Basal Cell Carcinoma: From Pathophysiology to Novel Therapeutic Approaches

 ,
,  , and
, and
Abstract
:1. Introduction
2. BCC Risk Factors
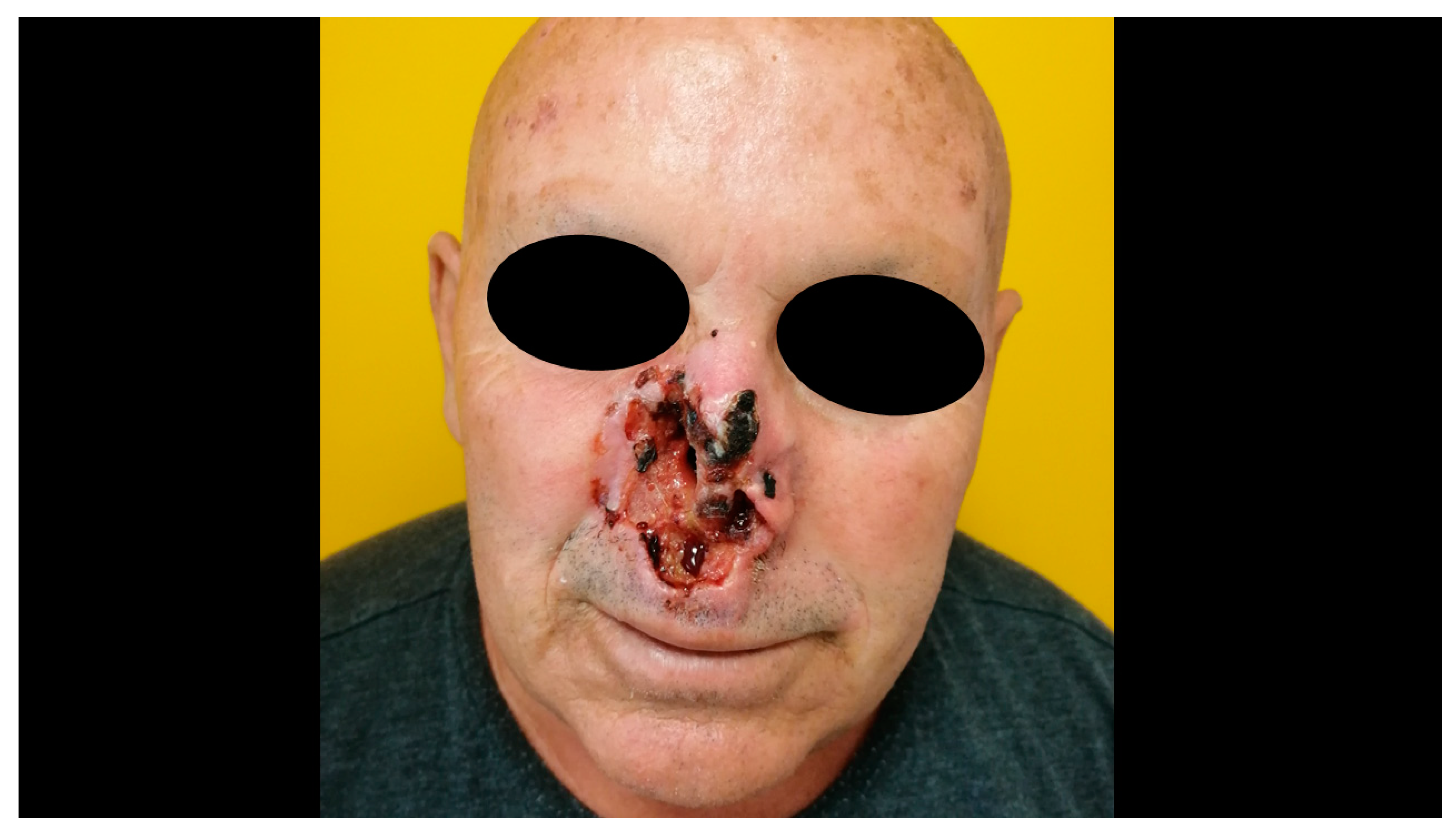
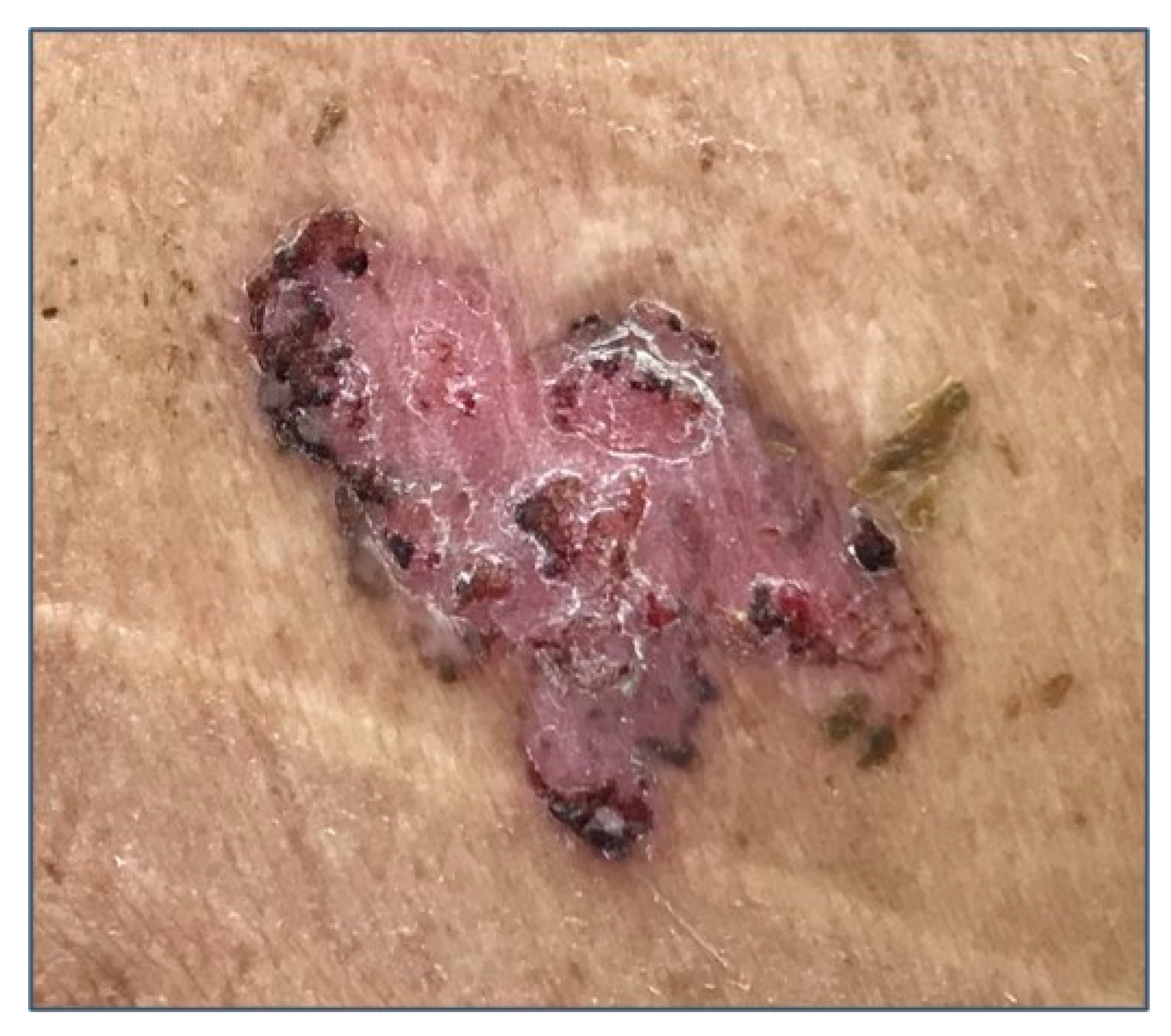
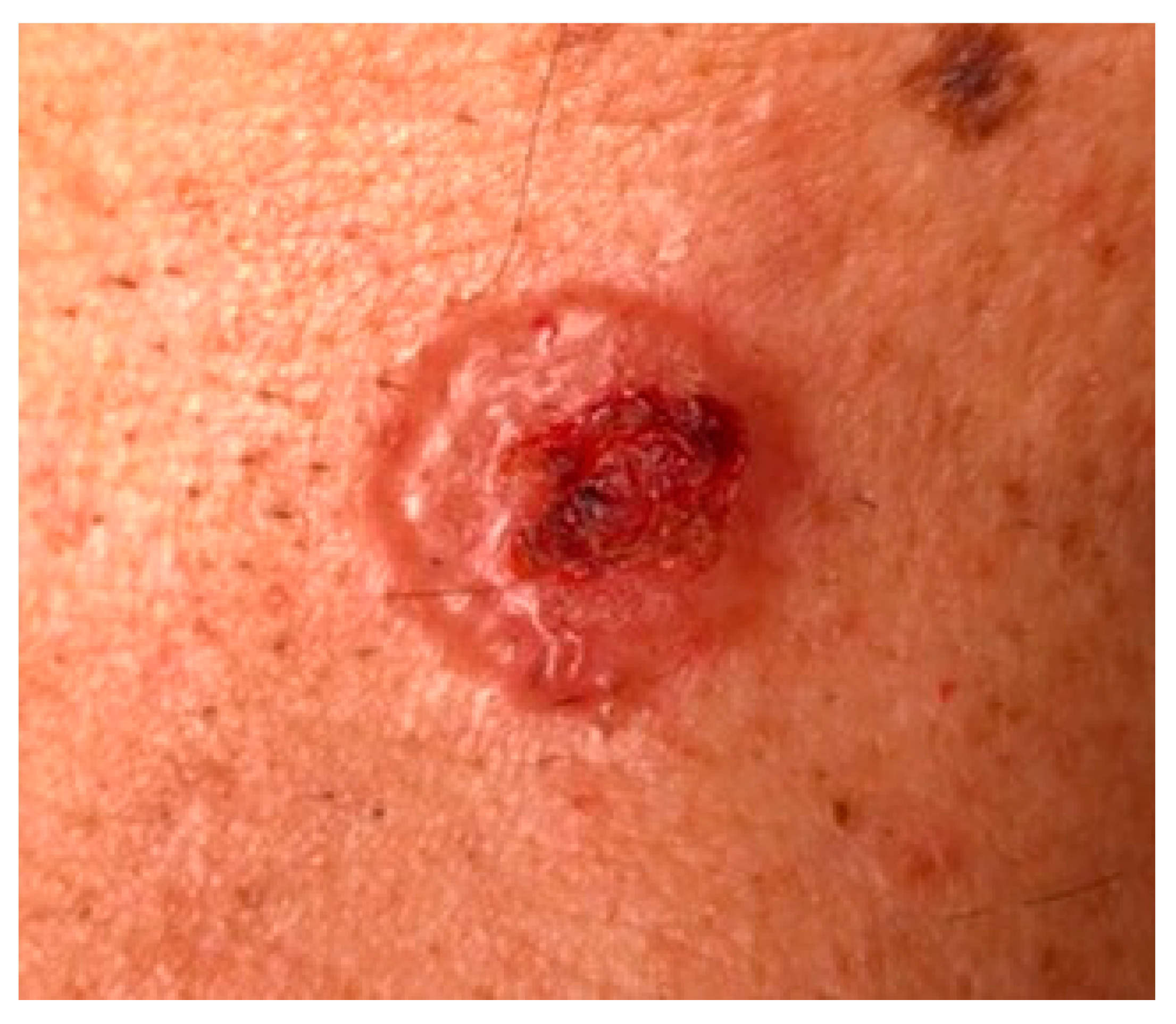
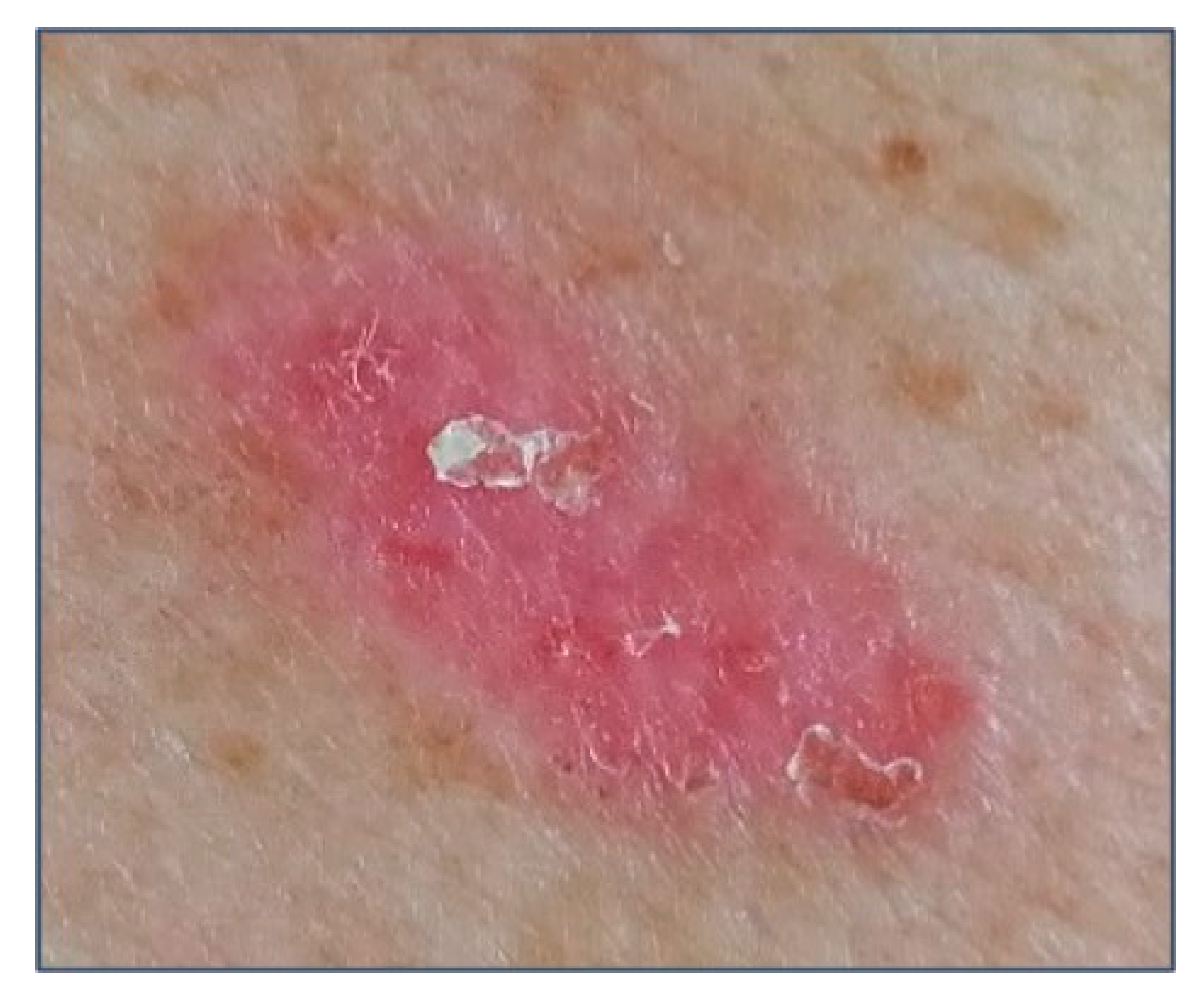
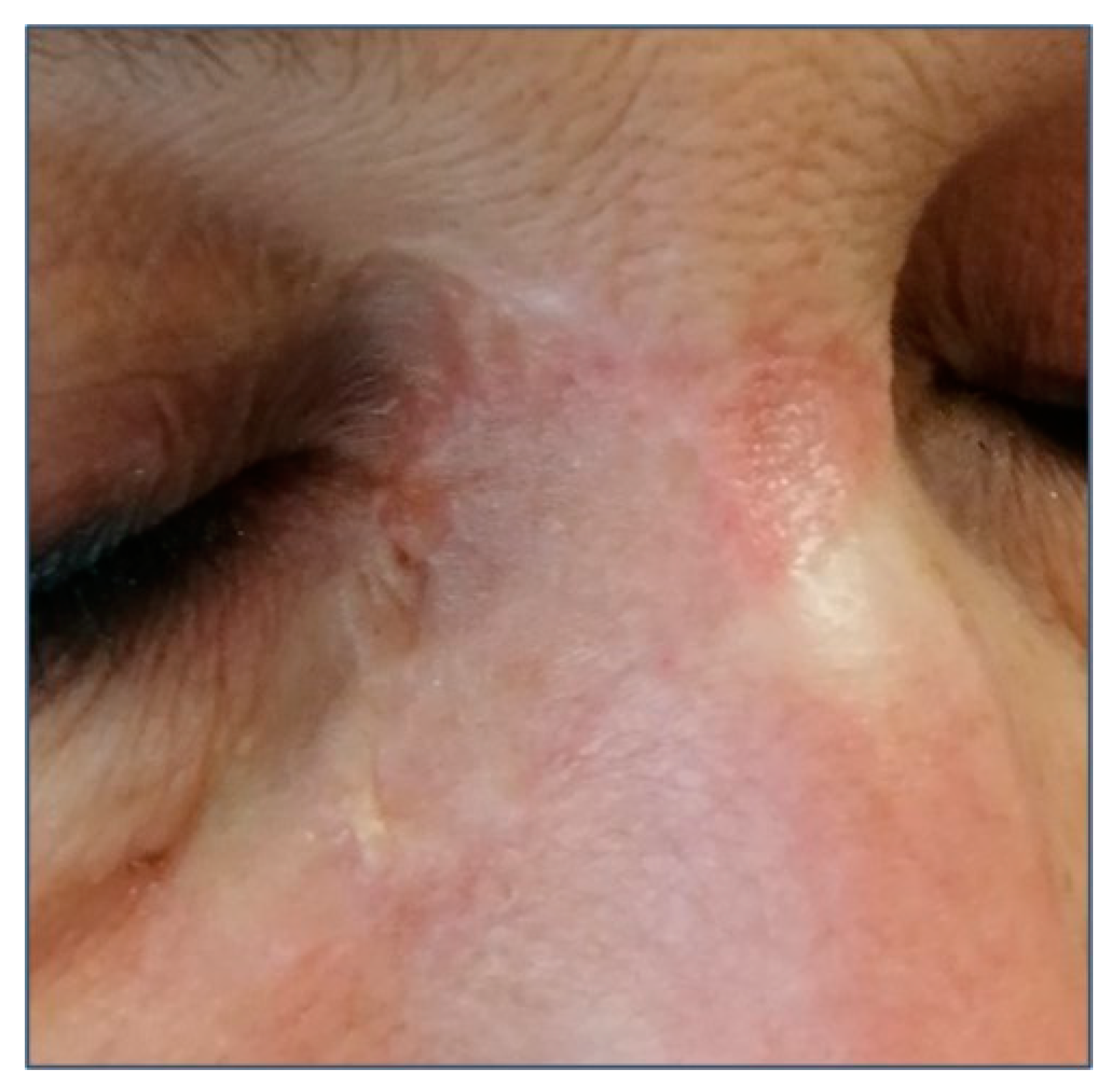
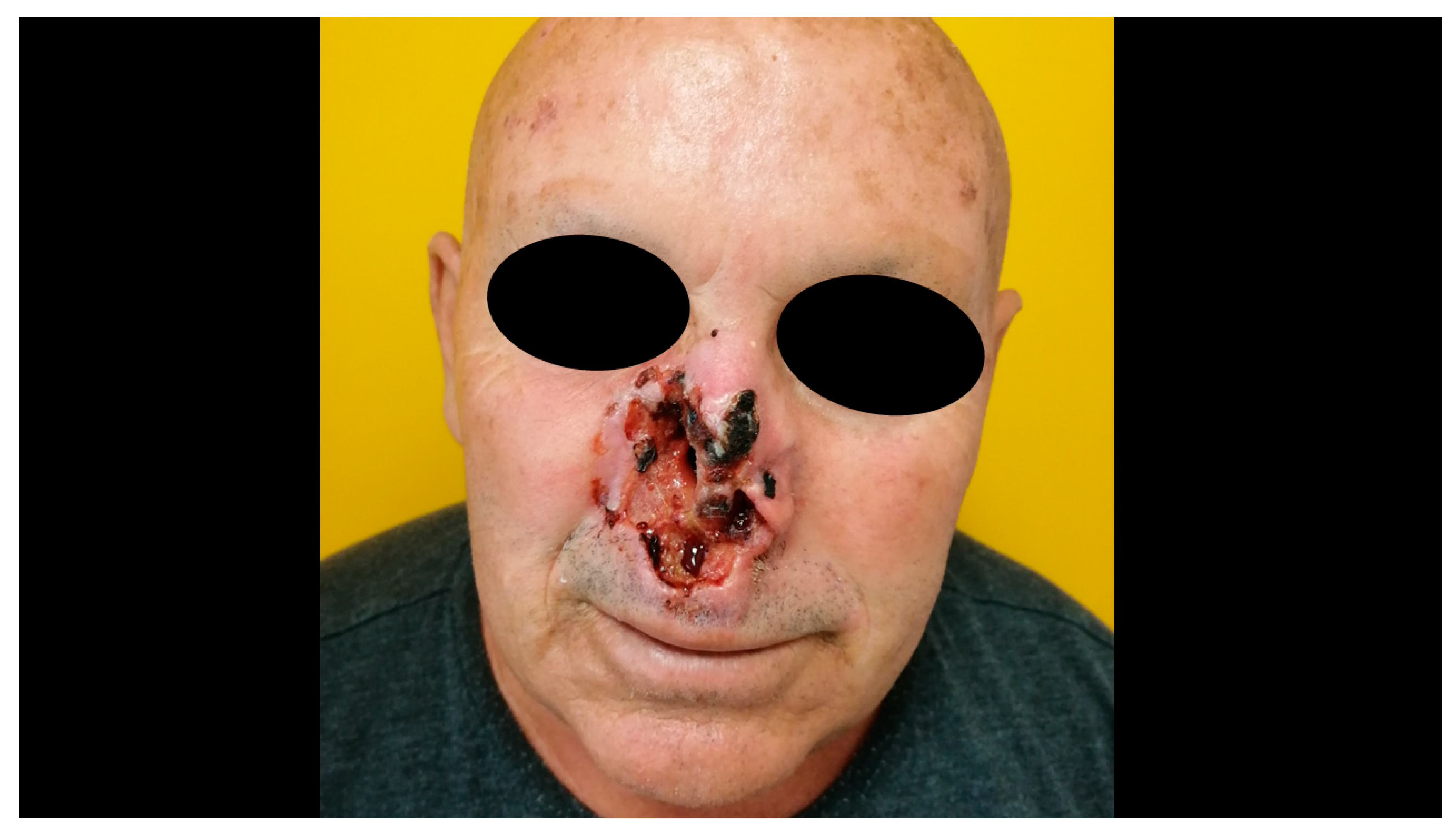
3. Clinical Features and Different Subtypes of BCCs
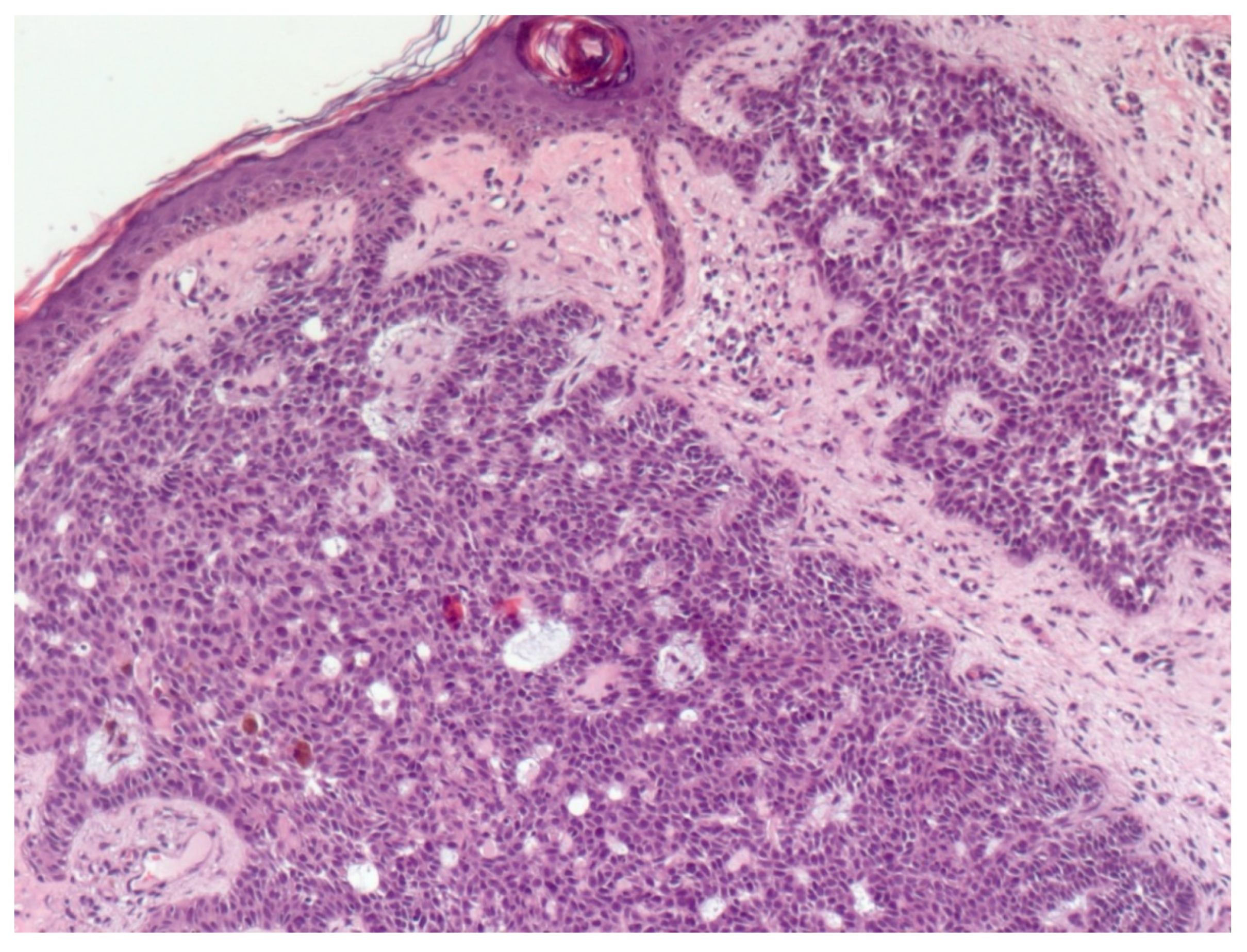
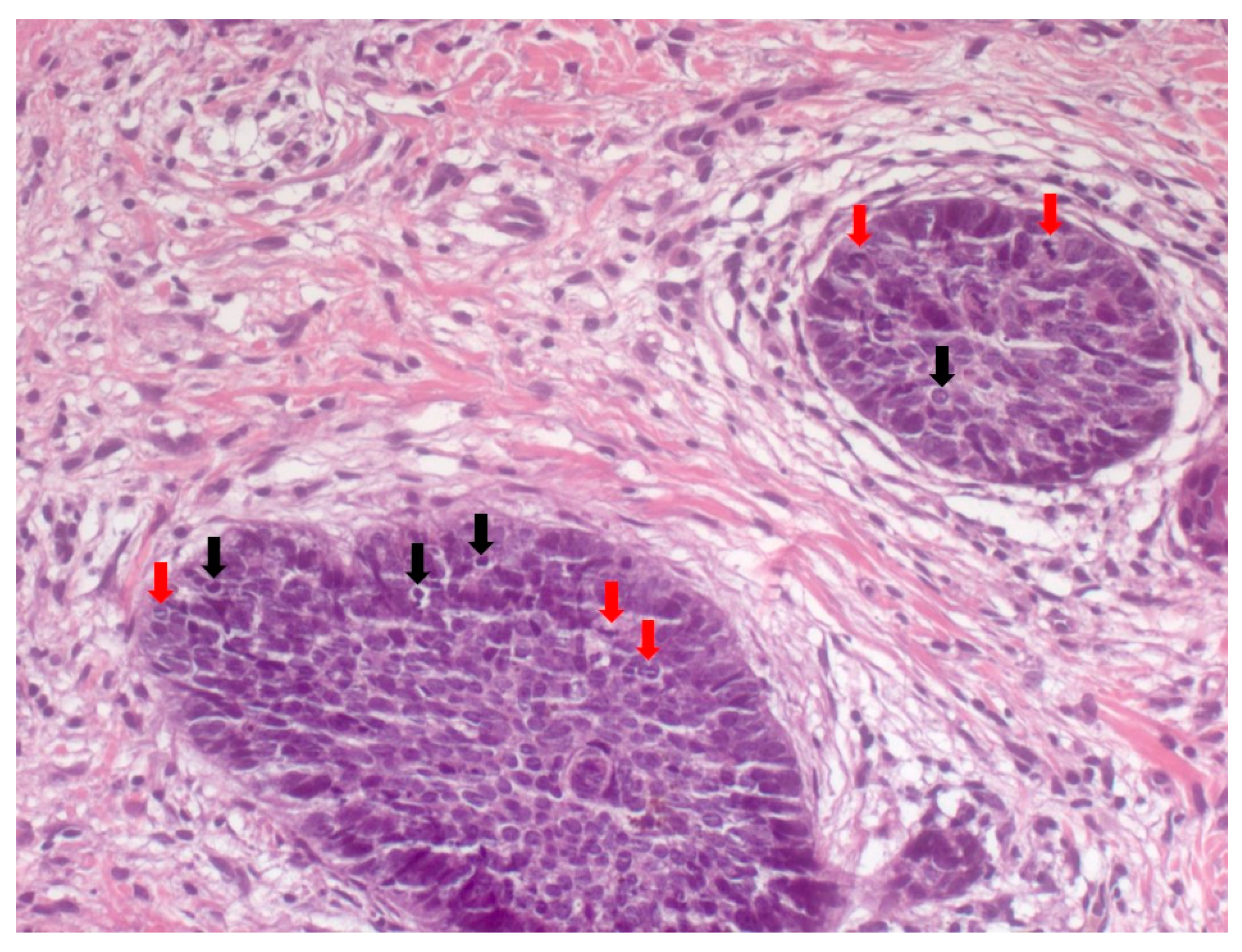
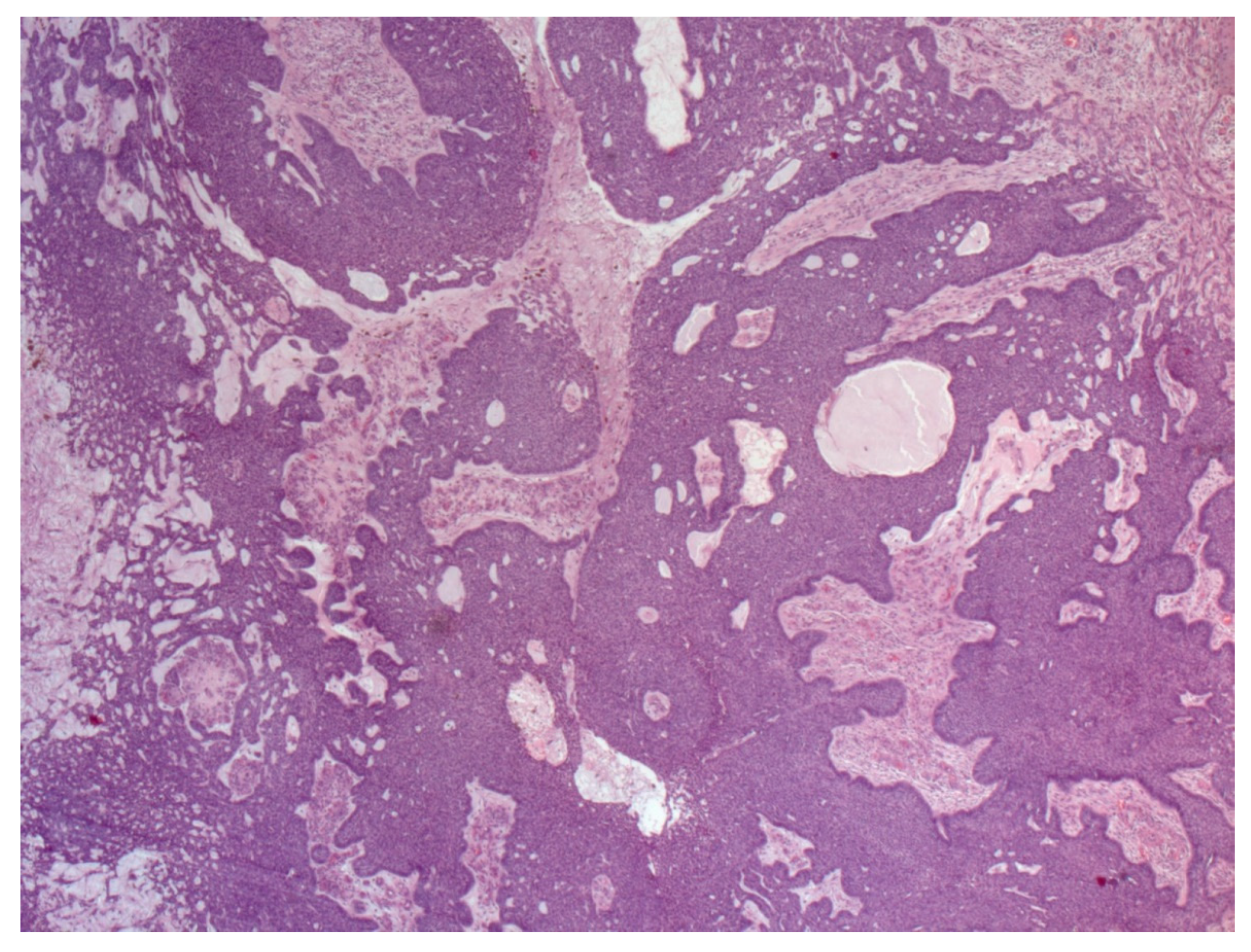
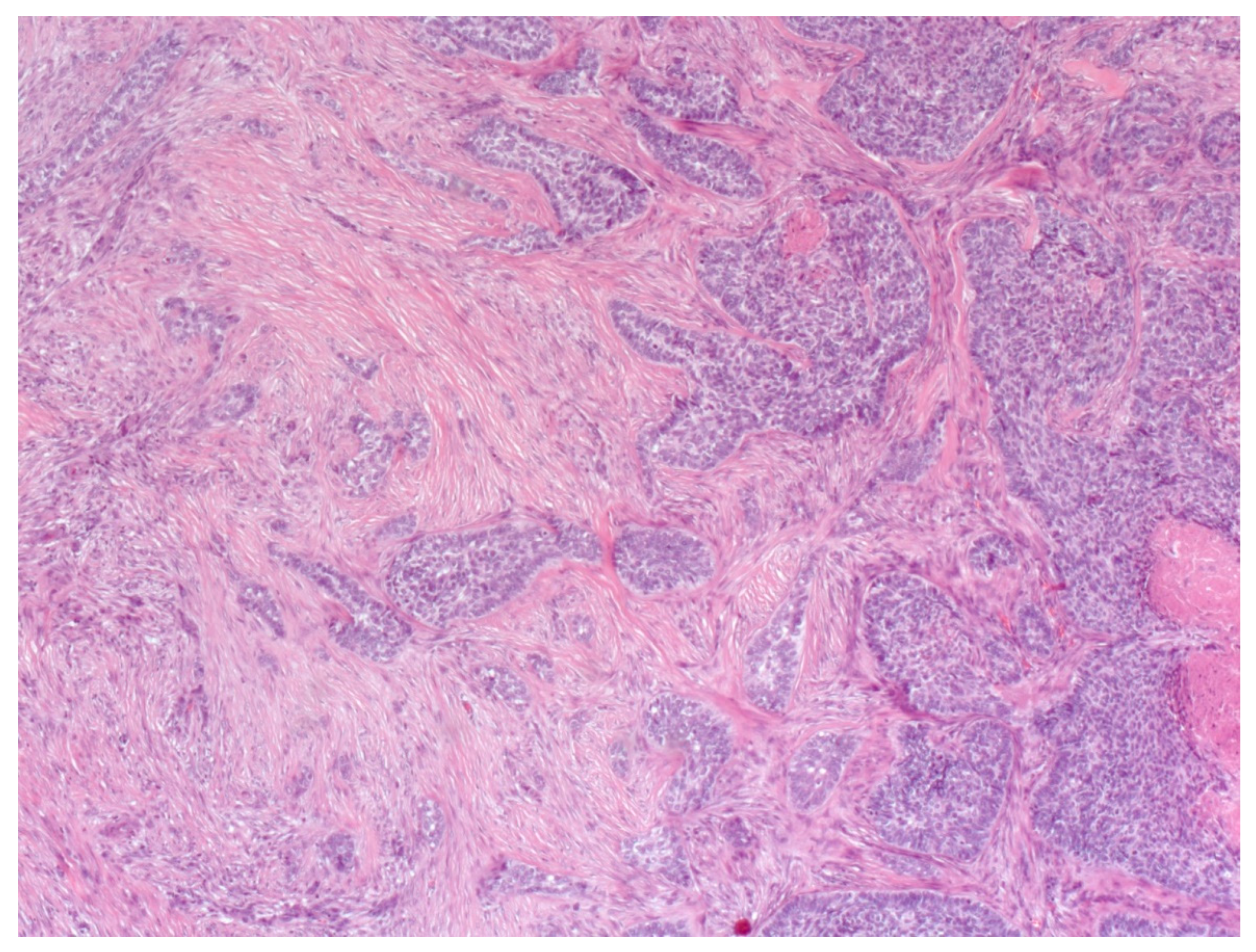
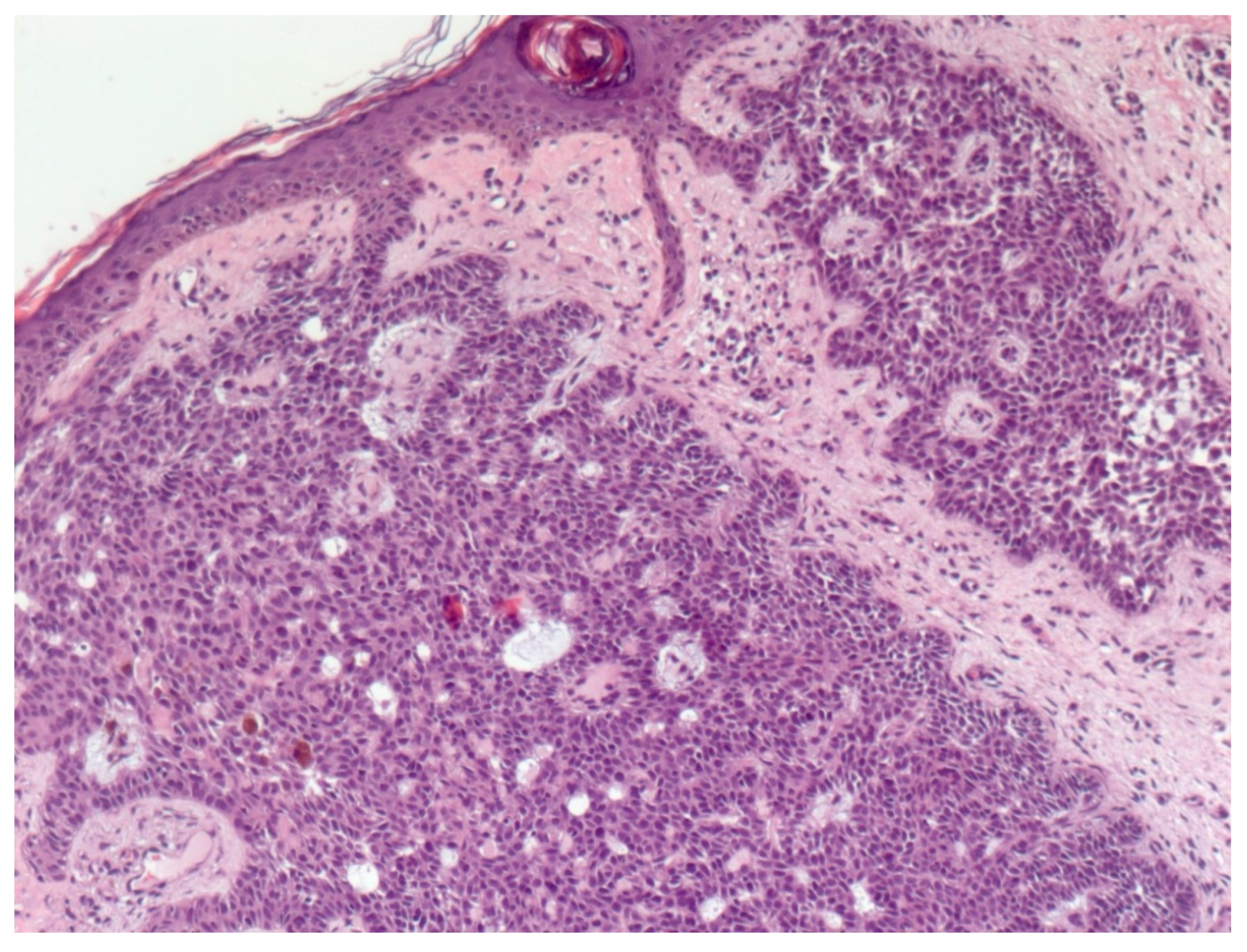
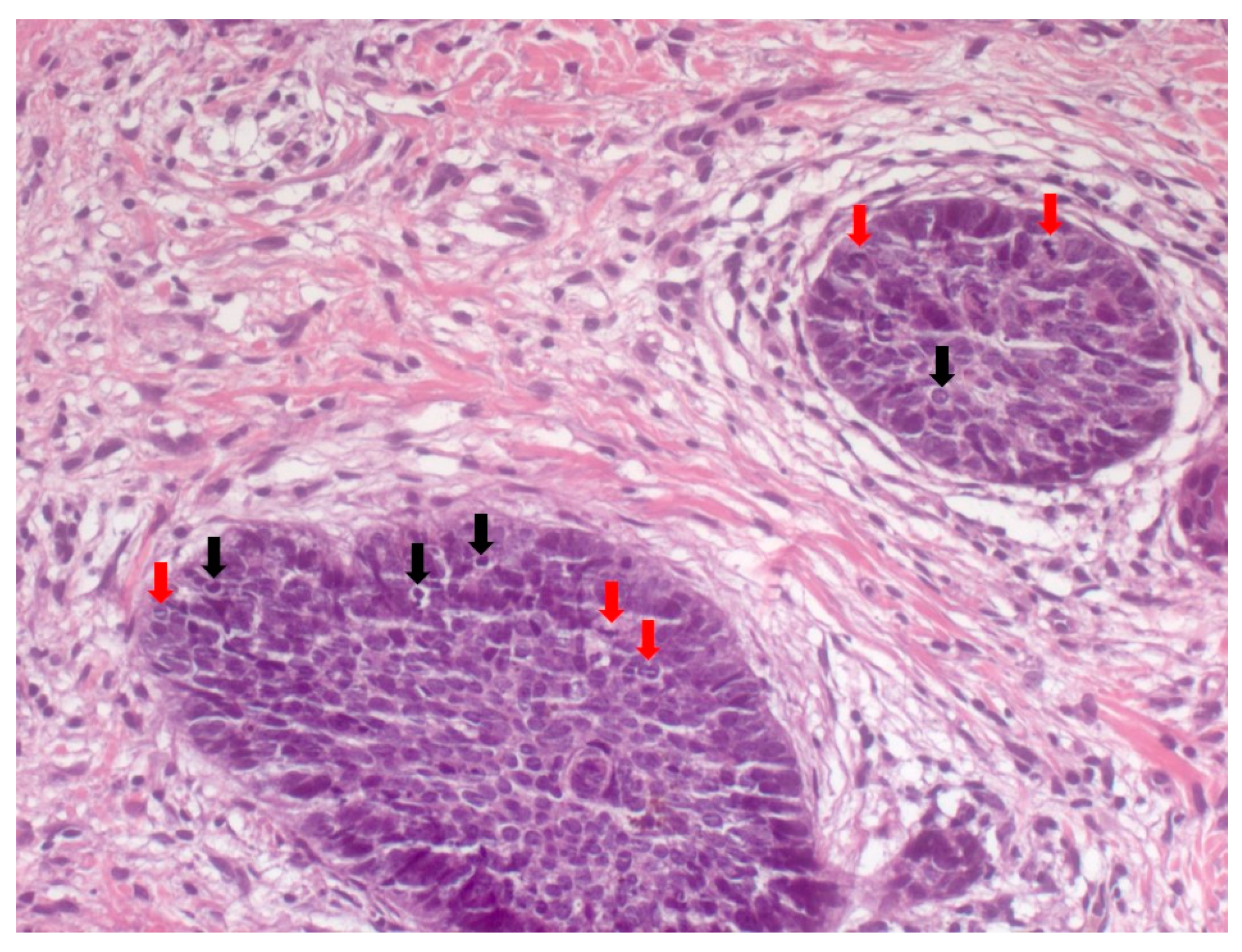
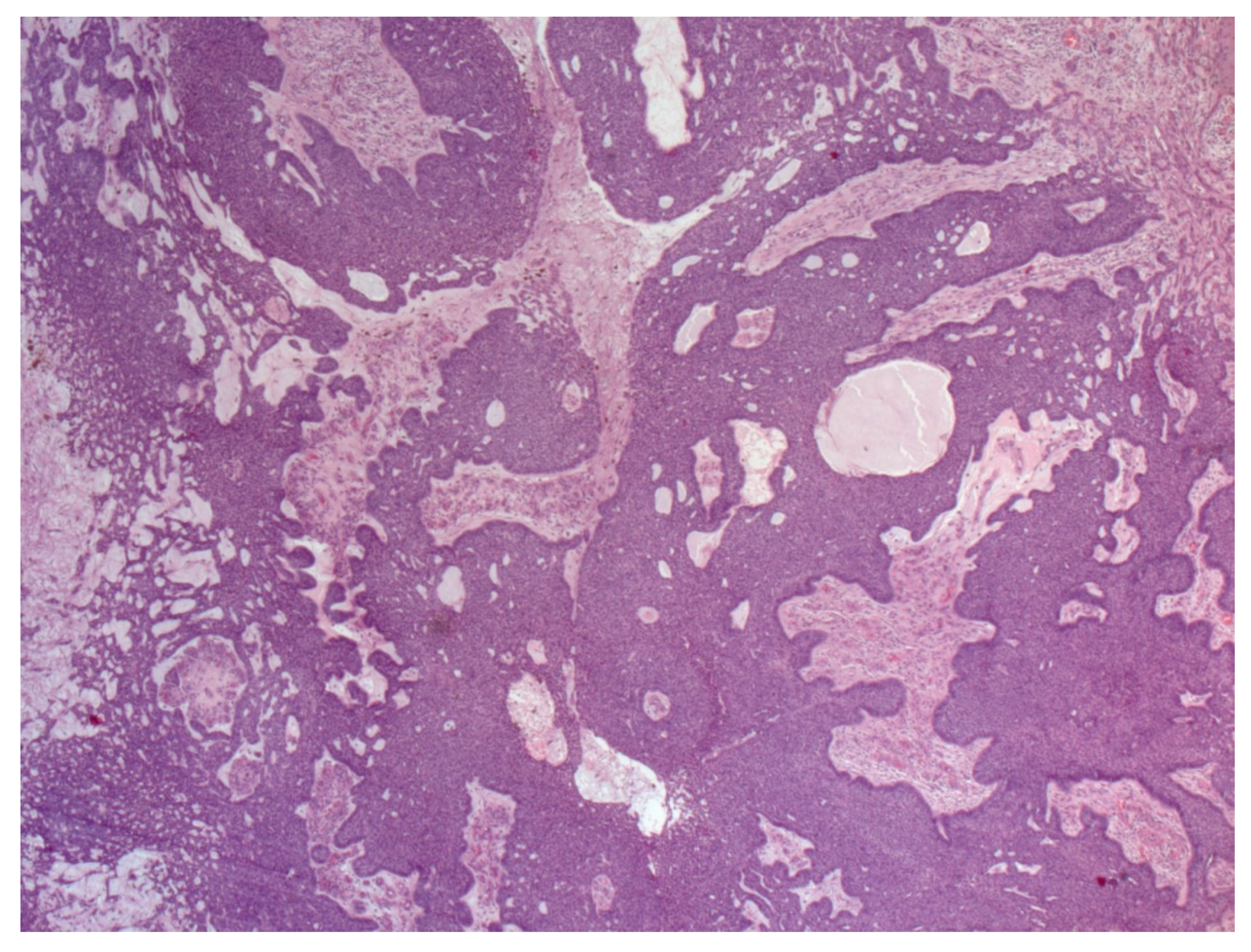
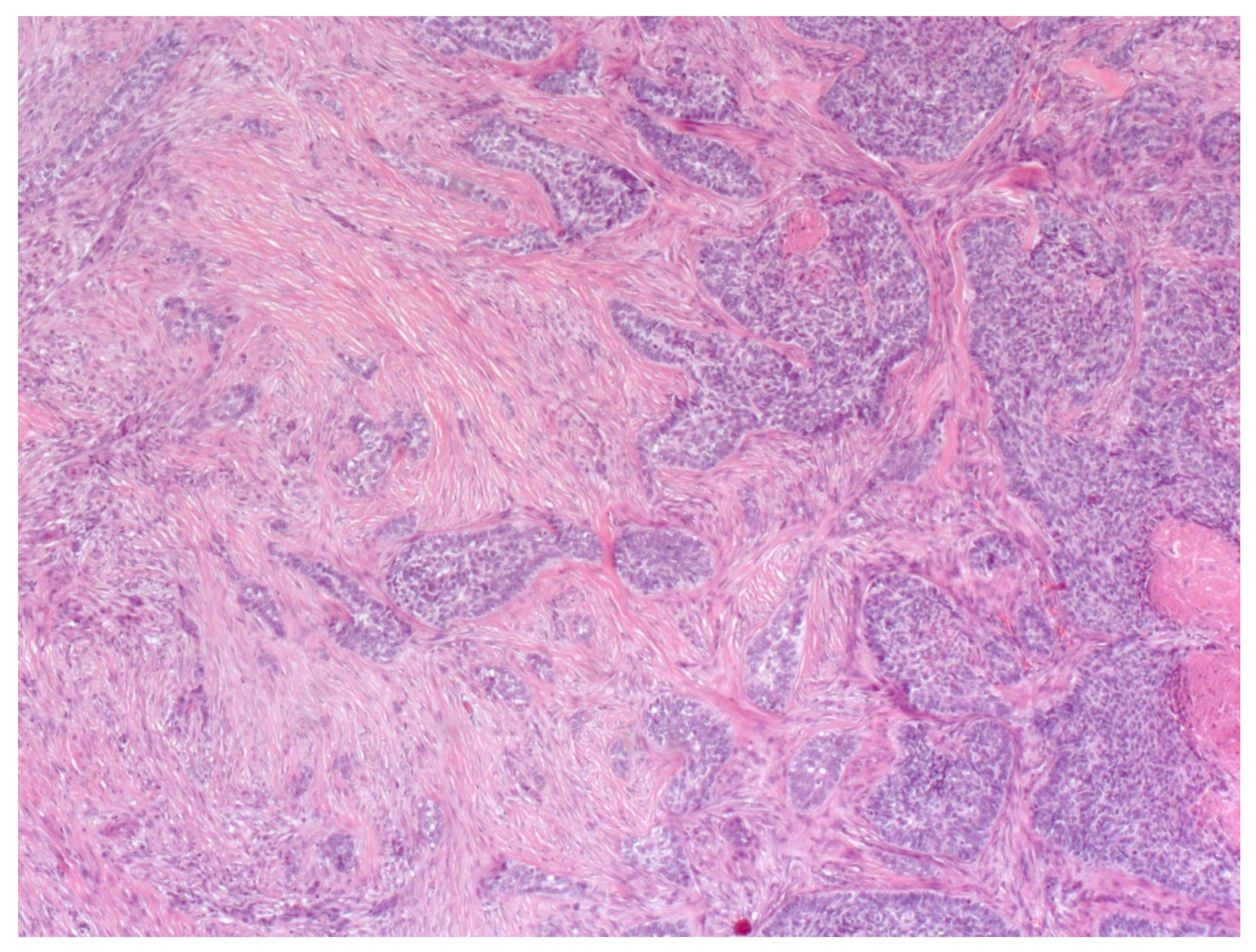
4. Histopathological Features of BCC
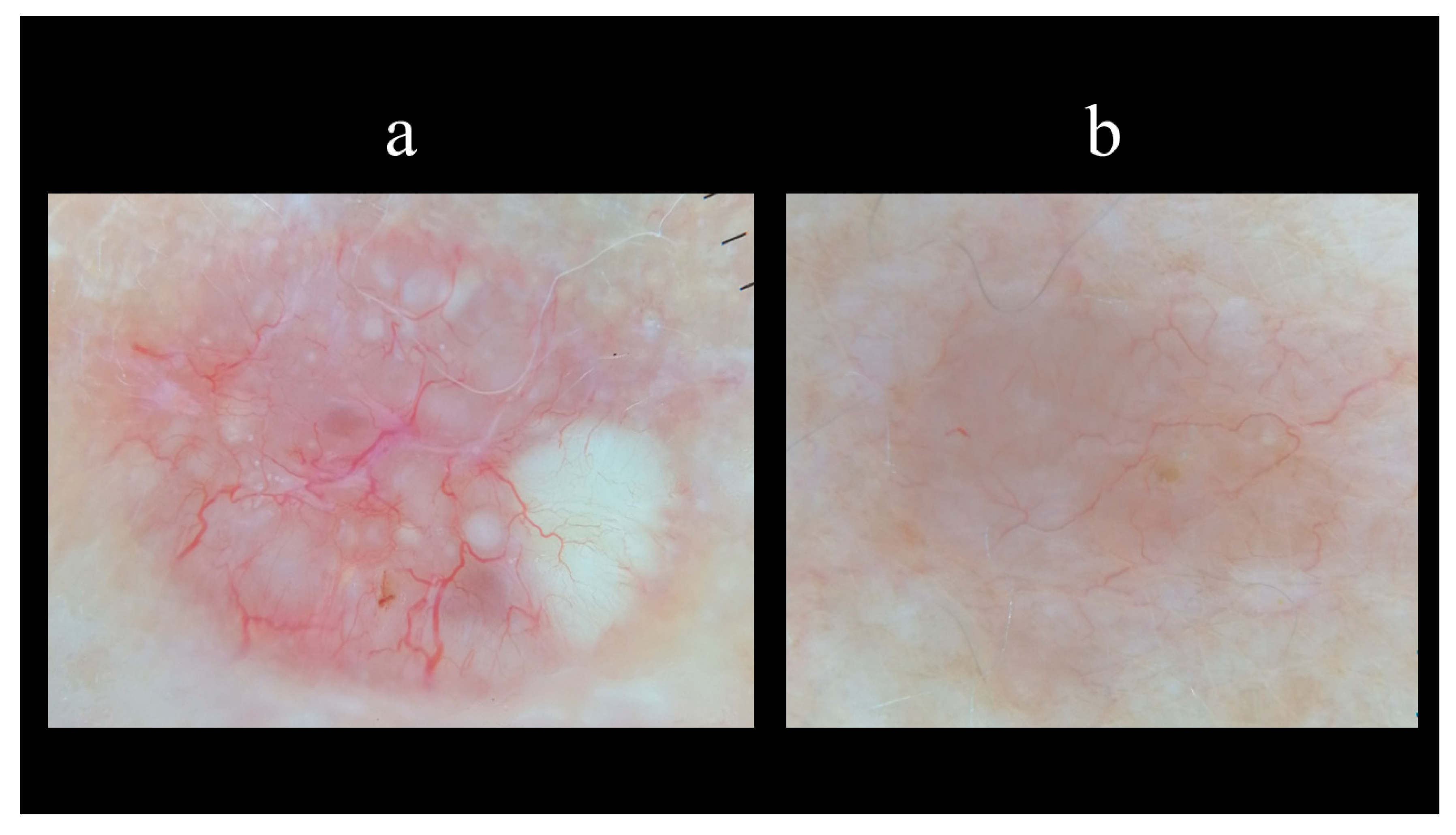
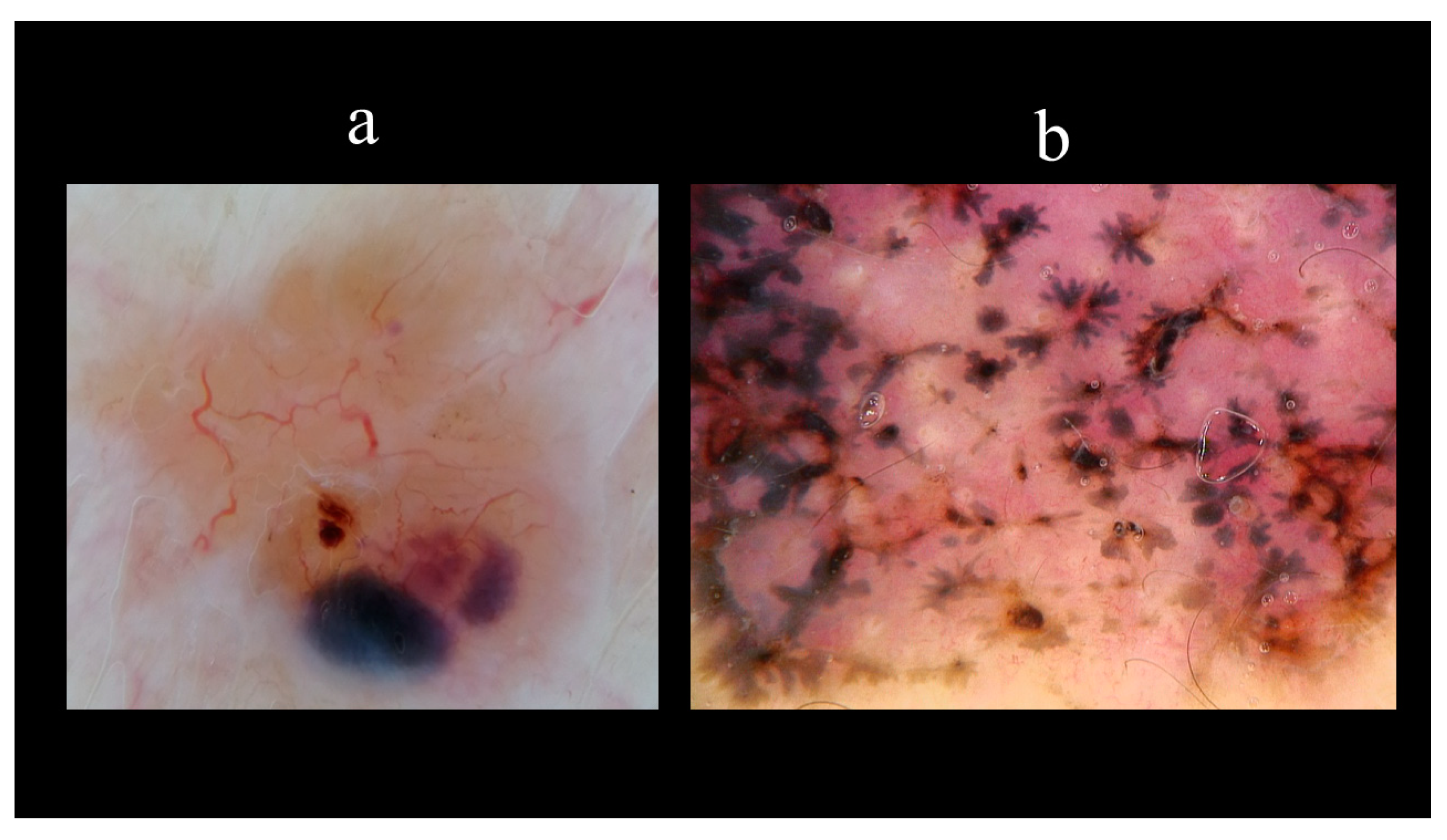
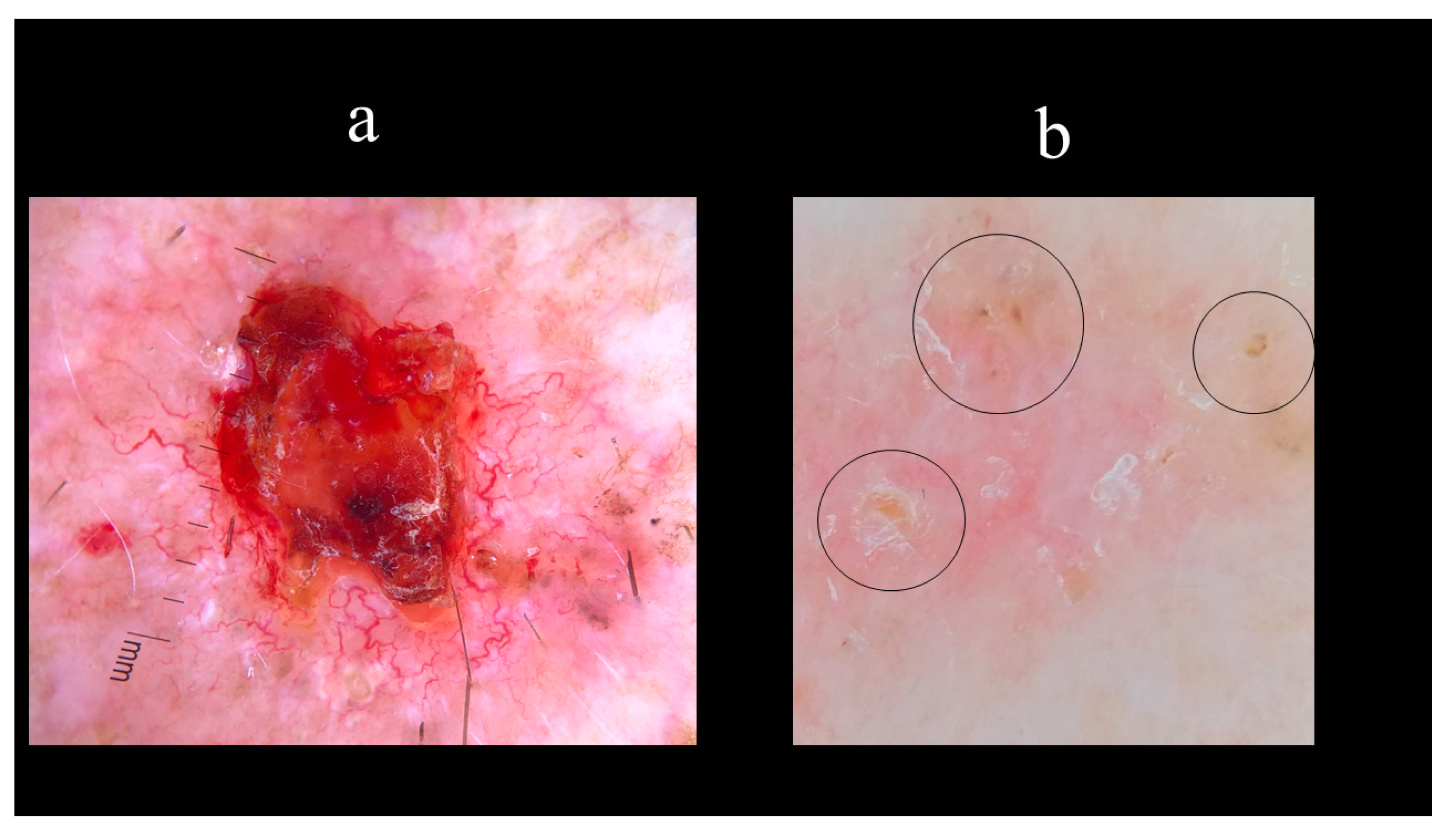
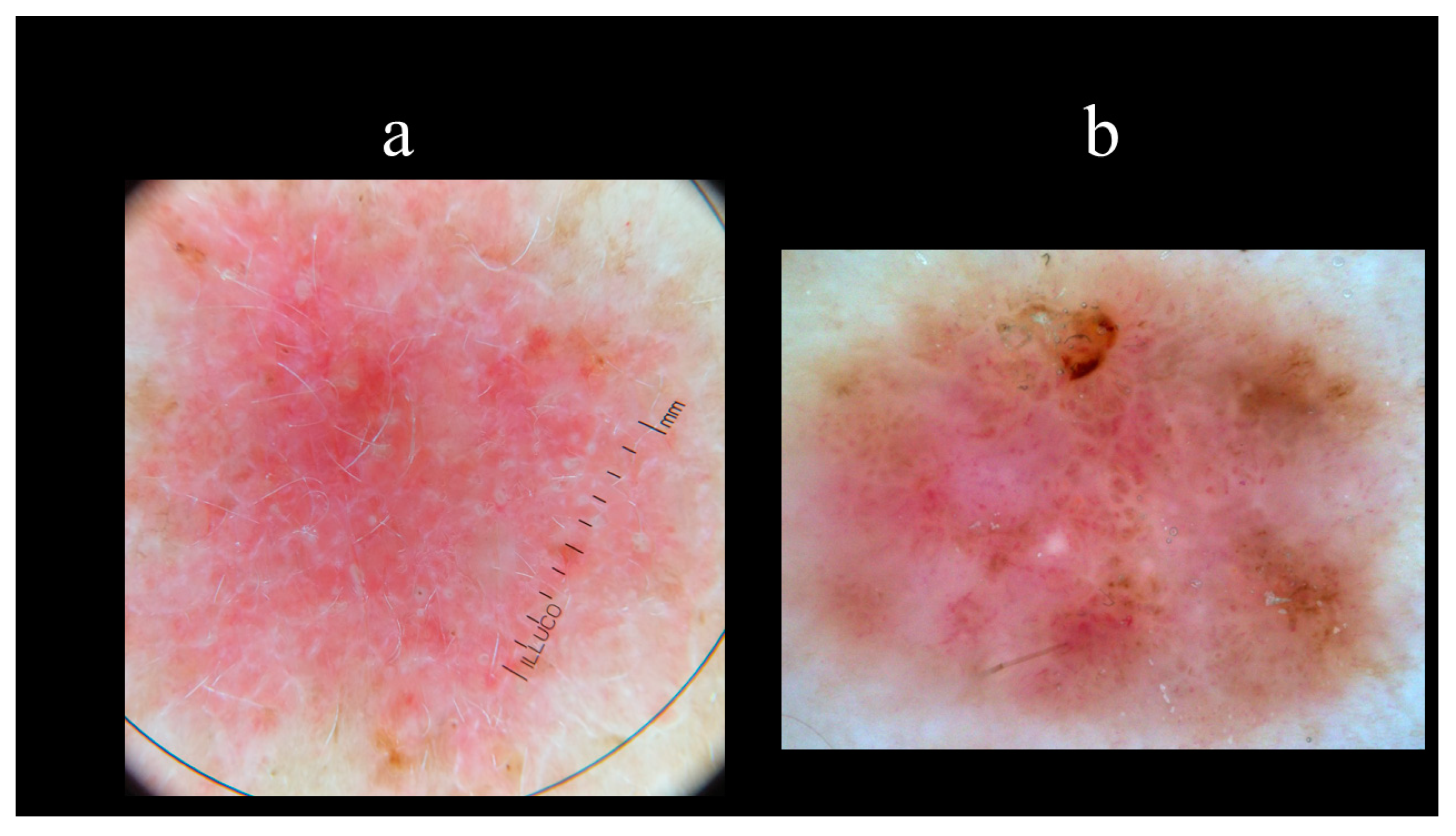
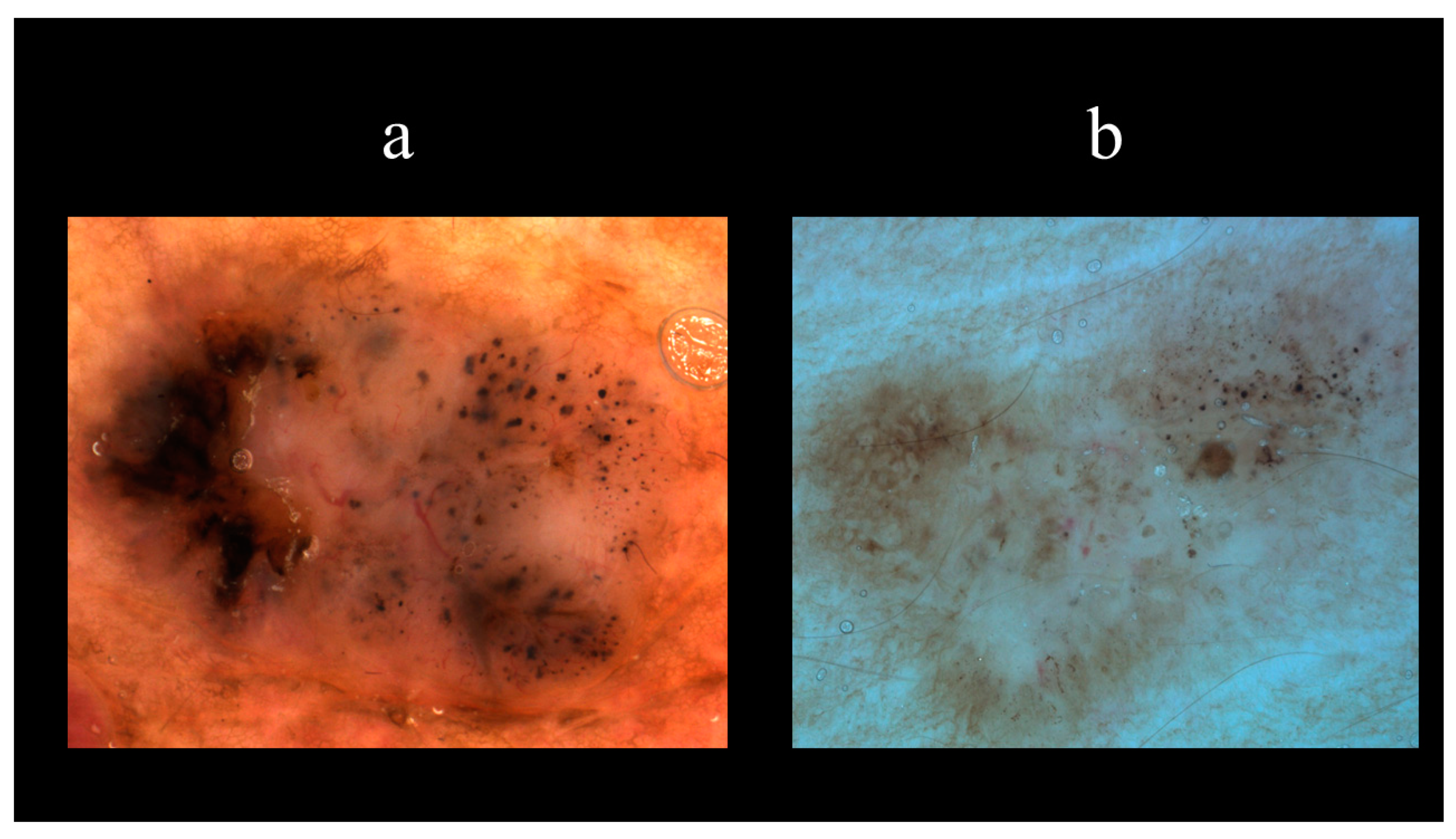
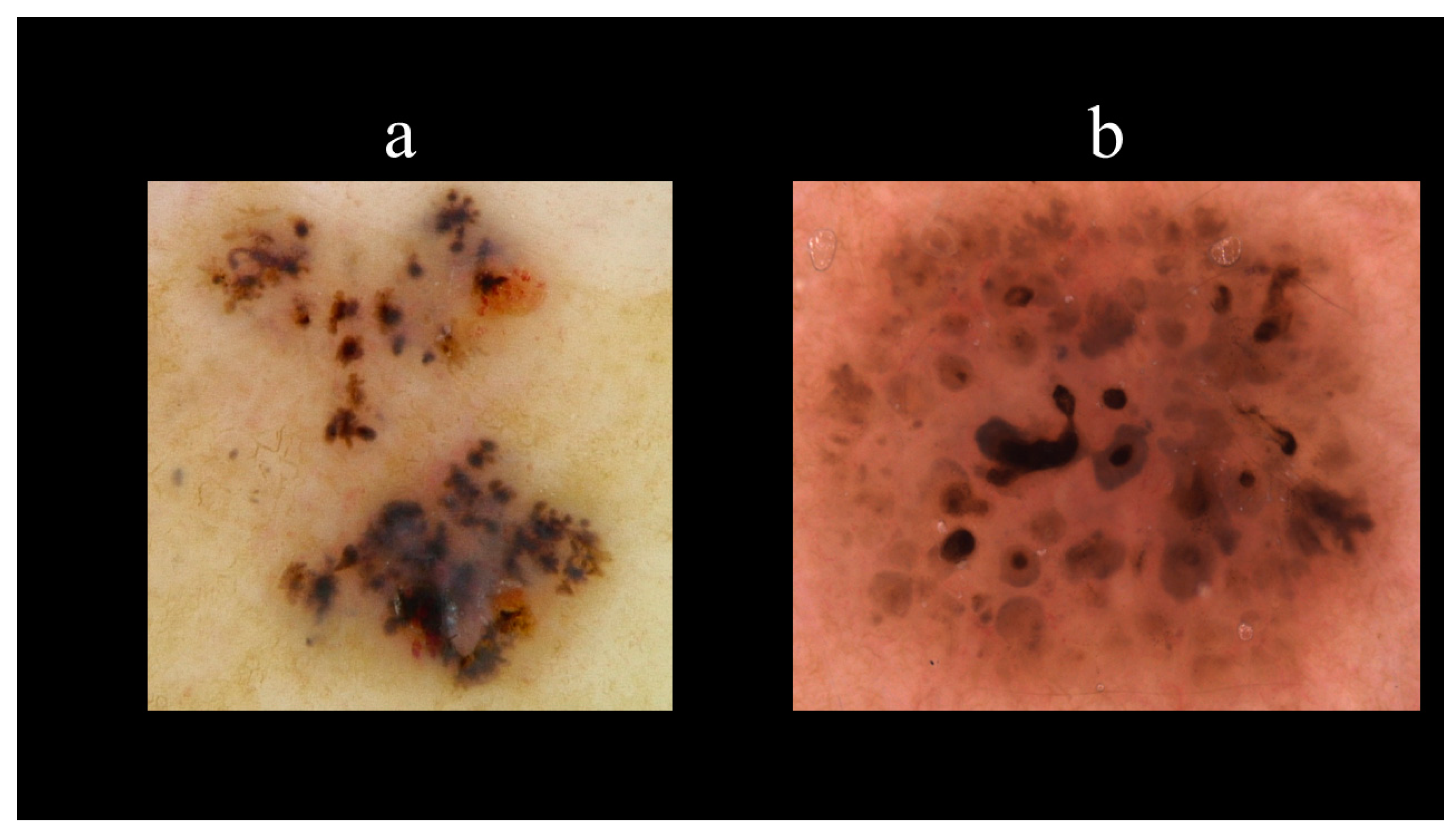
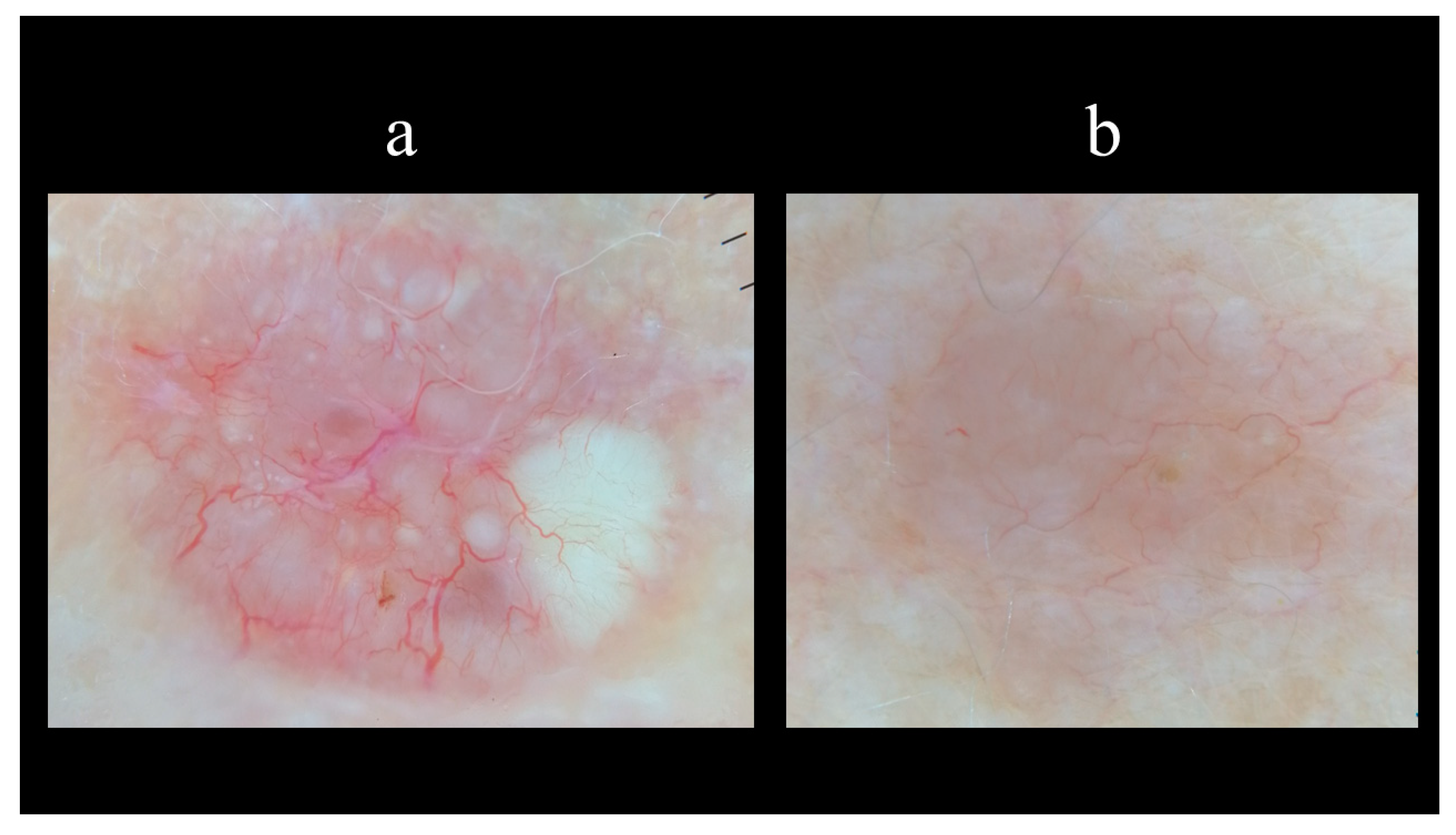
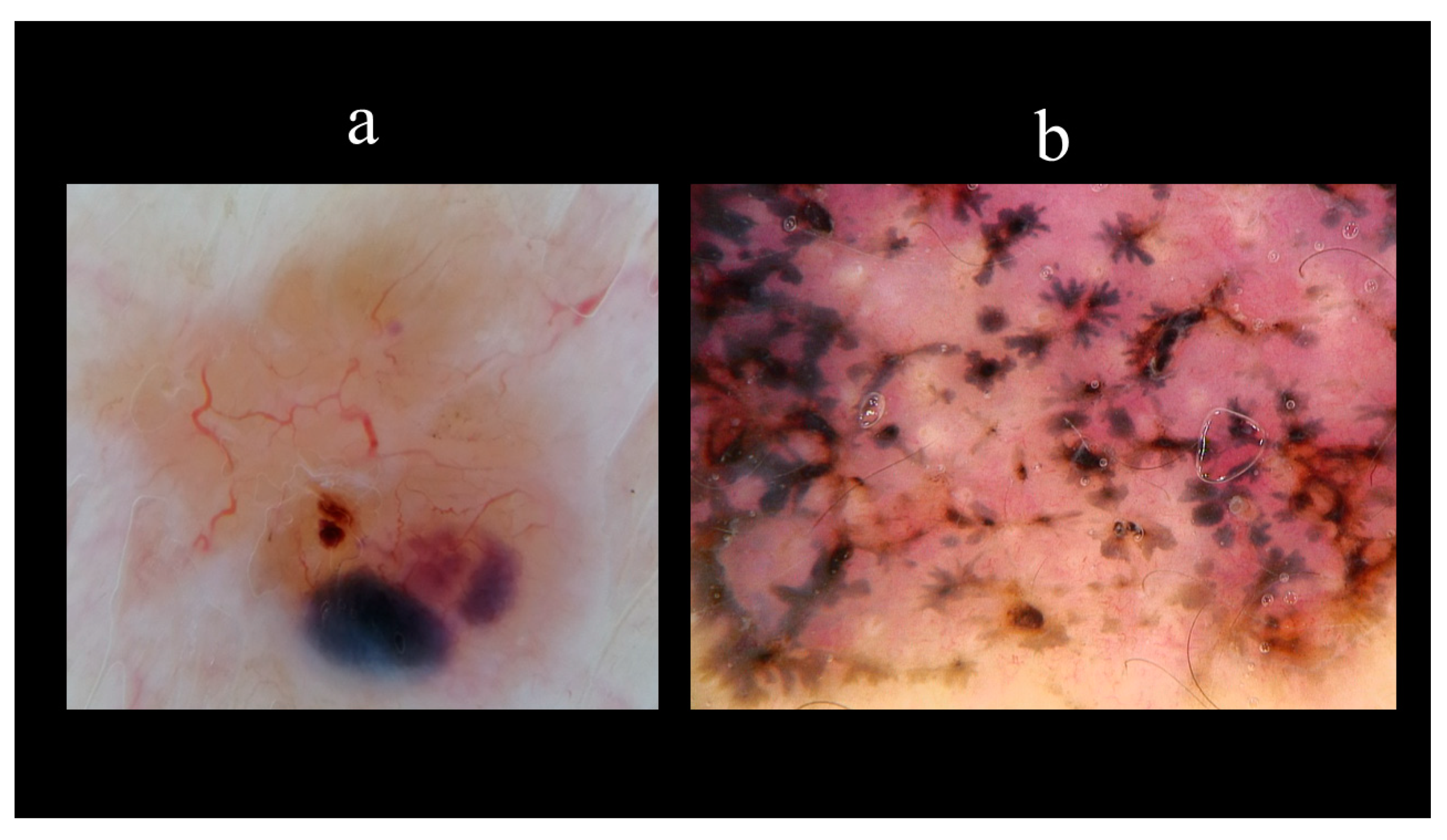
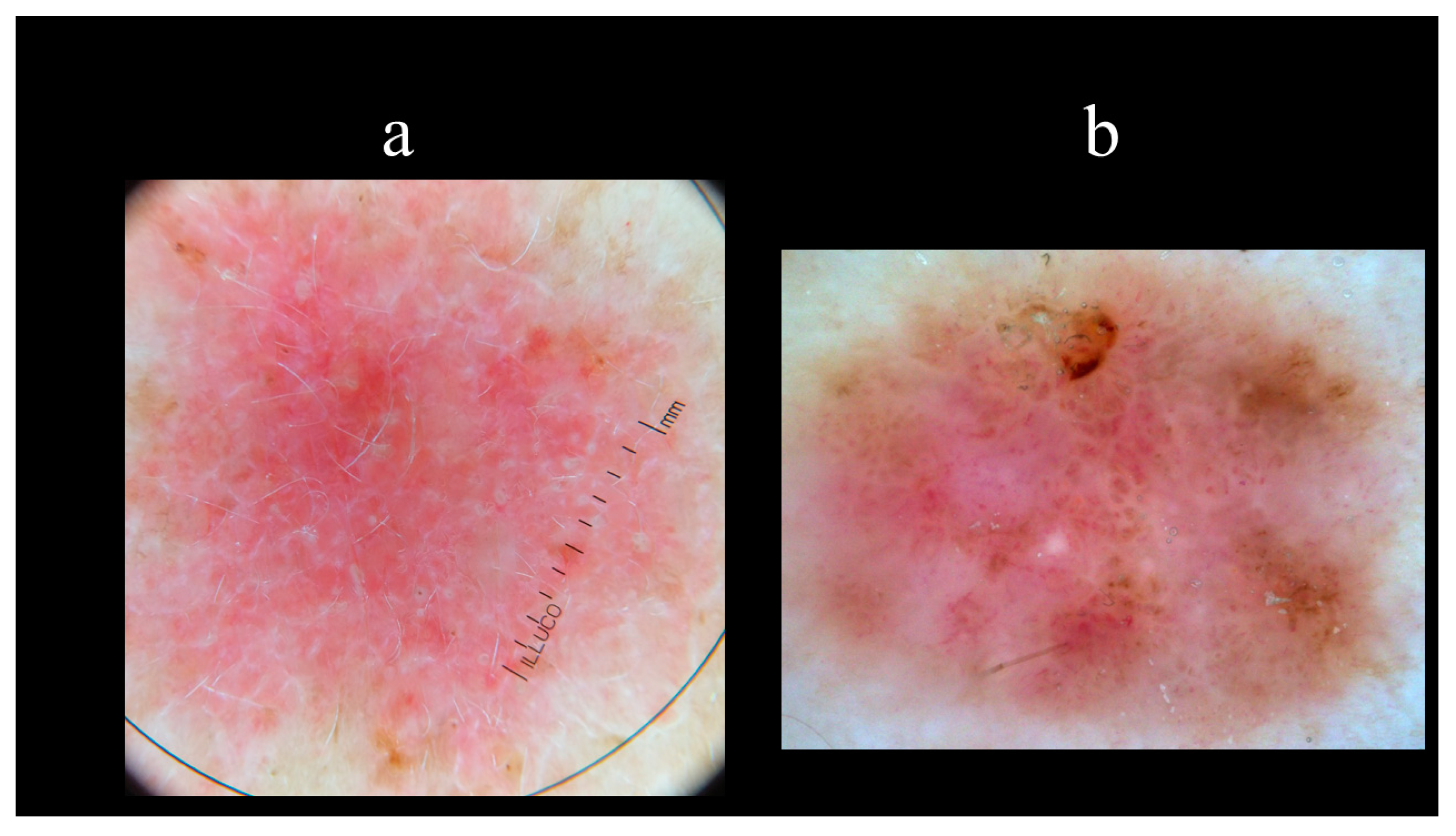
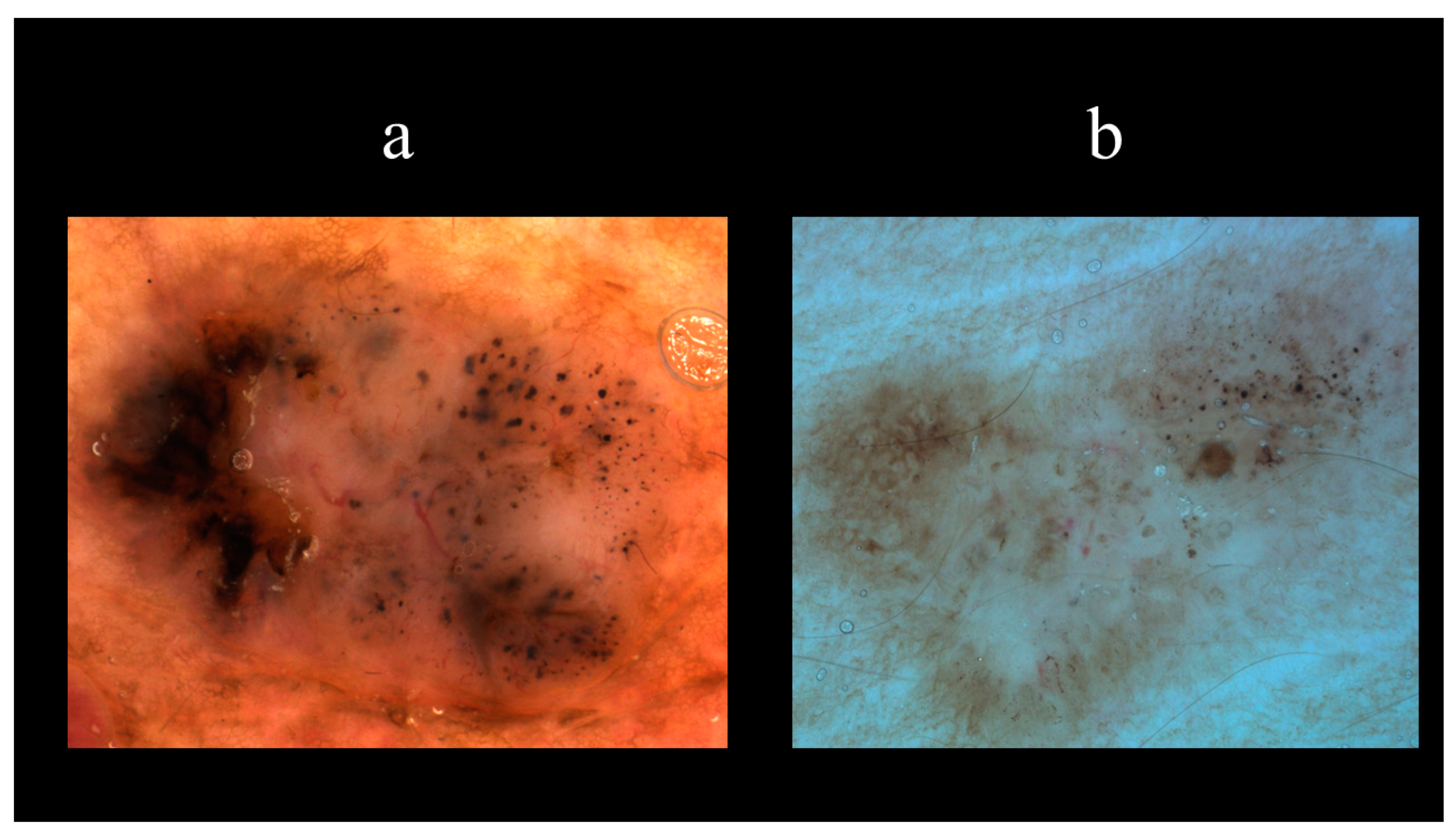
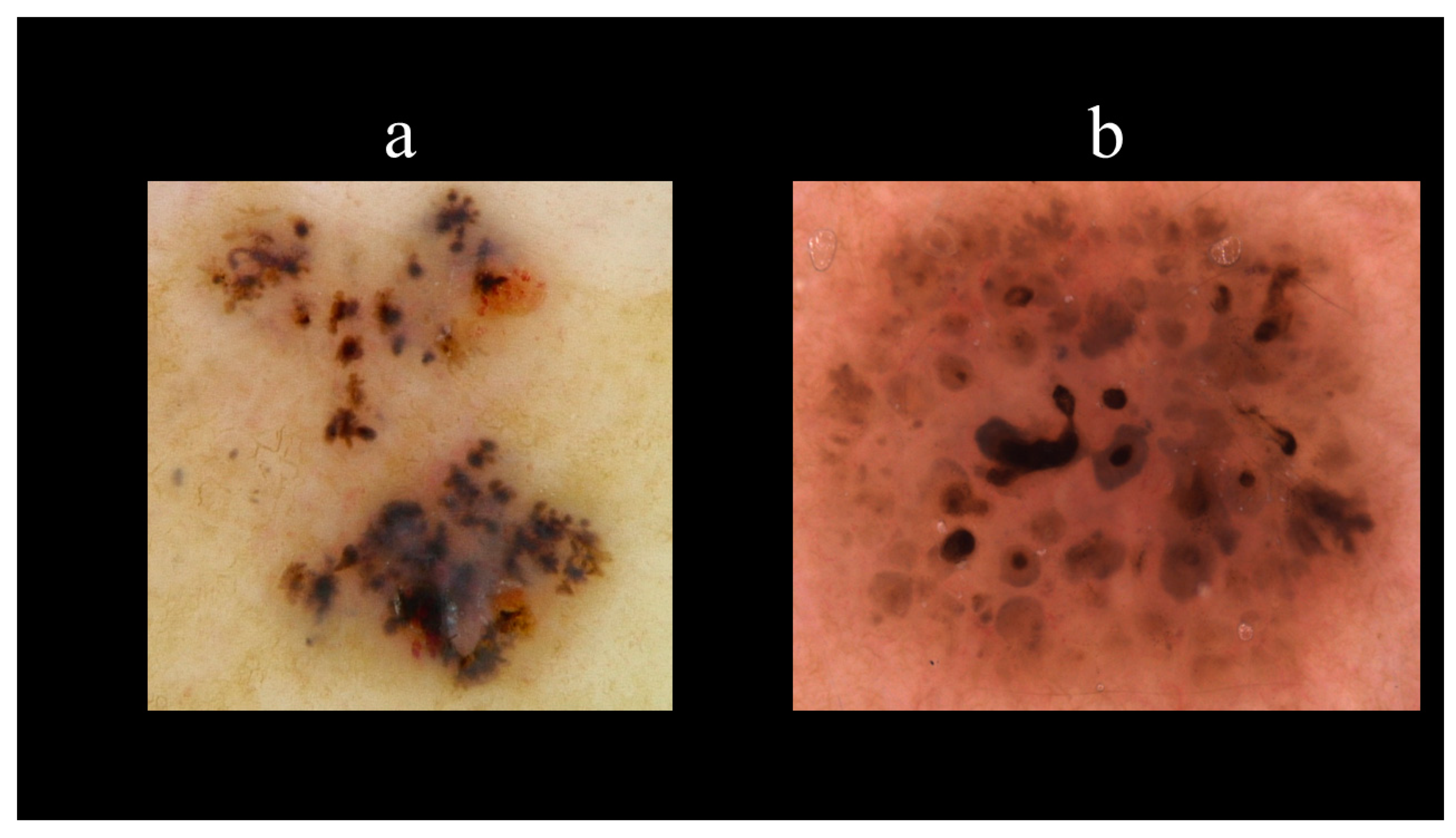
5. Dermoscopy
6. Reflectance Confocal Microscopy and Optical Coherence Tomography
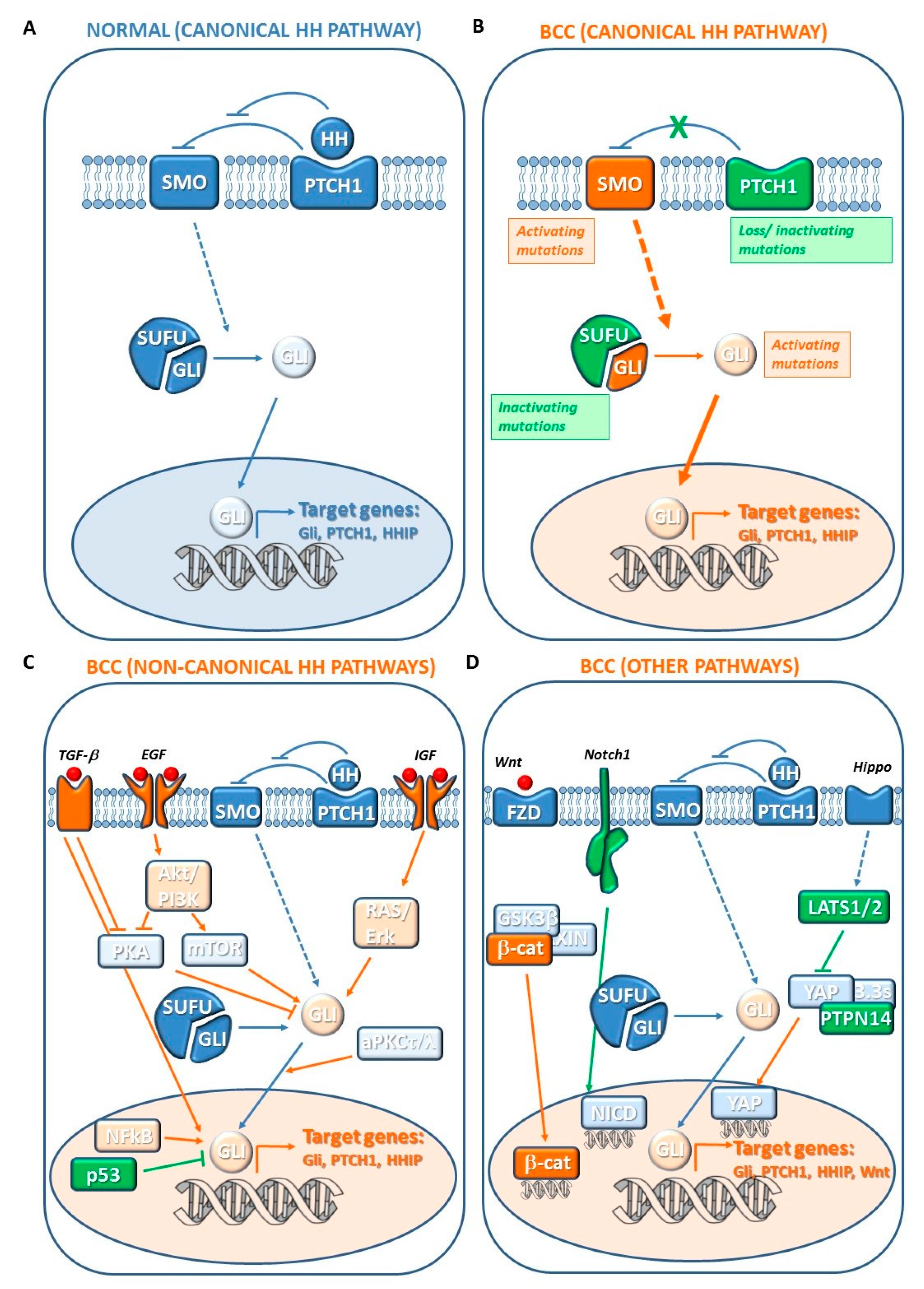
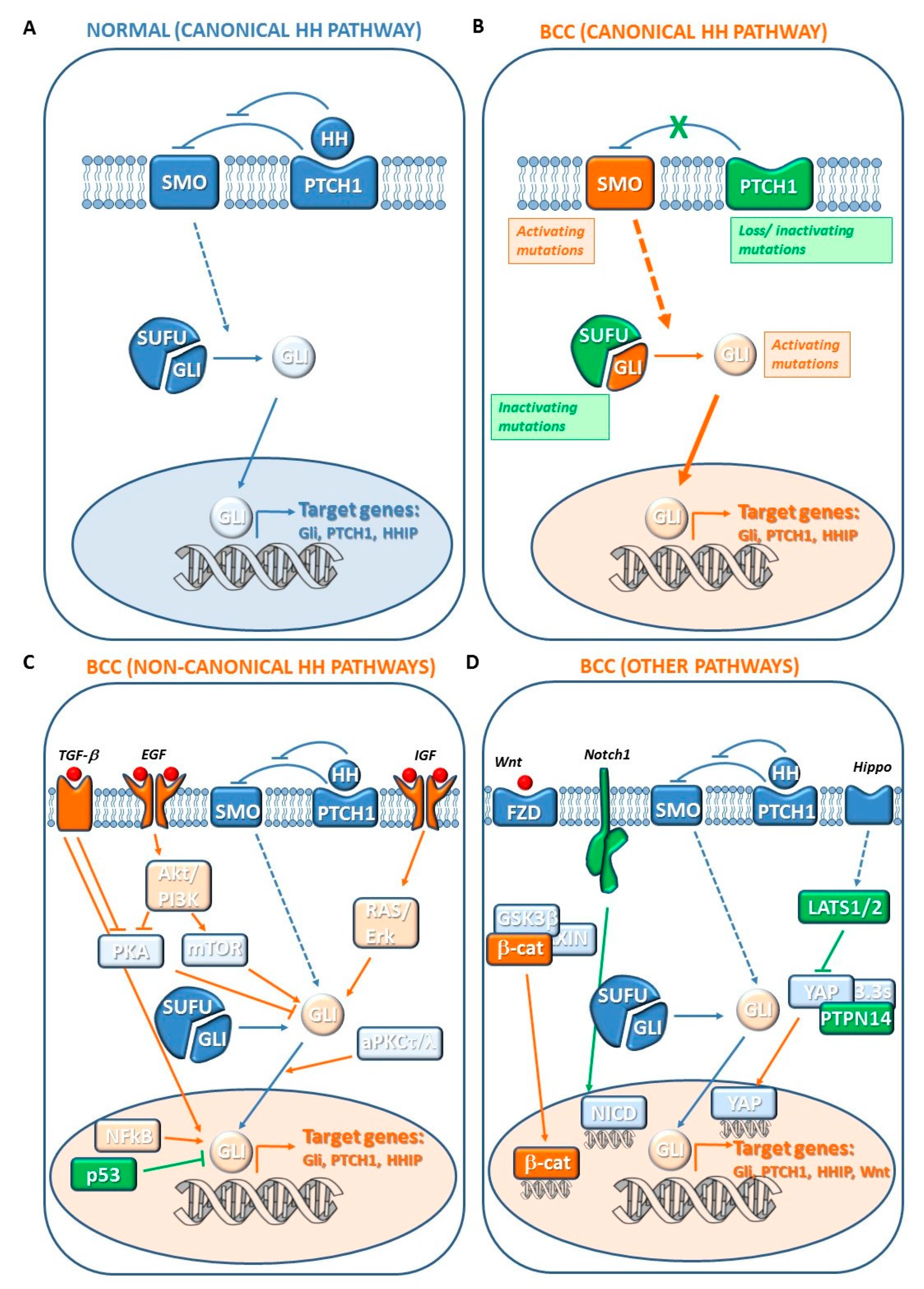
7. Pathogenesis of BCC
7.1. Canonical Hedgehog (HH) Pathway Genes
7.2. Noncanonical HH Pathway Genes
7.3. Other Genetic Changes
7.3.1. Hippo-YAP and WNT Signaling Genes
7.3.2. N-MYCN/FBXW7 Genes
7.3.3. NOTCH Signaling Genes
7.3.4. TERT-Promoter
7.3.5. DPH3-OXNAD1 Bidirectional Promoter
7.3.6. Other BCC-Associated Genes
7.3.7. Noncoding (nc) RNAs
7.4. Cellular Origin of BCC
8. Treatment of Localized BCC
8.1. Surgery
8.2. Mohs Micrographic Surgery (MMS)
8.3. Curettage and Electrodessication
8.4. Cryosurgery
8.5. Photodynamic Therapy (PDT)
8.6. Radiation (RT)
8.7. Topical Therapies
8.8. Intralesional Therapy
8.9. Laser Therapy
9. Targeted Therapy: Hedgehog Pathway Inhibitors (HPI)
9.1. Efficacy and Safety of Vismodegib in Advanced (a) BCC
9.2. Efficacy and Safety of Sonidegib in aBCC
9.3. Comparison between Sonidegib and Vismodegib in aBCC
10. BCC Prevention
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Apalla, Z.; Nashan, D.; Weller, R.B.; Castellsagué, X. Skin Cancer: Epidemiology, Disease Burden, Pathophysiology, Diagnosis, and Therapeutic Approaches. Dermatol. Ther. (Heidelb) 2017, 7, 5–19. [Google Scholar] [CrossRef] [Green Version]
- Miller, D.L.; Weinstock, M.A. Nonmelanoma skin cancer in the United States: Incidence. J. Am. Acad. Dermatol. 1994, 30, 774–778. [Google Scholar] [CrossRef]
- Sreekantaswamy, S.; Endo, J.; Chen, A.; Butler, D.; Morrison, L.; Linos, E. Aging and the treatment of basal cell carcinoma. Clin. Dermatol. 2019, 37, 373–378. [Google Scholar] [CrossRef]
- Fania, L.; Mazzanti, C.; Campione, E.; Candi, E.; Abeni, D.; Dellambra, E. Role of Nicotinamide in Genomic Stability and Skin Cancer Chemoprevention. Int. J. Mol. Sci. 2019, 20, 5946. [Google Scholar] [CrossRef] [Green Version]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Snaidr, V.A.; Damian, D.L.; Halliday, G.M. Nicotinamide for photoprotection and skin cancer chemoprevention: A review of efficacy and safety. Exp. Dermatol. 2019, 28 (Suppl. 1), 15–22. [Google Scholar] [CrossRef] [Green Version]
- Zanetti, R.; Rosso, S.; Martinez, C.; Nieto, A.; Miranda, A.; Mercier, M.; Loria, D.I.; Østerlind, A.; Greinert, R.; Navarro, C.; et al. Comparison of risk patterns in carcinoma and melanoma of the skin in men: A multi-centre case-case-control study. Br. J. Cancer 2006, 94, 743–751. [Google Scholar] [CrossRef]
- Kricker, A.; Armstrong, B.K.; English, D.R.; Heenan, P.J. Does intermittent sun exposure cause basal cell carcinoma? a case-control study in Western Australia. Int. J. Cancer 1995, 60, 489–494. [Google Scholar] [CrossRef]
- Gallagher, R.P.; Hill, G.B.; Bajdik, C.D.; Fincham, S.; Coldman, A.J.; McLean, D.I.; Threlfall, W.J. Sunlight exposure, pigmentary factors, and risk of nonmelanocytic skin cancer. I. Basal cell carcinoma. Arch. Dermatol. 1995, 131, 157–163. [Google Scholar] [CrossRef]
- Reizner, G.T.; Chuang, T.-Y.; Elpern, D.J.; Stone, J.L.; Farmer, E.R. Basal cell carcinoma in Kauai, Hawaii: The highest documented incidence in the United States. J. Am. Acad. Dermatol. 1993, 29, 184–189. [Google Scholar] [CrossRef]
- Chuang, T.-Y.; Popescu, A.; Su, W.D.; Chute, C.G. Basal cell carcinoma. J. Am. Acad. Dermatol. 1990, 22, 413–417. [Google Scholar] [CrossRef]
- Wehner, M.R.; Shive, M.L.; Chren, M.-M.; Han, J.; Qureshi, A.A.; Linos, E. Indoor tanning and non-melanoma skin cancer: Systematic review and meta-analysis. BMJ 2012, 345, e5909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrucci, L.M.; Cartmel, B.; Molinaro, A.M.; Leffell, D.J.; Bale, A.E.; Mayne, S.T. Indoor tanning and risk of early-onset basal cell carcinoma. J. Am. Acad. Dermatol. 2012, 67, 552–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyd, A.S.; Shyr, Y.; King, L.E. Basal cell carcinoma in young women: An evaluation of the association of tanning bed use and smoking. J. Am. Acad. Dermatol. 2002, 46, 706–709. [Google Scholar] [CrossRef]
- Bassukas, I.D.; Tatsioni, A. Male Sex is an Inherent Risk Factor for Basal Cell Carcinoma. J. Skin Cancer 2019, 2019, 8304271. [Google Scholar] [CrossRef] [Green Version]
- Huncharek, M. Lung function in coalworkers’ pneumoconiosis. Br. J. Ind. Med. 1987, 44, 215–216. [Google Scholar] [CrossRef] [Green Version]
- Stern, R.S. The risk of squamous cell and basal cell cancer associated with psoralen and ultraviolet A therapy: A 30-year prospective study. J. Am. Acad. Dermatol. 2012, 66, 553–562. [Google Scholar] [CrossRef]
- Schmidt, S.A.J.; Schmidt, M.; Mehnert, F.; Lemeshow, S.; Sørensen, H.T. Use of antihypertensive drugs and risk of skin cancer. J. Eur. Acad. Dermatol. Venereol. 2015, 29, 1545–1554. [Google Scholar] [CrossRef]
- Cassano, N.; Di Stefani, A.; Vena, G.A.; Peris, K. Antihypertensive drugs and risk of skin cancer. G. Ital. Dermatol. Venereol. 2018, 153, 672–684. [Google Scholar] [CrossRef]
- Ruiter, R.; Visser, L.E.; Eijgelsheim, M.; Rodenburg, E.M.; Hofman, A.; Coebergh, J.-W.W.; Nijsten, T.; Stricker, B.H.C. High-ceiling diuretics are associated with an increased risk of basal cell carcinoma in a population-based follow-up study. Eur. J. Cancer (Oxf. Engl.: 1990) 2010, 46, 2467–2472. [Google Scholar] [CrossRef]
- Marzuka, A.G.; Book, S.E. Basal cell carcinoma: Pathogenesis, epidemiology, clinical features, diagnosis, histopathology, and management. Yale J. Biol. Med. 2015, 88, 167–179. [Google Scholar] [PubMed]
- Karagas, M.R.; Tosteson, T.D.; Blum, J.; Morris, J.S.; Baron, J.A.; Klaue, B. Design of an epidemiologic study of drinking water arsenic exposure and skin and bladder cancer risk in a U.S. population. Environ. Health Perspect. 1998, 106 (Suppl. 4), 1047–1050. [Google Scholar] [CrossRef] [PubMed]
- Boaventura, P.; Oliveira, R.; Pereira, D.; Soares, P.; Teixeira-Gomes, J. Head and neck basal cell carcinoma prevalence in individuals submitted to childhood X-ray epilation for tinea capitis treatment. Eur. J. Dermatol. 2012, 22, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Lichter, M.D.; Karagas, M.R.; Mott, L.A.; Spencer, S.K.; Stukel, T.A.; Greenberg, E.R. Therapeutic ionizing radiation and the incidence of basal cell carcinoma and squamous cell carcinoma. The New Hampshire Skin Cancer Study Group. Arch. Dermatol. 2000, 136, 1007–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shore, R.E.; Moseson, M.; Xue, X.; Tse, Y.; Harley, N.; Pasternack, B.S. Skin Cancer after X-Ray Treatment for Scalp Ringworm. Radiat. Res. 2002, 157, 410–418. [Google Scholar] [CrossRef]
- Karagas, M.R.; McDonald, J.A.; Greenberg, E.R.; Stukel, T.A.; Weiss, J.E.; Baron, J.A.; Stevens, M.M. Risk of basal cell and squamous cell skin cancers after ionizing radiation therapy. J. Natl. Cancer Inst. 1996, 88, 1848–1853. [Google Scholar] [CrossRef] [Green Version]
