Forward-3′ | 5′-Reverse-3′ | Amplicon Size (bp) | Accession # |
|---|---|---|---|---|
| Atp12a | TCTTCGCCTTCACCACTCAG | GTTGAGGTGAGCCGGATAGG | 149 | NM_133517.2 |
| Atp6v0a2 | CTTCCGTAGCGAGAGCATGT | TTGAGGTCTCGGAACTGCAC | 111 | NM_053775.3 |
| Atp6v1e2 | GACCAAAGGCGGCTCCTG | ATGGCTGCTCTCAGGAATGG | 132 | NM_001108979.1 |
| Atp6v1g3 | TCCACCGAAGTTGCCTACAC | GCAGGTTCCTTCCTTTCCTCA | 134 | NM_001105991.1 |
| Cox15 | TGACATAGGCATTTTACTCTCCGA | TTCTGAGGTGAAGGGAGCCT | 81 | NM_001033699.4 |
| Cox8c | TCCCAGCTGCGTATGTGATG | CAAGACGTTCAAACGGGCAC | 148 | NM_183055.1 |
| Lhpp | CCTTCCGAGACAGTGGACG | ATCGGCAAAGCCTTGGGTAG | 102 | NM_001009706.1 |
| Ndufa5 | TCTACGTTCGATTGAGCGGG | CAATCCCACCAGGCCAGTAG | 112 | NM_012985.2 |
| Slc25a10 | CTCTAGACCTGCTCAAGGTGC | GAGGCACTCAGGCCATTGTA | 124 | NM_133418.1 |
| Slc25a15 | CTTGCTGTGTATCCGGTGGA | CTGACGTCATAGAGCCGTGA | 149 | NM_001047880.3 |
| Ucp1 | ATCCGGGCTTAAAGAGCGAG | CAGCCACCAGGGCTATTTGT | 70 | NM_012682.2 |
| Ucp2 | AGCAGTTCTACACCAAGGGC | TGGAAGCGGACCTTTACCAC | 124 | NM_019354.3 |
| Ucp3 | CGCCTGGAACAGAACAAAGC | TAACAGTGCAGGGTTCCGTC | 77 | NM_013167.2 |
| Uqcrfs1 | TGGCCATGTCGAAGATCGAG | TATGGCGCACAAACAGAGGT | 96 | NM_001008888.1 |
| Ldha | ACCCTCTGGGGAATCCAGAA | ACACAACTGGACCAACTGGA | 144 | NM_017025.1 |
| Gene | 5′-Forward-3′ | 5′-Reverse-3′ | Amplicon Size (bp) | Accession # |
|---|---|---|---|---|
| β-actin | CTATGAGGGTTACGCGCTCC | ATGTCACGCACGATTTCCCT | 141 | NM_031144.3 |
| B2m | CACTGAATTCACACCCACCG | TTACATGTCTCGGTCCCAGG | 100 | NM_012512.2 |
| Gapdh | TTGTGCAGTGCCAGCCTC | GGTAACCAGGCGTCCGATAC | 83 | NM_017008.4 |
| Hprt1 | ACAGGCCAGACTTTGTTGGA | TGCCGCTGTCTTTTAGGCTT | 149 | NM 012583.2 |
| Prlp0 | TTGAACATCTCCCCCTTCTCCT | CCACATTGCGGACACCCTCTA | 136 | NM 022402.2 |
| Gene | 5′-Forward-3′ | 5′-Reverse-3′ | Amplicon Size (bp) | Accession # |
|---|---|---|---|---|
| ATP6 | TAGGCTTCCGACACAAACTAAA | CTGCTAGTGCTATCGGTTGAATA | 129 | KF011917.1 |
| ATP8 | ATGCCACAACTAGACACAT | TTTGGGTGAGGGAGGTG | 120 | KF011917.1 |
| COXI | GCCAGTATTAGCAGCAGGTATC | GGTGGCCGAAGAATCAGAATAG | 125 | KF011917.1 |
| COXII | TCTCCCAGCTGTCATTCTTATTC | GCTTCAGTATCATTGGTGTCCTA | 121 | KF011917.1 |
| COXIII | GCTGACCTCCAACAGGAATTA | CCTTCTATTAGGCTGTGATGGG | 118 | KF011917.1 |
| Cyt B | CCTTCCTACCATTCCTGCATAC | TGGCCTCCGATTCATGTTAAG | 118 | KF011917.1 |
| GAPDH | ACTCCCATTCTTCCACCTTTG | CCCTGTTGCTGTAGCCATATT | 105 | NM_017008.4 |
| ND1 | GGCTCCTTCTCCCTACAAATAC | AAGGGAGCTCGATTTGTTTCT | 122 | KF011917.1 |
| ND2 | CCCAACTATCACCACCATTCTC | TCGTGTTTGGGTCTGGTTAAG | 79 | KF011917.1 |
| ND3 | TTCTGCACGCCTTCCTTT | GGTTGTTTGAATCGCTCATGG | 112 | KF011917.1 |
| ND4 | GATGAGGCAACCAAACAGAAC | GTGTTGTGAGGGAGAGGATTAG | 147 | KF011917.1 |
| ND4L | TCTCCTCTGCCTAGAAGGAATAA | TGGTAATTGGGATGGTTATGGAG | 101 | KF011917.1 |
| ND5 | GCCGCCACTATTATCTCCTTC | CTACTTCCTCCCACTCCATTTG | 112 | NM_133584.1 |
| ND6 | GGTGGGTTTGGATTGATTGTTAG | CCTCAGTAGCCATAGCAGTTG | 148 | NM_133584.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wen, J.J.; Cummins, C.B.; Williams, T.P.; Radhakrishnan, R.S. The Genetic Evidence of Burn-Induced Cardiac Mitochondrial Metabolism Dysfunction. Biomedicines 2020, 8, 566. https://doi.org/10.3390/biomedicines8120566
Wen JJ, Cummins CB, Williams TP, Radhakrishnan RS. The Genetic Evidence of Burn-Induced Cardiac Mitochondrial Metabolism Dysfunction. Biomedicines. 2020; 8(12):566. https://doi.org/10.3390/biomedicines8120566
Chicago/Turabian StyleWen, Jake J., Claire B. Cummins, Taylor P. Williams, and Ravi S. Radhakrishnan. 2020. "The Genetic Evidence of Burn-Induced Cardiac Mitochondrial Metabolism Dysfunction" Biomedicines 8, no. 12: 566. https://doi.org/10.3390/biomedicines8120566
APA StyleWen, J. J., Cummins, C. B., Williams, T. P., & Radhakrishnan, R. S. (2020). The Genetic Evidence of Burn-Induced Cardiac Mitochondrial Metabolism Dysfunction. Biomedicines, 8(12), 566. https://doi.org/10.3390/biomedicines8120566