- Boaventura, P.; Pereira, D.; Mendes, A.; Batista, R.; da Silva, A.F.; Guimarães, I.; Honavar, M.; Teixeira-Gomes, J.; Lopes, J.M.; Máximo, V.; et al. Mitochondrial D310 D-Loop instability and histological subtypes in radiation-induced cutaneous basal cell carcinomas. J. Dermatol. Sci. 2014, 73, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Karagas, M.R.; Stukel, T.A.; Greenberg, E.R.; Baron, J.A.; Mott, L.A.; Stern, R.S. Risk of subsequent basal cell carcinoma and squamous cell carcinoma of the skin among patients with prior skin cancer. JAMA 1992, 267, 3305–3310. [Google Scholar] [CrossRef]
- Marcil, I.; Stern, R.S. Risk of developing a subsequent nonmelanoma skin cancer in patients with a history of nonmelanoma skin cancer: A critical review of the literature and meta-analysis. Arch. Dermatol. 2000, 136, 1524–1530. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, S.; Rajaratnam, R.; Smith, A.G.; Lear, J.T.; Strange, R.C. Patients with both basal and squamous cell carcinomas are at a lower risk of further basal cell carcinomas than patients with only a basal cell carcinoma. J. Am. Acad. Dermatol. 2009, 61, 247–251. [Google Scholar] [CrossRef]
- Flohil, S.C.; van der Leest, R.J.T.; Arends, L.R.; de Vries, E.; Nijsten, T. Risk of subsequent cutaneous malignancy in patients with prior keratinocyte carcinoma: A systematic review and meta-analysis. Eur. J. Cancer (Oxf. Engl.: 1990) 2013, 49, 2365–2375. [Google Scholar] [CrossRef] [PubMed]
- Wehner, M.R.; Linos, E.; Parvataneni, R.; Stuart, S.E.; Boscardin, W.J.; Chren, M.-M. Timing of subsequent new tumors in patients who present with basal cell carcinoma or cutaneous squamous cell carcinoma. JAMA Dermatol. 2015, 151, 382–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiiski, V.; de Vries, E.; Flohil, S.C.; Bijl, M.J.; Hofman, A.; Stricker, B.H.C.; Nijsten, T. Risk factors for single and multiple basal cell carcinomas. Arch. Dermatol. 2010, 146, 848–855. [Google Scholar] [CrossRef] [PubMed]
- Ricci, F.; Paradisi, A.; Fania, L.; Pallotta, S.; Di Lella, G.; Sobrino, L.; Panebianco, A.; Abeni, D. High melanoma risk in non-melanoma skin cancer patients under age 40: A large retrospective cohort study. G. Ital. Dermatol. Venereol. 2019. [Google Scholar] [CrossRef]
- Correia de Sá, T.R.; Silva, R.; Lopes, J.M. Basal cell carcinoma of the skin (part 1): Epidemiology, pathology and genetic syndromes. Future Oncol. 2015, 11, 3011–3021. [Google Scholar] [CrossRef]
- Kim, D.P.; Kus, K.J.B.; Ruiz, E. Basal Cell Carcinoma Review. Hematol./Oncol. Clin. N. Am. 2019, 33, 13–24. [Google Scholar] [CrossRef]
- Gandhi, S.A.; Kampp, J. Skin Cancer Epidemiology, Detection, and Management. Med. Clin. N. Am. 2015, 99, 1323–1335. [Google Scholar] [CrossRef]
- Scrivener, Y.; Grosshans, E.; Cribier, B. Variations of basal cell carcinomas according to gender, age, location and histopathological subtype. Br. J. Dermatol. 2002, 147, 41–47. [Google Scholar] [CrossRef]
- McCormack, C.J.; Kelly, J.W.; Dorevitch, A.P. Differences in age and body site distribution of the histological subtypes of basal cell carcinoma. A possible indicator of differing causes. Arch. Dermatol. 1997, 133, 593–596. [Google Scholar] [CrossRef]
- Zoccali, G.; Pajand, R.; Papa, P.; Orsini, G.; Lomartire, N.; Giuliani, M. Giant basal cell carcinoma of the skin: Literature review and personal experience. J. Eur. Acad. Dermatol. Venereol. 2012, 26, 942–952. [Google Scholar] [CrossRef]
- Crowson, A.N. Basal cell carcinoma: Biology, morphology and clinical implications. Mod. Pathol. 2006, 19 (Suppl. 2), S127–S147. [Google Scholar] [CrossRef] [PubMed]
- Trakatelli, M.; Morton, C.; Nagore, E.; Ulrich, C.; Del Marmol, V.; Peris, K.; Basset-Seguin, N. Update of the European guidelines for basal cell carcinoma management. Eur. J. Dermatol. 2014, 24, 312–329. [Google Scholar] [CrossRef] [PubMed]
- Dandurand, M.; Petit, T.; Martel, P.; Guillot, B. Management of basal cell carcinoma in adults Clinical practice guidelines. Eur. J. Dermatol. 2006, 16, 394–401. [Google Scholar] [PubMed]
- Peris, K.; Fargnoli, M.C.; Garbe, C.; Kaufmann, R.; Bastholt, L.; Seguin, N.B.; Bataille, V.; Marmol, V.D.; Dummer, R.; Harwood, C.A.; et al. Diagnosis and treatment of basal cell carcinoma: European consensus-based interdisciplinary guidelines. Eur. J. Cancer (Oxf. Engl.: 1990) 2019, 118, 10–34. [Google Scholar] [CrossRef] [Green Version]
- Paolino, G.; Donati, M.; Didona, D.; Mercuri, S.R.; Cantisani, C. Histology of Non-Melanoma Skin Cancers: An Update. Biomedicines 2017, 5, 71. [Google Scholar] [CrossRef] [Green Version]
- Mavrikakis, I.; Malhotra, R.; Selva, D.; Huilgol, S.C.; Barlow, R. Linear basal cell carcinoma: A distinct clinical entity. J. Plast. Reconstr. Aesthet. Surg. 2006, 59, 419–423. [Google Scholar] [CrossRef]
- Leblebici, C.; Bambul Sığırcı, B.; Kelten Talu, C.; Koca, S.B.; Huq, G.E. CD10, TDAG51, CK20, AR, INSM1, and Nestin Expression in the Differential Diagnosis of Trichoblastoma and Basal Cell Carcinoma. Int. J. Surg. Pathol. 2019, 27, 19–27. [Google Scholar] [CrossRef]
- Yada, K.; Kashima, K.; Daa, T.; Kitano, S.; Fujiwara, S.; Yokoyama, S. Expression of CD10 in basal cell carcinoma. Am. J. Dermatopathol. 2004, 26, 463–471. [Google Scholar] [CrossRef]
- Di Stefani, A.; Chimenti, S. Basal cell carcinoma: Clinical and pathological features. G. Ital. Dermatol. Venereol. 2015, 150, 385–391. [Google Scholar]
- Lubeek, S.F.K.; van Vugt, L.J.; Aben, K.K.H.; van de Kerkhof, P.C.M.; Gerritsen, M.-J.P. The Epidemiology and Clinicopathological Features of Basal Cell Carcinoma in Patients 80 Years and Older: A Systematic Review. JAMA Dermatol. 2017, 153, 71–78. [Google Scholar] [CrossRef]
- Jetley, S.; Jairajpuri, Z.S.; Rana, S.; Talikoti, M.A. Adenoid basal cell carcinoma and its mimics. Indian J. Dermatol. 2013, 58, 244. [Google Scholar] [CrossRef] [PubMed]
- Elston, D.M.; Bergfeld, W.F.; Petroff, N. Basal cell carcinoma with monster cells. J. Cutan. Pathol. 1993, 20, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Sexton, M.; Jones, D.B.; Maloney, M.E. Histologic pattern analysis of basal cell carcinoma. J. Am. Acad. Dermatol. 1990, 23, 1118–1126. [Google Scholar] [CrossRef]
- Dreier, J.; Cheng, P.F.; Bogdan Alleman, I.; Gugger, A.; Hafner, J.; Tschopp, A.; Goldinger, S.M.; Levesque, M.P.; Dummer, R. Basal cell carcinomas in a tertiary referral centre: A systematic analysis. Br. J. Dermatol. 2014, 171, 1066–1072. [Google Scholar] [CrossRef]
- Liersch, J.; Schaller, J. Das Basalzellkarzinom und seine seltenen Formvarianten. Pathologe 2014, 35, 433–442. [Google Scholar] [CrossRef]
- Abesamis-Cubillan, E.; El-Shabrawi-Caelen, L.; LeBoit, P.E. Merked cells and sclerosing epithelial neoplasms. Am. J. Dermatopathol. 2000, 22, 311–315. [Google Scholar] [CrossRef]
- Goldberg, L.H. Basal cell carcinoma. Lancet 1996, 347, 663–667. [Google Scholar] [CrossRef]
- Faria, J. de. Basal cell carcinoma of the skin with areas of squamous cell carcinoma: A basosquamous cell carcinoma? J. Clin. Pathol. 1985, 38, 1273–1277. [Google Scholar] [CrossRef] [Green Version]
- Annessi, G.; Baliva, G. Basal cell and squamous cell carcinomas. Clinico-histological features. Ann. Ist. Super. Sanita 1996, 32, 29–36. [Google Scholar]
- Garcia, C.; Poletti, E.; Crowson, A.N. Basosquamous carcinoma. J. Am. Acad. Dermatol. 2009, 60, 137–143. [Google Scholar] [CrossRef]
- Menzies, S.W.; Westerhoff, K.; Rabinovitz, H.; Kopf, A.W.; McCarthy, W.H.; Katz, B. Surface microscopy of pigmented basal cell carcinoma. Arch. Dermatol. 2000, 136, 1012–1016. [Google Scholar] [CrossRef] [Green Version]
- Altamura, D.; Menzies, S.W.; Argenziano, G.; Zalaudek, I.; Soyer, H.P.; Sera, F.; Avramidis, M.; DeAmbrosis, K.; Fargnoli, M.C.; Peris, K. Dermatoscopy of basal cell carcinoma: Morphologic variability of global and local features and accuracy of diagnosis. J. Am. Acad. Dermatol. 2010, 62, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Lallas, A.; Argenziano, G.; Zendri, E.; Moscarella, E.; Longo, C.; Grenzi, L.; Pellacani, G.; Zalaudek, I. Update on non-melanoma skin cancer and the value of dermoscopy in its diagnosis and treatment monitoring. Expert Rev. Anticancer Ther. 2013, 13, 541–558. [Google Scholar] [CrossRef]
- Lallas, A.; Apalla, Z.; Ioannides, D.; Argenziano, G.; Castagnetti, F.; Moscarella, E.; Longo, C.; Palmieri, T.; Ramundo, D.; Zalaudek, I. Dermoscopy in the diagnosis and management of basal cell carcinoma. Future Oncol. 2015, 11, 2975–2984. [Google Scholar] [CrossRef]
- Lallas, A.; Apalla, Z.; Argenziano, G.; Longo, C.; Moscarella, E.; Specchio, F.; Raucci, M.; Zalaudek, I. The dermatoscopic universe of basal cell carcinoma. Dermatol. Pract. Concept. 2014, 4, 11–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.; Chamberlain, A.J.; Bailey, M.; Chong, A.H.; Haskett, M.; Kelly, J.W. Dermatoscopy aids in the diagnosis of the solitary red scaly patch or plaque-features distinguishing superficial basal cell carcinoma, intraepidermal carcinoma, and psoriasis. J. Am. Acad. Dermatol. 2008, 59, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Micantonio, T.; Gulia, A.; Altobelli, E.; Di Cesare, A.; Fidanza, R.; Riitano, A.; Fargnoli, M.C.; Peris, K. Vascular patterns in basal cell carcinoma. J. Eur. Acad. Dermatol. Venereol. 2011, 25, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Zalaudek, I.; Kreusch, J.; Giacomel, J.; Ferrara, G.; Catricalà, C.; Argenziano, G. How to diagnose nonpigmented skin tumors: A review of vascular structures seen with dermoscopy: Part II. Nonmelanocytic skin tumors. J. Am. Acad. Dermatol. 2010, 63, 377–386. [Google Scholar] [CrossRef]
- Argenziano, G.; Zalaudek, I.; Corona, R.; Sera, F.; Cicale, L.; Petrillo, G.; Ruocco, E.; Hofmann-Wellenhof, R.; Soyer, H.P. Vascular structures in skin tumors: A dermoscopy study. Arch. Dermatol. 2004, 140, 1485–1489. [Google Scholar] [CrossRef]
- Giacomel, J.; Zalaudek, I. Dermoscopy of superficial basal cell carcinoma. Dermatol. Surg. Off. Publ. Am. Soc. Dermatol. Surg. 2005, 31, 1710–1713. [Google Scholar] [CrossRef]
- Suppa, M.; Micantonio, T.; Di Stefani, A.; Soyer, H.P.; Chimenti, S.; Fargnoli, M.C.; Peris, K. Dermoscopic variability of basal cell carcinoma according to clinical type and anatomic location. J. Eur. Acad. Dermatol. Venereol. 2015, 29, 1732–1741. [Google Scholar] [CrossRef] [PubMed]
- Marghoob, A.A.; Cowell, L.; Kopf, A.W.; Scope, A. Observation of chrysalis structures with polarized dermoscopy. Arch. Dermatol. 2009, 145, 618. [Google Scholar] [CrossRef]
- Shitara, D.; Ishioka, P.; Alonso-Pinedo, Y.; Palacios-Bejarano, L.; Carrera, C.; Malvehy, J.; Puig, S. Shiny white streaks: A sign of malignancy at dermoscopy of pigmented skin lesions. Acta Derm. Venereol. 2014, 94, 132–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajadhyaksha, M.; Grossman, M.; Esterowitz, D.; Webb, R.H.; Anderson, R.R. In vivo confocal scanning laser microscopy of human skin: Melanin provides strong contrast. J. Investig. Dermatol. 1995, 104, 946–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nwaneshiudu, A.; Kuschal, C.; Sakamoto, F.H.; Anderson, R.R.; Schwarzenberger, K.; Young, R.C. Introduction to confocal microscopy. J. Investig. Dermatol. 2012, 132, e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nori, S.; Rius-Díaz, F.; Cuevas, J.; Goldgeier, M.; Jaen, P.; Torres, A.; González, S. Sensitivity and specificity of reflectance-mode confocal microscopy for in vivo diagnosis of basal cell carcinoma: A multicenter study. J. Am. Acad. Dermatol. 2004, 51, 923–930. [Google Scholar] [CrossRef]
- Segura, S.; Puig, S.; Carrera, C.; Palou, J.; Malvehy, J. Dendritic cells in pigmented basal cell carcinoma: A relevant finding by reflectance-mode confocal microscopy. Arch. Dermatol. 2007, 143, 883–886. [Google Scholar] [CrossRef]
- Ulrich, M.; Roewert-Huber, J.; González, S.; Rius-Diaz, F.; Stockfleth, E.; Kanitakis, J. Peritumoral clefting in basal cell carcinoma: Correlation of in vivo reflectance confocal microscopy and routine histology. J. Cutan. Pathol. 2011, 38, 190–195. [Google Scholar] [CrossRef]
- Wurm, E.M.T.; Curchin, C.E.S.; Lambie, D.; Longo, C.; Pellacani, G.; Soyer, H.P. Confocal features of equivocal facial lesions on severely sun-damaged skin: Four case studies with dermatoscopic, confocal, and histopathologic correlation. J. Am. Acad. Dermatol. 2012, 66, 463–473. [Google Scholar] [CrossRef]
- Casari, A.; Pellacani, G.; Seidenari, S.; Cesinaro, A.M.; Beretti, F.; Pepe, P.; Longo, C. Pigmented nodular Basal cell carcinomas in differential diagnosis with nodular melanomas: Confocal microscopy as a reliable tool for in vivo histologic diagnosis. J. Skin Cancer 2011, 2011, 406859. [Google Scholar] [CrossRef] [Green Version]
- Guitera, P.; Menzies, S.W.; Longo, C.; Cesinaro, A.M.; Scolyer, R.A.; Pellacani, G. In vivo confocal microscopy for diagnosis of melanoma and basal cell carcinoma using a two-step method: Analysis of 710 consecutive clinically equivocal cases. J. Investig. Dermatol. 2012, 132, 2386–2394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longo, C.; Casari, A.; Pepe, P.; Moscarella, E.; Zalaudek, I.; Argenziano, G.; Pellacani, G. Confocal microscopy insights into the treatment and cellular immune response of Basal cell carcinoma to photodynamic therapy. Dermatology (Basel) 2012, 225, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Longo, C.; Borsari, S.; Pampena, R.; Benati, E.; Bombonato, C.; Raucci, M.; Mirra, M.; Di Stefani, A.; Peris, K.; Pellacani, G. Basal cell carcinoma: The utility of in vivo and ex vivo confocal microscopy. J. Eur. Acad. Dermatol. Venereol. 2018, 32, 2090–2096. [Google Scholar] [CrossRef] [PubMed]
- Moscarella, E.; Rabinovitz, H.; Oliviero, M.C.; Brown, L.; Longo, C.; Zalaudek, I.; Piana, S.; Farnetani, F.; Lallas, A.; Argenziano, G.; et al. The role of reflectance confocal microscopy as an aid in the diagnosis of collision tumors. Dermatology (Basel) 2013, 227, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Peppelman, M.; Wolberink, E.A.W.; Blokx, W.A.M.; van de Kerkhof, P.C.M.; van Erp, P.E.J.; Gerritsen, M.-J.P. In vivo diagnosis of basal cell carcinoma subtype by reflectance confocal microscopy. Dermatology (Basel) 2013, 227, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Longo, C.; Lallas, A.; Kyrgidis, A.; Rabinovitz, H.; Moscarella, E.; Ciardo, S.; Zalaudek, I.; Oliviero, M.; Losi, A.; Gonzalez, S.; et al. Classifying distinct basal cell carcinoma subtype by means of dermatoscopy and reflectance confocal microscopy. J. Am. Acad. Dermatol. 2014, 71, 716–724. [Google Scholar] [CrossRef]
- Cheng, H.M.; Guitera, P. Systematic review of optical coherence tomography usage in the diagnosis and management of basal cell carcinoma. Br. J. Dermatol. 2015, 173, 1371–1380. [Google Scholar] [CrossRef]
- Cheng, H.M.; Lo, S.; Scolyer, R.; Meekings, A.; Carlos, G.; Guitera, P. Accuracy of optical coherence tomography for the diagnosis of superficial basal cell carcinoma: A prospective, consecutive, cohort study of 168 cases. Br. J. Dermatol. 2016, 175, 1290–1300. [Google Scholar] [CrossRef]
- Ulrich, M.; von Braunmuehl, T.; Kurzen, H.; Dirschka, T.; Kellner, C.; Sattler, E.; Berking, C.; Welzel, J.; Reinhold, U. The sensitivity and specificity of optical coherence tomography for the assisted diagnosis of nonpigmented basal cell carcinoma: An observational study. Br. J. Dermatol. 2015, 173, 428–435. [Google Scholar] [CrossRef] [Green Version]
- Lui, H.; Zhao, J.; McLean, D.; Zeng, H. Real-time Raman spectroscopy for in vivo skin cancer diagnosis. Cancer Res. 2012, 72, 2491–2500. [Google Scholar] [CrossRef] [Green Version]
- Woodward, R.M.; Wallace, V.P.; Pye, R.J.; Cole, B.E.; Arnone, D.D.; Linfield, E.H.; Pepper, M. Terahertz pulse imaging of ex vivo basal cell carcinoma. J. Investig. Dermatol. 2003, 120, 72–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Lui, H.; Kalia, S.; Zeng, H. Real-time Raman spectroscopy for automatic in vivo skin cancer detection: An independent validation. Anal. Bioanal. Chem. 2015, 407, 8373–8379. [Google Scholar] [CrossRef] [PubMed]
- Bens, G.; Binois, R.; Roussel, A.; Kerdraon, R.; Estève, É. Échographie cutanée haute résolution dans le diagnostic différentiel entre carcinome basocellulaire nodulaire et hyperplasie sébacée du visage: Une étude pilote. Ann. Dermatol. Venereol. 2015, 142, 646–652. [Google Scholar] [CrossRef]
- Ikehata, H.; Ono, T. The mechanisms of UV mutagenesis. J. Radiat. Res. 2011, 52, 115–125. [Google Scholar] [CrossRef] [Green Version]
- Didona, D.; Paolino, G.; Bottoni, U.; Cantisani, C. Non Melanoma Skin Cancer Pathogenesis Overview. Biomedicines 2018, 6, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epstein, E.H. Basal cell carcinomas: Attack of the hedgehog. Nat. Rev. Cancer 2008, 8, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varjosalo, M.; Taipale, J. Hedgehog: Functions and mechanisms. Genes Dev. 2008, 22, 2454–2472. [Google Scholar] [CrossRef] [Green Version]
- Niyaz, M.; Khan, M.S.; Mudassar, S. Hedgehog Signaling: An Achilles’ Heel in Cancer. Transl. Oncol. 2019, 12, 1334–1344. [Google Scholar] [CrossRef]
- Pellegrini, C.; Maturo, M.G.; Di Nardo, L.; Ciciarelli, V.; Gutiérrez García-Rodrigo, C.; Fargnoli, M.C. Understanding the Molecular Genetics of Basal Cell Carcinoma. Int. J. Mol. Sci. 2017, 18, 2485. [Google Scholar] [CrossRef] [Green Version]
- Bakshi, A.; Chaudhary, S.C.; Rana, M.; Elmets, C.A.; Athar, M. Basal cell carcinoma pathogenesis and therapy involving hedgehog signaling and beyond. Mol. Carcinog. 2017, 56, 2543–2557. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Platt, K.A.; Censullo, P.; Ruiz i Altaba, A. Gli1 is a target of Sonic hedgehog that induces ventral neural tube development. Development 1997, 124, 2537–2552. [Google Scholar] [PubMed]
- Xie, J.; Murone, M.; Luoh, S.M.; Ryan, A.; Gu, Q.; Zhang, C.; Bonifas, J.M.; Lam, C.W.; Hynes, M.; Goddard, A.; et al. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature 1998, 391, 90–92. [Google Scholar] [CrossRef] [PubMed]
- Reifenberger, J.; Wolter, M.; Weber, R.G.; Megahed, M.; Ruzicka, T.; Lichter, P.; Reifenberger, G. Missense mutations in SMOH in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer Res. 1998, 58, 1798–1803. [Google Scholar] [PubMed]
- Gailani, M.R.; Ståhle-Bäckdahl, M.; Leffell, D.J.; Glynn, M.; Zaphiropoulos, P.G.; Pressman, C.; Undén, A.B.; Dean, M.; Brash, D.E.; Bale, A.E.; et al. The role of the human homologue of Drosophila patched in sporadic basal cell carcinomas. Nat. Genet. 1996, 14, 78–81. [Google Scholar] [CrossRef]
- Aszterbaum, M.; Rothman, A.; Johnson, R.L.; Fisher, M.; Xie, J.; Bonifas, J.M.; Zhang, X.; Scott, M.P.; Epstein, E.H. Identification of mutations in the human PATCHED gene in sporadic basal cell carcinomas and in patients with the basal cell nevus syndrome. J. Investig. Dermatol. 1998, 110, 885–888. [Google Scholar] [CrossRef] [Green Version]
- Danhof, R.; Lewis, K.; Brown, M. Small Molecule Inhibitors of the Hedgehog Pathway in the Treatment of Basal Cell Carcinoma of the Skin. Am. J. Clin. Dermatol. 2018, 19, 195–207. [Google Scholar] [CrossRef]
- Nilsson, M.; Undèn, A.B.; Krause, D.; Malmqwist, U.; Raza, K.; Zaphiropoulos, P.G.; Toftgård, R. Induction of basal cell carcinomas and trichoepitheliomas in mice overexpressing GLI-1. Proc. Natl. Acad. Sci. USA 2000, 97, 3438–3443. [Google Scholar] [CrossRef]
- Sheng, H.; Goich, S.; Wang, A.; Grachtchouk, M.; Lowe, L.; Mo, R.; Lin, K.; de Sauvage, F.J.; Sasaki, H.; Hui, C.; et al. Dissecting the oncogenic potential of Gli2: Deletion of an NH(2)-terminal fragment alters skin tumor phenotype. Cancer Res. 2002, 62, 5308–5316. [Google Scholar]
- Grachtchouk, M.; Mo, R.; Yu, S.; Zhang, X.; Sasaki, H.; Hui, C.C.; Dlugosz, A.A. Basal cell carcinomas in mice overexpressing Gli2 in skin. Nat. Genet. 2000, 24, 216–217. [Google Scholar] [CrossRef] [PubMed]
- Di Magno, L.; Coni, S.; Di Marcotullio, L.; Canettieri, G. Digging a hole under Hedgehog: Downstream inhibition as an emerging anticancer strategy. Biochim. Biophys. Acta 2015, 1856, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Smyth, I.; Narang, M.A.; Evans, T.; Heimann, C.; Nakamura, Y.; Chenevix-Trench, G.; Pietsch, T.; Wicking, C.; Wainwright, B.J. Isolation and characterization of human patched 2 (PTCH2), a putative tumour suppressor gene inbasal cell carcinoma and medulloblastoma on chromosome 1p32. Hum. Mol. Genet. 1999, 8, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Couvé-Privat, S.; Le Bret, M.; Traiffort, E.; Queille, S.; Coulombe, J.; Bouadjar, B.; Avril, M.F.; Ruat, M.; Sarasin, A.; Daya-Grosjean, L. Functional analysis of novel sonic hedgehog gene mutations identified in basal cell carcinomas from xeroderma pigmentosum patients. Cancer Res. 2004, 64, 3559–3565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oro, A.E.; Higgins, K.M.; Hu, Z.; Bonifas, J.M.; Epstein, E.H.; Scott, M.P. Basal cell carcinomas in mice overexpressing sonic hedgehog. Science 1997, 276, 817–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, R.L.; Rothman, A.L.; Xie, J.; Goodrich, L.V.; Bare, J.W.; Bonifas, J.M.; Quinn, A.G.; Myers, R.M.; Cox, D.R.; Epstein, E.H.; et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science 1996, 272, 1668–1671. [Google Scholar] [CrossRef] [Green Version]
- Hahn, H.; Wicking, C.; Zaphiropoulos, P.G.; Gailani, M.R.; Shanley, S.; Chidambaram, A.; Vorechovsky, I.; Holmberg, E.; Unden, A.B.; Gillies, S.; et al. Mutations of the Human Homolog of Drosophila patched in the Nevoid Basal Cell Carcinoma Syndrome. Cell 1996, 85, 841–851. [Google Scholar] [CrossRef] [Green Version]
- Fan, H.; Oro, A.E.; Scott, M.P.; Khavari, P.A. Induction of basal cell carcinoma features in transgenic human skin expressing Sonic Hedgehog. Nat. Med. 1997, 3, 788–792. [Google Scholar] [CrossRef]
- Hutchin, M.E.; Kariapper, M.S.T.; Grachtchouk, M.; Wang, A.; Wei, L.; Cummings, D.; Liu, J.; Michael, L.E.; Glick, A.; Dlugosz, A.A. Sustained Hedgehog signaling is required for basal cell carcinoma proliferation and survival: Conditional skin tumorigenesis recapitulates the hair growth cycle. Genes Dev. 2005, 19, 214–223. [Google Scholar] [CrossRef] [Green Version]
- Pietrobono, S.; Gagliardi, S.; Stecca, B. Non-canonical Hedgehog Signaling Pathway in Cancer: Activation of GLI Transcription Factors Beyond Smoothened. Front. Genet. 2019, 10, 556. [Google Scholar] [CrossRef] [Green Version]
- Kasper, M.; Schnidar, H.; Neill, G.W.; Hanneder, M.; Klingler, S.; Blaas, L.; Schmid, C.; Hauser-Kronberger, C.; Regl, G.; Philpott, M.P.; et al. Selective modulation of Hedgehog/GLI target gene expression by epidermal growth factor signaling in human keratinocytes. Mol. Cell. Biol. 2006, 26, 6283–6298. [Google Scholar] [CrossRef] [Green Version]
- Dennler, S.; André, J.; Verrecchia, F.; Mauviel, A. Cloning of the human GLI2 Promoter: Transcriptional activation by transforming growth factor-beta via SMAD3/beta-catenin cooperation. J. Biol. Chem. 2009, 284, 31523–31531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atwood, S.X.; Chang, A.L.S.; Oro, A.E. Hedgehog pathway inhibition and the race against tumor evolution. J. Cell Biol. 2012, 199, 193–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riobó, N.A.; Lu, K.; Ai, X.; Haines, G.M.; Emerson, C.P. Phosphoinositide 3-kinase and Akt are essential for Sonic Hedgehog signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 4505–4510. [Google Scholar] [CrossRef] [Green Version]
- DiDonato, J.A.; Mercurio, F.; Karin, M. NF-κB and the link between inflammation and cancer. Immunol. Rev. 2012, 246, 379–400. [Google Scholar] [CrossRef]
- Colavito, S.A.; Zou, M.R.; Yan, Q.; Nguyen, D.X.; Stern, D.F. Significance of glioma-associated oncogene homolog 1 (GLI1) expression in claudin-low breast cancer and crosstalk with the nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) pathway. Breast Cancer Res. 2014, 16, 444. [Google Scholar] [CrossRef] [Green Version]
- Mazzà, D.; Infante, P.; Colicchia, V.; Greco, A.; Alfonsi, R.; Siler, M.; Antonucci, L.; Po, A.; de Smaele, E.; Ferretti, E.; et al. PCAF ubiquitin ligase activity inhibits Hedgehog/Gli1 signaling in p53-dependent response to genotoxic stress. Cell Death Differ. 2013, 20, 1688–1697. [Google Scholar] [CrossRef] [Green Version]
- Stecca, B.; Ruiz i Altaba, A. A GLI1-p53 inhibitory loop controls neural stem cell and tumour cell numbers. EMBO J. 2009, 28, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Atwood, S.X.; Sarin, K.Y.; Whitson, R.J.; Li, J.R.; Kim, G.; Rezaee, M.; Ally, M.S.; Kim, J.; Yao, C.; Chang, A.L.S.; et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell 2015, 27, 342–353. [Google Scholar] [CrossRef] [Green Version]
- Wolff, F.; Loipetzberger, A.; Gruber, W.; Esterbauer, H.; Aberger, F.; Frischauf, A.M. Imiquimod directly inhibits Hedgehog signalling by stimulating adenosine receptor/protein kinase A-mediated GLI phosphorylation. Oncogene 2013, 32, 5574–5581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenstein, B.S.; Phelps, R.G.; Weinstock, M.A.; Bernstein, J.L.; Gordon, M.L.; Rudikoff, D.; Kantor, I.; Shelton, R.; Lebwohl, M.G. p53 mutations in basal cell carcinomas arising in routine users of sunscreens. Photochem. Photobiol. 1999, 70, 798–806. [Google Scholar] [CrossRef]
- Bonilla, X.; Parmentier, L.; King, B.; Bezrukov, F.; Kaya, G.; Zoete, V.; Seplyarskiy, V.B.; Sharpe, H.J.; McKee, T.; Letourneau, A.; et al. Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nat. Genet. 2016, 48, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Pasolli, H.A.; Fuchs, E. Yes-associated protein (YAP) transcriptional coactivator functions in balancing growth and differentiation in skin. Proc. Natl. Acad. Sci. USA 2011, 108, 2270–2275. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Li, L.; Lei, Q.; Guan, K.-L. The Hippo-YAP pathway in organ size control and tumorigenesis: An updated version. Genes Dev. 2010, 24, 862–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smirnov, A.; Anemona, L.; Novelli, F.; Piro, C.M.; Annicchiarico-Petruzzelli, M.; Melino, G.; Candi, E. p63 Is a Promising Marker in the Diagnosis of Unusual Skin Cancer. Int. J. Mol. Sci. 2019, 20, 5781. [Google Scholar] [CrossRef] [Green Version]
- Saladi, S.V.; Ross, K.; Karaayvaz, M.; Tata, P.R.; Mou, H.; Rajagopal, J.; Ramaswamy, S.; Ellisen, L.W. ACTL6A Is Co-Amplified with p63 in Squamous Cell Carcinoma to Drive YAP Activation, Regenerative Proliferation, and Poor Prognosis. Cancer Cell 2017, 31, 35–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markov, B.A.; Zol’nikov, Z.I.; Atakulov, D.O.; Zhakova, I.I. Rentenologicheskaia kharakteristika obodochnoĭ kishki do i posle obshirnykh rezektsiĭ po povodu bolezni Girshprunga u deteĭ. Vestn. Rentgenol. Radiol. 1988, 5, 81–83. [Google Scholar]
- Furth, N.; Aylon, Y.; Oren, M. p53 shades of Hippo. Cell Death Differ. 2018, 25, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Debaugnies, M.; Sánchez-Danés, A.; Rorive, S.; Raphaël, M.; Liagre, M.; Parent, M.-A.; Brisebarre, A.; Salmon, I.; Blanpain, C. YAP and TAZ are essential for basal and squamous cell carcinoma initiation. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef]
- van Amerongen, R.; Nusse, R. Towards an integrated view of Wnt signaling in development. Development 2009, 136, 3205–3214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo Muzio, L.; Pannone, G.; Staibano, S.; Mignogna, M.D.; Grieco, M.; Ramires, P.; Romito, A.M.; de Rosa, G.; Piattelli, A. WNT-1 expression in basal cell carcinoma of head and neck. An immunohistochemical and confocal study with regard to the intracellular distribution of beta-catenin. Anticancer Res. 2002, 22, 565–576. [Google Scholar]
- Mullor, J.L.; Dahmane, N.; Sun, T.; i Altaba, A.R. Wnt signals are targets and mediators of Gli function. Curr. Biol. 2001, 11, 769–773. [Google Scholar] [CrossRef] [Green Version]
- Doglioni, C.; Piccinin, S.; Demontis, S.; Cangi, M.G.; Pecciarini, L.; Chiarelli, C.; Armellin, M.; Vukosavljevic, T.; Boiocchi, M.; Maestro, R. Alterations of β-Catenin Pathway in Non-Melanoma Skin Tumors. Am. J. Pathol. 2003, 163, 2277–2287. [Google Scholar] [CrossRef]
- Yamazaki, F.; Aragane, Y.; Kawada, A.; Tezuka, T. Immunohistochemical detection for nuclear beta-catenin in sporadic basal cell carcinoma. Br. J. Dermatol. 2001, 145, 771–777. [Google Scholar] [CrossRef]
- Saldanha, G.; Ghura, V.; Potter, L.; Fletcher, A. Nuclear beta-catenin in basal cell carcinoma correlates with increased proliferation. Br. J. Dermatol. 2004, 151, 157–164. [Google Scholar] [CrossRef] [PubMed]
- El-Bahrawy, M.; El-Masry, N.; Alison, M.; Poulsom, R.; Fallowfield, M. Expression of beta-catenin in basal cell carcinoma. Br. J. Dermatol. 2003, 148, 964–970. [Google Scholar] [CrossRef]
- Yang, S.H.; Andl, T.; Grachtchouk, V.; Wang, A.; Liu, J.; Syu, L.-J.; Ferris, J.; Wang, T.S.; Glick, A.B.; Millar, S.E.; et al. Pathological responses to oncogenic Hedgehog signaling in skin are dependent on canonical Wnt/beta3-catenin signaling. Nat. Genet. 2008, 40, 1130–1135. [Google Scholar] [CrossRef]
- Carmon, K.S.; Lin, Q.; Gong, X.; Thomas, A.; Liu, Q. LGR5 interacts and cointernalizes with Wnt receptors to modulate Wnt/β-catenin signaling. Mol. Cell. Biol. 2012, 32, 2054–2064. [Google Scholar] [CrossRef] [Green Version]
- Hatton, B.A.; Knoepfler, P.S.; Kenney, A.M.; Rowitch, D.H.; de Alborán, I.M.; Olson, J.M.; Eisenman, R.N. N-myc is an essential downstream effector of Shh signaling during both normal and neoplastic cerebellar growth. Cancer Res. 2006, 66, 8655–8661. [Google Scholar] [CrossRef] [Green Version]
- Watt, F.M.; Estrach, S.; Ambler, C.A. Epidermal Notch signalling: Differentiation, cancer and adhesion. Curr. Opin. Cell Biol. 2008, 20, 171–179. [Google Scholar] [CrossRef] [Green Version]
- Jayaraman, S.S.; Rayhan, D.J.; Hazany, S.; Kolodney, M.S. Mutational landscape of basal cell carcinomas by whole-exome sequencing. J. Investig. Dermatol. 2014, 134, 213–220. [Google Scholar] [CrossRef] [Green Version]
- Martincorena, I.; Roshan, A.; Gerstung, M.; Ellis, P.; van Loo, P.; McLaren, S.; Wedge, D.C.; Fullam, A.; Alexandrov, L.B.; Tubio, J.M.; et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 2015, 348, 880–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolas, M.; Wolfer, A.; Raj, K.; Kummer, J.A.; Mill, P.; van Noort, M.; Hui, C.; Clevers, H.; Dotto, G.P.; Radtke, F. Notch1 functions as a tumor suppressor in mouse skin. Nat. Genet. 2003, 33, 416–421. [Google Scholar] [CrossRef]
- Scott, G.A.; Laughlin, T.S.; Rothberg, P.G. Mutations of the TERT promoter are common in basal cell carcinoma and squamous cell carcinoma. Mod. Pathol. 2014, 27, 516–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griewank, K.G.; Murali, R.; Schilling, B.; Schimming, T.; Möller, I.; Moll, I.; Schwamborn, M.; Sucker, A.; Zimmer, L.; Schadendorf, D.; et al. TERT promoter mutations are frequent in cutaneous basal cell carcinoma and squamous cell carcinoma. PLoS ONE 2013, 8, e80354. [Google Scholar] [CrossRef] [PubMed]
- Stern, J.L.; Hibshman, G.; Hu, K.; Ferrara, S.E.; Costello, J.C.; Kim, W.; Tamayo, P.; Cech, T.R.; Huang, F.W. Mesenchymal and MAPK Expression Signatures Associate with Telomerase Promoter Mutations in Multiple Cancers. Mol. Cancer Res. 2020, 18, 1050–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pópulo, H.; Boaventura, P.; Vinagre, J.; Batista, R.; Mendes, A.; Caldas, R.; Pardal, J.; Azevedo, F.; Honavar, M.; Guimarães, I.; et al. TERT promoter mutations in skin cancer: The effects of sun exposure and X-irradiation. J. Investig. Dermatol. 2014, 134, 2251–2257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verkouteren, B.J.A.; Wakkee, M.; van Geel, M.; van Doorn, R.; Winnepenninckx, V.J.; Korpershoek, E.; Mooyaart, A.L.; Reyners, A.K.L.; Terra, J.B.; Aarts, M.J.B.; et al. Molecular testing in metastatic basal cell carcinoma. J. Am. Acad. Dermatol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Maturo, M.G.; Rachakonda, S.; Heidenreich, B.; Pellegrini, C.; Srinivas, N.; Requena, C.; Serra-Guillen, C.; Llombart, B.; Sanmartin, O.; Guillen, C.; et al. Coding and noncoding somatic mutations in candidate genes in basal cell carcinoma. Sci. Rep. 2020, 10, 8005. [Google Scholar] [CrossRef]
- Denisova, E.; Heidenreich, B.; Nagore, E.; Rachakonda, P.S.; Hosen, I.; Akrap, I.; Traves, V.; García-Casado, Z.; López-Guerrero, J.A.; Requena, C.; et al. Frequent DPH3 promoter mutations in skin cancers. Oncotarget 2015, 6, 35922–35930. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Shi, Y.; Ju, P.; Liu, R.; Yeo, S.P.; Xia, Y.; Owlanj, H.; Feng, Z. Silencing of diphthamide synthesis 3 (Dph3) reduces metastasis of murine melanoma. PLoS ONE 2012, 7, e49988. [Google Scholar] [CrossRef] [Green Version]
- Stefansson, B.; Brautigan, D.L. Protein phosphatase PP6 N terminal domain restricts G1 to S phase progression in human cancer cells. Cell Cycle 2007, 6, 1386–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sand, M.; Sand, D.; Altmeyer, P.; Bechara, F.G. MicroRNA in non-melanoma skin cancer. Cancer Biomark. 2012, 11, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Heffelfinger, C.; Ouyang, Z.; Engberg, A.; Leffell, D.J.; Hanlon, A.M.; Gordon, P.B.; Zheng, W.; Zhao, H.; Snyder, M.P.; Bale, A.E. Correlation of Global MicroRNA Expression with Basal Cell Carcinoma Subtype. G3 (Bethesda) 2012, 2, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Saydam, O.; Shen, Y.; Würdinger, T.; Senol, O.; Boke, E.; James, M.F.; Tannous, B.A.; Stemmer-Rachamimov, A.O.; Yi, M.; Stephens, R.M.; et al. Downregulated microRNA-200a in meningiomas promotes tumor growth by reducing E-cadherin and activating the Wnt/beta-catenin signaling pathway. Mol. Cell. Biol. 2009, 29, 5923–5940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonkoly, E.; Lovén, J.; Xu, N.; Meisgen, F.; Wei, T.; Brodin, P.; Jaks, V.; Kasper, M.; Shimokawa, T.; Harada, M.; et al. MicroRNA-203 functions as a tumor suppressor in basal cell carcinoma. Oncogenesis 2012, 1, e3. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Jiang, P. MicroRNA-451a acts as tumor suppressor in cutaneous basal cell carcinoma. Mol. Genet. Genom. Med. 2018, 6, 1001–1009. [Google Scholar] [CrossRef]
- Sand, M.; Skrygan, M.; Sand, D.; Georgas, D.; Hahn, S.A.; Gambichler, T.; Altmeyer, P.; Bechara, F.G. Expression of microRNAs in basal cell carcinoma. Br. J. Dermatol. 2012, 167, 847–855. [Google Scholar] [CrossRef]
- Sand, M.; Bechara, F.G.; Sand, D.; Gambichler, T.; Hahn, S.A.; Bromba, M.; Stockfleth, E.; Hessam, S. Long-noncoding RNAs in basal cell carcinoma. Tumour Biol. 2016, 37, 10595–10608. [Google Scholar] [CrossRef]
- Youssef, K.K.; van Keymeulen, A.; Lapouge, G.; Beck, B.; Michaux, C.; Achouri, Y.; Sotiropoulou, P.A.; Blanpain, C. Identification of the cell lineage at the origin of basal cell carcinoma. Nat. Cell Biol. 2010, 12, 299–305. [Google Scholar] [CrossRef]
- Seykora, J.T.; Cotsarelis, G. Keratin 15-positive stem cells give rise to basal cell carcinomas in irradiated Ptch1(+/-) mice. Cancer Cell 2011, 19, 5–6. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.Y.; Wang, J.; Mancianti, M.-L.; Epstein, E.H. Basal cell carcinomas arise from hair follicle stem cells in Ptch1(+/-) mice. Cancer Cell 2011, 19, 114–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grachtchouk, M.; Pero, J.; Yang, S.H.; Ermilov, A.N.; Michael, L.E.; Wang, A.; Wilbert, D.; Patel, R.M.; Ferris, J.; Diener, J.; et al. Basal cell carcinomas in mice arise from hair follicle stem cells and multiple epithelial progenitor populations. J. Clin. Investig. 2011, 121, 1768–1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Danés, A.; Hannezo, E.; Larsimont, J.-C.; Liagre, M.; Youssef, K.K.; Simons, B.D.; Blanpain, C. Defining the clonal dynamics leading to mouse skin tumour initiation. Nature 2016, 536, 298–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, Y.-C.; Li, L.; Fuchs, E. Emerging interactions between skin stem cells and their niches. Nat. Med. 2014, 20, 847–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bath-Hextall, F.; Bong, J.; Perkins, W.; Williams, H. Interventions for basal cell carcinoma of the skin: Systematic review. BMJ 2004, 329, 705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology. In Guidelines, Basal Cell Skin Cancer; Version 1; 2020; Available online: https://jnccn.org/view/journals/jnccn/14/5/article-p574.xml (accessed on 6 May 2016).
- Wolf, D.J.; Zitelli, J.A. Surgical margins for basal cell carcinoma. Arch. Dermatol. 1987, 123, 340–344. [Google Scholar] [CrossRef]
- Codazzi, D.; van der Velden, J.; Carminati, M.; Bruschi, S.; Bocchiotti, M.A.; Di Serio, C.; Barberis, M.; Robotti, E. Positive compared with negative margins in a single-centre retrospective study on 3957 consecutive excisions of basal cell carcinomas. Associated risk factors and preferred surgical management. J. Plast. Surg. Hand Surg. 2014, 48, 38–43. [Google Scholar] [CrossRef]
- Rowe, D.E.; Carroll, R.J.; Day, C.L. Mohs surgery is the treatment of choice for recurrent (previously treated) basal cell carcinoma. J. Dermatol. Surg. Oncol. 1989, 15, 424–431. [Google Scholar] [CrossRef]
- van Loo, E.; Mosterd, K.; Krekels, G.A.M.; Roozeboom, M.H.; Ostertag, J.U.; Dirksen, C.D.; Steijlen, P.M.; Neumann, H.A.M.; Nelemans, P.J.; Kelleners-Smeets, N.W.J. Surgical excision versus Mohs’ micrographic surgery for basal cell carcinoma of the face: A randomised clinical trial with 10 year follow-up. Eur. J. Cancer (Oxf. Engl.: 1990) 2014, 50, 3011–3020. [Google Scholar] [CrossRef]
- Walker, P.; Hill, D. Surgical treatment of basal cell carcinomas using standard postoperative histological assessment. Australas. J. Dermatol. 2006, 47, 1–12. [Google Scholar] [CrossRef]
- Kuflik, E.G. Cryosurgery for skin cancer: 30-year experience and cure rates. Dermatol. Surg. Off. Publ. Am. Soc. Dermatol. Surg. 2004, 30, 297–300. [Google Scholar] [CrossRef]
- Wang, I.; Bendsoe, N.; Klinteberg, C.A.; Enejder, A.M.; Andersson-Engels, S.; Svanberg, S.; Svanberg, K. Photodynamic therapy vs. cryosurgery of basal cell carcinomas: Results of a phase III clinical trial. Br. J. Dermatol. 2001, 144, 832–840. [Google Scholar] [CrossRef]
- Savoia, P.; Deboli, T.; Previgliano, A.; Broganelli, P. Usefulness of Photodynamic Therapy as a Possible Therapeutic Alternative in the Treatment of Basal Cell Carcinoma. Int. J. Mol. Sci. 2015, 16, 23300–23317. [Google Scholar] [CrossRef] [Green Version]
- Roozeboom, M.H.; Aardoom, M.A.; Nelemans, P.J.; Thissen, M.R.T.M.; Kelleners-Smeets, N.W.J.; Kuijpers, D.I.M.; Mosterd, K. Fractionated 5-aminolevulinic acid photodynamic therapy after partial debulking versus surgical excision for nodular basal cell carcinoma: A randomized controlled trial with at least 5-year follow-up. J. Am. Acad. Dermatol. 2013, 69, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Fantini, F.; Greco, A.; Del Giovane, C.; Cesinaro, A.M.; Venturini, M.; Zane, C.; Surrenti, T.; Peris, K.; Calzavara-Pinton, P.G. Photodynamic therapy for basal cell carcinoma: Clinical and pathological determinants of response. J. Eur. Acad. Dermatol. Venereol. 2011, 25, 896–901. [Google Scholar] [CrossRef] [PubMed]
- Horn, M.; Wolf, P.; Wulf, H.C.; Warloe, T.; Fritsch, C.; Rhodes, L.E.; Kaufmann, R.; de Rie, M.; Legat, F.J.; Stender, I.M.; et al. Topical methyl aminolaevulinate photodynamic therapy in patients with basal cell carcinoma prone to complications and poor cosmetic outcome with conventional treatment. Br. J. Dermatol. 2003, 149, 1242–1249. [Google Scholar] [CrossRef]
- Cameron, M.C.; Lee, E.; Hibler, B.P.; Giordano, C.N.; Barker, C.A.; Mori, S.; Cordova, M.; Nehal, K.S.; Rossi, A.M. Basal cell carcinoma: Contemporary approaches to diagnosis, treatment, and prevention. J. Am. Acad. Dermatol. 2019, 80, 321–339. [Google Scholar] [CrossRef]
- Neville, J.A.; Welch, E.; Leffell, D.J. Management of nonmelanoma skin cancer in 2007. Nat. Clin. Pract. Oncol. 2007, 4, 462–469. [Google Scholar] [CrossRef]
- Hall, V.L.; Leppard, B.J.; McGill, J.; Kesseler, M.E.; White, J.E.; Goodwin, P. Treatment of basal-cell carcinoma: Comparison of radiotherapy and cryotherapy. Clin. Radiol. 1986, 37, 33–34. [Google Scholar] [CrossRef]
- Avril, M.F.; Auperin, A.; Margulis, A.; Gerbaulet, A.; Duvillard, P.; Benhamou, E.; Guillaume, J.C.; Chalon, R.; Petit, J.Y.; Sancho-Garnier, H.; et al. Basal cell carcinoma of the face: Surgery or radiotherapy? Results of a randomized study. Br. J. Cancer 1997, 76, 100–106. [Google Scholar] [CrossRef] [Green Version]
- Schulze, H.J.; Cribier, B.; Requena, L.; Reifenberger, J.; Ferrándiz, C.; Garcia Diez, A.; Tebbs, V.; McRae, S. Imiquimod 5% cream for the treatment of superficial basal cell carcinoma: Results from a randomized vehicle-controlled phase III study in Europe. Br. J. Dermatol. 2005, 152, 939–947. [Google Scholar] [CrossRef] [PubMed]
- Geisse, J.; Caro, I.; Lindholm, J.; Golitz, L.; Stampone, P.; Owens, M. Imiquimod 5% cream for the treatment of superficial basal cell carcinoma: Results from two phase III, randomized, vehicle-controlled studies. J. Am. Acad. Dermatol. 2004, 50, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Geisse, J.K.; Rich, P.; Pandya, A.; Gross, K.; Andres, K.; Ginkel, A.; Owens, M. Imiquimod 5% cream for the treatment of superficial basal cell carcinoma: A double-blind, randomized, vehicle-controlled study. J. Am. Acad. Dermatol. 2002, 47, 390–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micali, G.; Lacarrubba, F.; Nasca, M.R.; Ferraro, S.; Schwartz, R.A. Topical pharmacotherapy for skin cancer: Part II. Clinical applications. J. Am. Acad. Dermatol. 2014, 70, 979.e1–979.e12. [Google Scholar] [CrossRef] [PubMed]
- Paolino, G.; Didona, D.; Scarnò, M.; Tallarico, M.; Cantoresi, F.; Calvieri, S.; Mercuri, S.R.; Piccolo, D.; Bottoni, U.; Kyriakou, A.; et al. Sequential treatment of daylight photodynamic therapy and imiquimod 5% cream for the treatment of superficial basal cell carcinoma on sun exposed areas. Dermatol. Ther. 2019, 32, e12788. [Google Scholar] [CrossRef]
- Quirk, C.; Gebauer, K.; De’Ambrosis, B.; Slade, H.B.; Meng, T.-C. Sustained clearance of superficial basal cell carcinomas treated with imiquimod cream 5%: Results of a prospective 5-year study. Cutis 2010, 85, 318–324. [Google Scholar]
- Gollnick, H.; Barona, C.G.; Frank, R.G.; Ruzicka, T.; Megahed, M.; Maus, J.; Munzel, U. Recurrence rate of superficial basal cell carcinoma following treatment with imiquimod 5% cream: Conclusion of a 5-year long-term follow-up study in Europe. Eur. J. Dermatol. 2008, 18, 677–682. [Google Scholar] [CrossRef]
- Shumack, S.; Robinson, J.; Kossard, S.; Golitz, L.; Greenway, H.; Schroeter, A.; Andres, K.; Amies, M.; Owens, M. Efficacy of topical 5% imiquimod cream for the treatment of nodular basal cell carcinoma: Comparison of dosing regimens. Arch. Dermatol. 2002, 138, 1165–1171. [Google Scholar] [CrossRef]
- Micali, G.; de Pasquale, R.; Caltabiano, R.; Impallomeni, R.; Lacarrubba, F. Topical imiquimod treatment of superficial and nodular basal cell carcinomas in patients affected by basal cell nevus syndrome: A preliminary report. J. Dermatolog. Treat. 2002, 13, 123–127. [Google Scholar] [CrossRef]
- Micali, G.; Lacarrubba, F.; Nasca, M.R.; Pasquale, R. de. The use of imiquimod 5% cream for the treatment of basal cell carcinoma as observed in Gorlin’s syndrome. Clin. Exp. Dermatol. 2003, 28 (Suppl. 1), 19–23. [Google Scholar] [CrossRef]
- Arits, A.H.; Mosterd, K.; Essers, B.A.B.; Spoorenberg, E.; Sommer, A.; de Rooij, M.J.M.; van Pelt, H.P.A.; Quaedvlieg, P.J.F.; Krekels, G.A.M.; van Neer, P.A.; et al. Photodynamic therapy versus topical imiquimod versus topical fluorouracil for treatment of superficial basal-cell carcinoma: A single blind, non-inferiority, randomised controlled trial. Lancet Oncol. 2013, 14, 647–654. [Google Scholar] [CrossRef]
- Roozeboom, M.H.; Arits, A.H.M.M.; Mosterd, K.; Sommer, A.; Essers, B.A.B.; de Rooij, M.J.M.; Quaedvlieg, P.J.F.; Steijlen, P.M.; Nelemans, P.J.; Kelleners-Smeets, N.W.J. Three-Year Follow-Up Results of Photodynamic Therapy vs. Imiquimod vs. Fluorouracil for Treatment of Superficial Basal Cell Carcinoma: A Single-Blind, Noninferiority, Randomized Controlled Trial. J. Investig. Dermatol. 2016, 136, 1568–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Čeović, R.; Smolković, N.; Pašić, A.; Kostović, K.; Hrsan, D. Multiple basal cell carcinomas of lower legs with stasis dermatitis: A therapeutic challenge. Acta Dermatovenerol. Croat. 2012, 20, 191–196. [Google Scholar] [PubMed]
- Shelley, W.B.; Wood, M.G. Nodular superficial pigmented basal cell epitheliomas. Arch. Dermatol. 1982, 118, 928–930. [Google Scholar] [CrossRef] [PubMed]
- Love, W.E.; Bernhard, J.D.; Bordeaux, J.S. Topical imiquimod or fluorouracil therapy for basal and squamous cell carcinoma: A systematic review. Arch. Dermatol. 2009, 145, 1431–1438. [Google Scholar] [CrossRef]
- Good, L.M.; Miller, M.D.; High, W.A. Intralesional agents in the management of cutaneous malignancy: A review. J. Am. Acad. Dermatol. 2011, 64, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Lanoue, J.; Goldenberg, G. Basal Cell Carcinoma: A Comprehensive Review of Existing and Emerging Nonsurgical Therapies. J. Clin. Aesthet. Dermatol. 2016, 9, 26–36. [Google Scholar] [PubMed]
- Mercuri, S.R.; Brianti, P.; Paolino, G.; Rizzo, N.; Bartolucci, M.; Dattola, A.; Didona, D.; Nisticò, S.P. Basal cell carcinoma in post-traumatic scar successfully treated with thulium laser and photodynamic therapy. G. Ital. Dermatol. Venereol. 2020, 155, 107–108. [Google Scholar] [CrossRef]
- Campolmi, P.; Brazzini, B.; Urso, C.; Ghersetich, I.; Mavilia, L.; Hercogova, J.; Lotti, T. Superpulsed CO2 laser treatment of basal cell carcinoma with intraoperatory histopathologic and cytologic examination. Dermatol. Surg. Off. Publ. Am. Soc. Dermatol. Surg. 2002, 28, 909–911, discussion 912. [Google Scholar] [CrossRef]
- Moskalik, K.; Kozlov, A.; Demin, E.; Boiko, E. The efficacy of facial skin cancer treatment with high-energy pulsed neodymium and Nd:YAG lasers. Photomed. Laser Surg. 2009, 27, 345–349. [Google Scholar] [CrossRef]
- Meiss, F.; Andrlová, H.; Zeiser, R. Vismodegib. Recent Results Cancer Res. 2018, 211, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Zito, P.M.; Nassereddin, A.; Scharf, R. StatPearls. Vismodegib; Treasure Island: Petersburg, FL, USA, 2020. [Google Scholar]
- Gupta, A.K.; Mays, R.R.; Abramovits, W.; Vincent, K.D. Odomzo® (Sonidegib). Skinmed 2018, 16, 35–38. [Google Scholar] [PubMed]
- Casey, D.; Demko, S.; Shord, S.; Zhao, H.; Chen, H.; He, K.; Putman, A.; Helms, W.; Keegan, P.; Pazdur, R. FDA Approval Summary: Sonidegib for Locally Advanced Basal Cell Carcinoma. Clin. Cancer Res. 2017, 23, 2377–2381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahid, M.; Jawed, A.; Mandal, R.K.; Dar, S.A.; Khan, S.; Akhter, N.; Haque, S. Vismodegib, itraconazole and sonidegib as hedgehog pathway inhibitors and their relative competencies in the treatment of basal cell carcinomas. Crit. Rev. Oncol. Hematol. 2016, 98, 235–241. [Google Scholar] [CrossRef]
- Peer, E.; Tesanovic, S.; Aberger, F. Next-Generation Hedgehog/GLI Pathway Inhibitors for Cancer Therapy. Cancers (Basel) 2019, 11, 538. [Google Scholar] [CrossRef] [Green Version]
- Sekulic, A.; Migden, M.R.; Oro, A.E.; Dirix, L.; Lewis, K.D.; Hainsworth, J.D.; Solomon, J.A.; Yoo, S.; Arron, S.T.; Friedlander, P.A.; et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N. Engl. J. Med. 2012, 366, 2171–2179. [Google Scholar] [CrossRef] [Green Version]
- Sekulic, A.; Migden, M.R.; Basset-Seguin, N.; Garbe, C.; Gesierich, A.; Lao, C.D.; Miller, C.; Mortier, L.; Murrell, D.F.; Hamid, O.; et al. Long-term safety and efficacy of vismodegib in patients with advanced basal cell carcinoma: Final update of the pivotal ERIVANCE BCC study. BMC Cancer 2017, 17, 332. [Google Scholar] [CrossRef]
- Lear, J.T.; Migden, M.R.; Lewis, K.D.; Chang, A.L.S.; Guminski, A.; Gutzmer, R.; Dirix, L.; Combemale, P.; Stratigos, A.; Plummer, R.; et al. Long-term efficacy and safety of sonidegib in patients with locally advanced and metastatic basal cell carcinoma: 30-month analysis of the randomized phase 2 BOLT study. J. Eur. Acad. Dermatol. Venereol. 2018, 32, 372–381. [Google Scholar] [CrossRef]
- Basset-Séguin, N.; Hauschild, A.; Kunstfeld, R.; Grob, J.; Dréno, B.; Mortier, L.; Ascierto, P.A.; Licitra, L.; Dutriaux, C.; Thomas, L.; et al. Vismodegib in patients with advanced basal cell carcinoma: Primary analysis of STEVIE, an international, open-label trial. Eur. J. Cancer (Oxf. Engl.: 1990) 2017, 86, 334–348. [Google Scholar] [CrossRef]
- Migden, M.R.; Guminski, A.; Gutzmer, R.; Dirix, L.; Lewis, K.D.; Combemale, P.; Herd, R.M.; Kudchadkar, R.; Trefzer, U.; Gogov, S.; et al. Treatment with two different doses of sonidegib in patients with locally advanced or metastatic basal cell carcinoma (BOLT): A multicentre, randomised, double-blind phase 2 trial. Lancet Oncol. 2015, 16, 716–728. [Google Scholar] [CrossRef]
- Dummer, R.; Ascierto, P.A.; Basset-Seguin, N.; Dréno, B.; Garbe, C.; Gutzmer, R.; Hauschild, A.; Krattinger, R.; Lear, J.T.; Malvehy, J.; et al. Sonidegib and vismodegib in the treatment of patients with locally advanced basal cell carcinoma: A joint expert opinion. J. Eur. Acad. Dermatol. Venereol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Therasse, P.; Arbuck, S.G.; Eisenhauer, E.A.; Wanders, J.; Kaplan, R.S.; Rubinstein, L.; Verweij, J.; van Glabbeke, M.; van Oosterom, A.T.; Christian, M.C.; et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J. Natl. Cancer Inst. 2000, 92, 205–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Office of the Surgeon General (US). The Surgeon General’s Call to Action to Prevent Skin Cancer; Office of the Surgeon General: Washington, DC, USA, 2014.
- Rowan-Robinson, K. Sun Protection for Preventing Basal Cell and Squamous Cell Skin Cancers. Public Health Nurs. 2017, 34, 312–313. [Google Scholar] [CrossRef] [PubMed]
- Bichakjian, C.K.; Olencki, T.; Aasi, S.Z.; Alam, M.; Andersen, J.S.; Berg, D.; Bowen, G.M.; Cheney, R.T.; Daniels, G.A.; Glass, L.F.; et al. Basal Cell Skin Cancer, Version 1.2016, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc. Netw. 2016, 14, 574–597. [Google Scholar] [CrossRef] [PubMed]
- Damian, D.L. Photoprotective effects of nicotinamide. Photochem. Photobiol. Sci. 2010, 9, 578–585. [Google Scholar] [CrossRef]
- Karthikeyan, K.; Thappa, D.M. Pellagra and skin. Int. J. Dermatol. 2002, 41, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Petley, A.; Macklin, B.; Renwick, A.G.; Wilkin, T.J. The pharmacokinetics of nicotinamide in humans and rodents. Diabetes 1995, 44, 152–155. [Google Scholar] [CrossRef]
- Forbat, E.; Al-Niaimi, F.; Ali, F.R. Use of nicotinamide in dermatology. Clin. Exp. Dermatol. 2017, 42, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Gensler, H.L.; Williams, T.; Huang, A.C.; Jacobson, E.L. Oral niacin prevents photocarcinogenesis and photoimmunosuppression in mice. Nutr. Cancer 1999, 34, 36–41. [Google Scholar] [CrossRef]
- Gensler, H.L. Prevention of photoimmunosuppression and photocarcinogenesis by topical nicotinamide. Nutr. Cancer 1997, 29, 157–162. [Google Scholar] [CrossRef]
- Damian, D.L.; Patterson, C.R.S.; Stapelberg, M.; Park, J.; Barnetson, R.S.C.; Halliday, G.M. UV radiation-induced immunosuppression is greater in men and prevented by topical nicotinamide. J. Investig. Dermatol. 2008, 128, 447–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, A.C.; Martin, A.J.; Choy, B.; Fernández-Peñas, P.; Dalziell, R.A.; McKenzie, C.A.; Scolyer, R.A.; Dhillon, H.M.; Vardy, J.L.; Kricker, A.; et al. A Phase 3 Randomized Trial of Nicotinamide for Skin-Cancer Chemoprevention. N. Engl. J. Med. 2015, 373, 1618–1626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surjana, D.; Halliday, G.M.; Martin, A.J.; Moloney, F.J.; Damian, D.L. Oral nicotinamide reduces actinic keratoses in phase II double-blinded randomized controlled trials. J. Investig. Dermatol. 2012, 132, 1497–1500. [Google Scholar] [CrossRef] [Green Version]
- Drago, F.; Ciccarese, G.; Cogorno, L.; Calvi, C.; Marsano, L.A.; Parodi, A. Prevention of non-melanoma skin cancers with nicotinamide in transplant recipients: A case-control study. Eur. J. Dermatol. 2017, 27, 382–385. [Google Scholar] [CrossRef]
- Chen, A.C.; Martin, A.J.; Dalziell, R.A.; McKenzie, C.A.; Lowe, P.M.; Eris, J.M.; Scolyer, R.A.; Dhillon, H.M.; Vardy, J.L.; Bielski, V.A.; et al. A phase II randomized controlled trial of nicotinamide for skin cancer chemoprevention in renal transplant recipients. Br. J. Dermatol. 2016, 175, 1073–1075. [Google Scholar] [CrossRef] [PubMed]
- Lang, B.M.; Balermpas, P.; Bauer, A.; Blum, A.; Brölsch, G.F.; Dirschka, T.; Follmann, M.; Frank, J.; Frerich, B.; Fritz, K.; et al. S2k Guidelines for Cutaneous Basal Cell Carcinoma—Part 2: Treatment, Prevention and Follow-up. J. Dtsch. Dermatol. Ges. 2019, 17, 214–230. [Google Scholar] [CrossRef]
- Elmets, C.A.; Viner, J.L.; Pentland, A.P.; Cantrell, W.; Lin, H.-Y.; Bailey, H.; Kang, S.; Linden, K.G.; Heffernan, M.; Duvic, M.; et al. Chemoprevention of nonmelanoma skin cancer with celecoxib: A randomized, double-blind, placebo-controlled trial. J. Natl. Cancer Inst. 2010, 102, 1835–1844. [Google Scholar] [CrossRef]
- Muranushi, C.; Olsen, C.M.; Green, A.C.; Pandeya, N. Can oral nonsteroidal antiinflammatory drugs play a role in the prevention of basal cell carcinoma? A systematic review and metaanalysis. J. Am. Acad. Dermatol. 2016, 74, 108–119.e1. [Google Scholar] [CrossRef]
- Soltani-Arabshahi, R.; Tristani-Firouzi, P. Chemoprevention of nonmelanoma skin cancer. Facial Plast. Surg. 2013, 29, 373–383. [Google Scholar] [CrossRef]
- Wolfe, C.M.; Green, W.H.; Cognetta, A.B.; Hatfield, H.K. A possible chemopreventive role for photodynamic therapy in Gorlin syndrome: A report of basal cell carcinoma reduction and review of literature. Australas. J. Dermatol. 2013, 54, 64–68. [Google Scholar] [CrossRef]
- Kim, J.Y.S.; Kozlow, J.H.; Mittal, B.; Moyer, J.; Olencki, T.; Rodgers, P. Guidelines of care for the management of basal cell carcinoma. J. Am. Acad. Dermatol. 2018, 78, 540–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
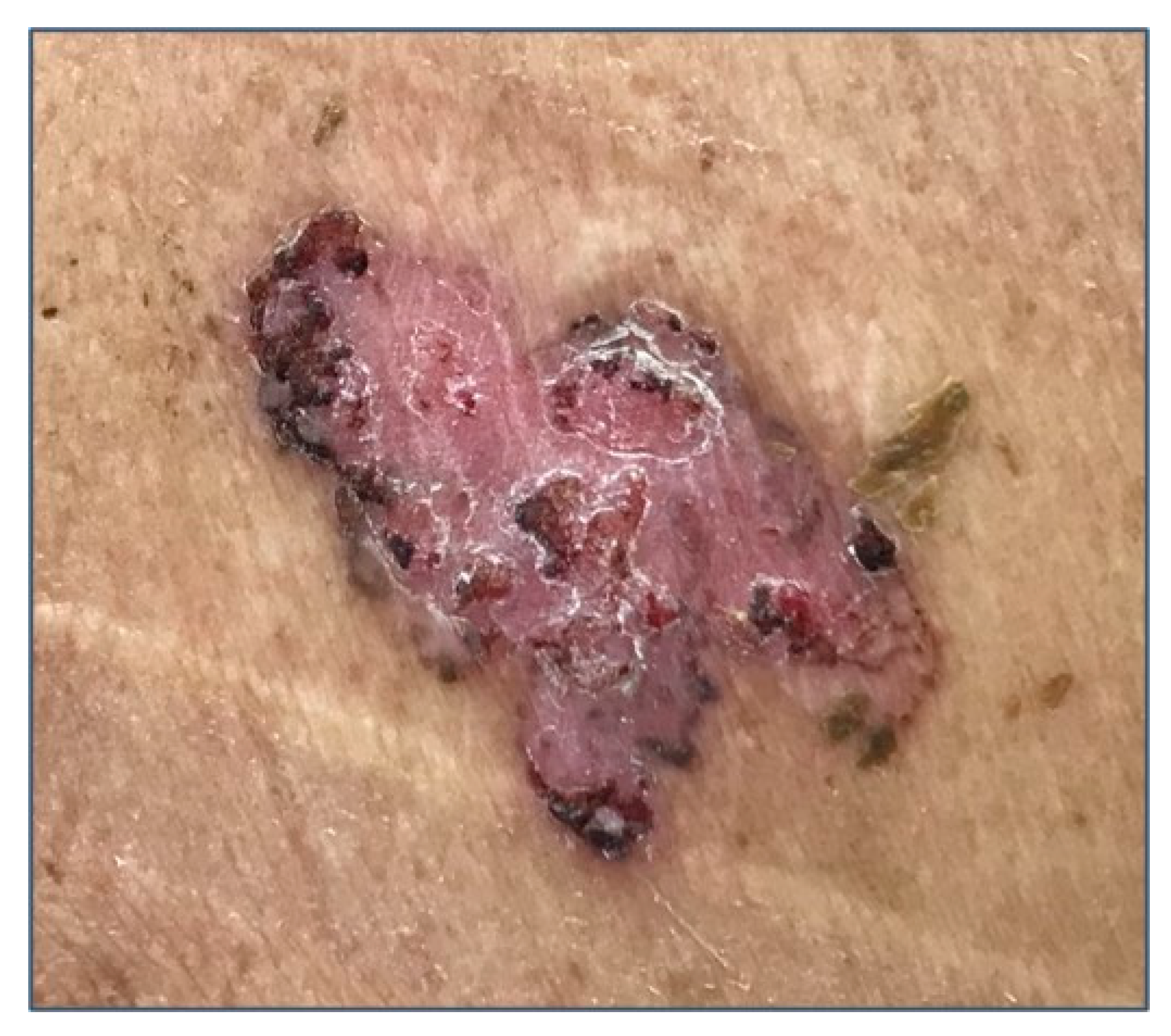
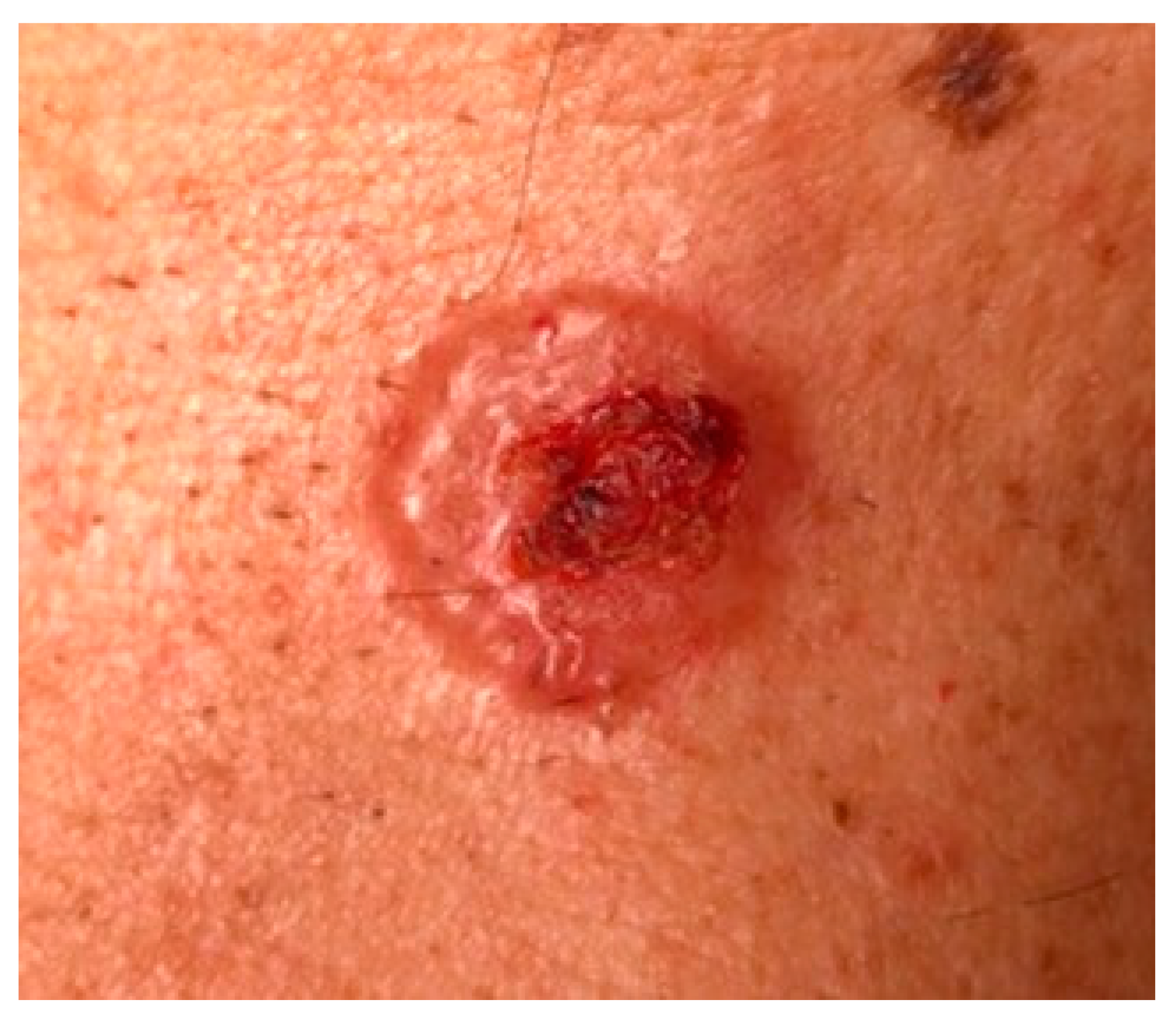
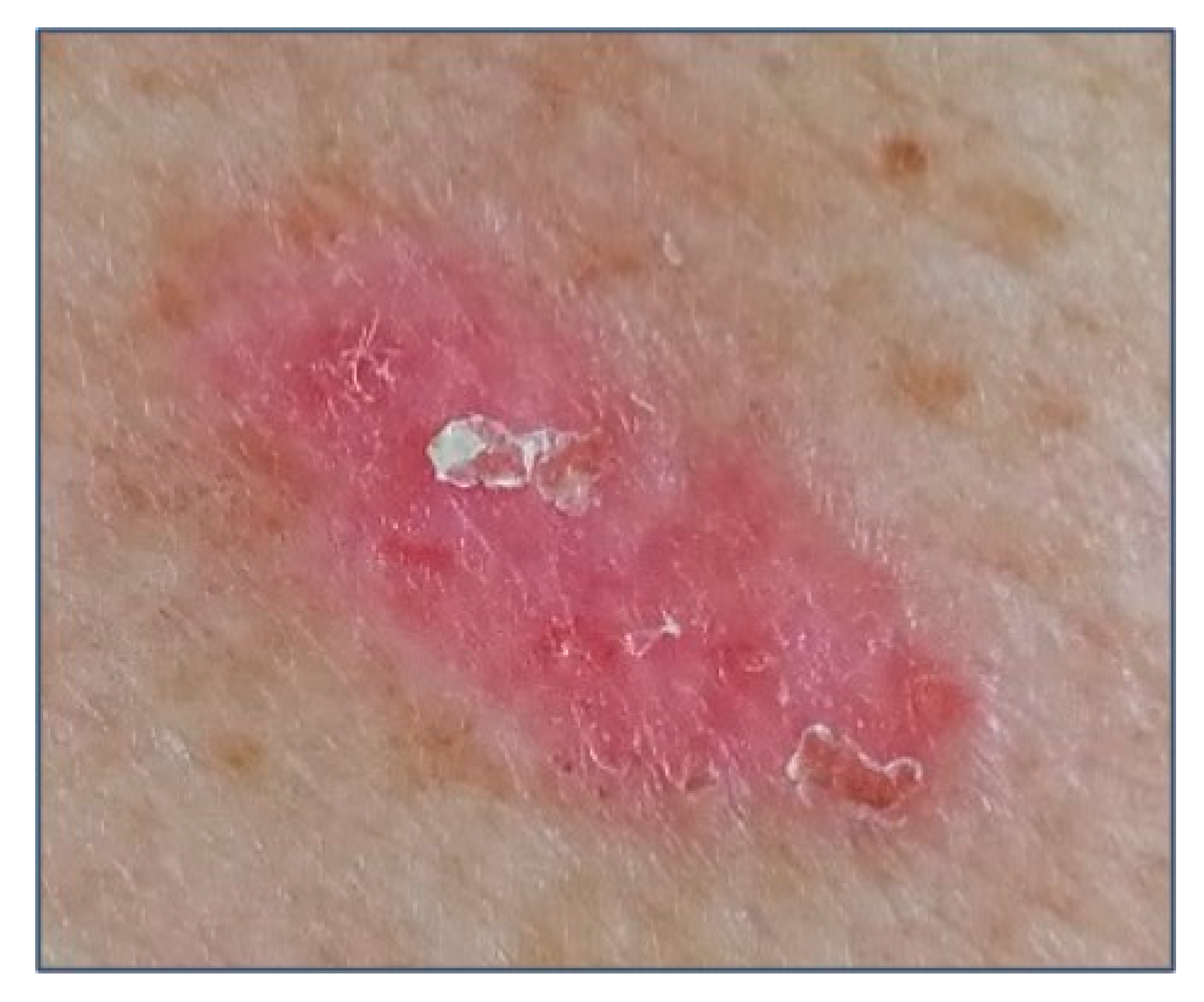
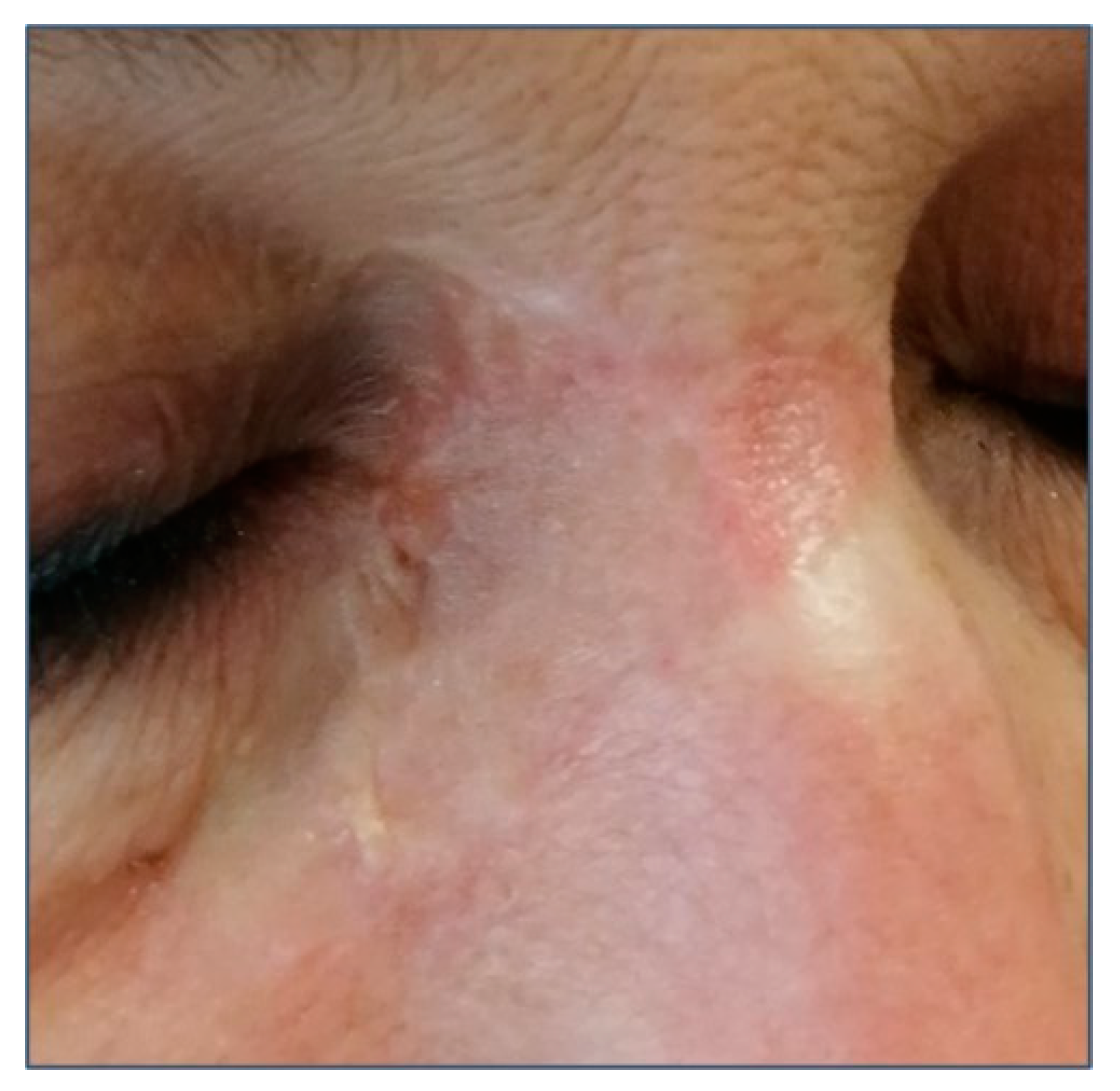


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Low Risk BCC | Intermediate Risk BCC | High Risk BCC | |
|---|---|---|---|
| Superficial primary BCC | Superficial recurrent BCC | Morpheaform or poor-defined | |
| Nodular primary BCC when: <1 cm in intermediate risk area <2 cm in low risk area | Nodular primary BCC when: <1 cm in high risk area >1 cm in intermediate risk area >2 cm in low risk area | Nodular primary BCC when: >1 cm in high risk area | |
| Pinkus tumor | Histological forms: aggressive | Recurrent forms (apart from superficial BCC) | |
| Vascular Structures | Structures Related to Pigment | Nonvascular/Nonpigmented Structures |
|---|---|---|
| Arborizing vessels | Maple leaf-like areas | Ulcerations |
| Short fine telangiectasias | Spoke-wheel areas | Multiple small erosions |
| Blue–grey nests and globules | Shiny white–red structureless areas | |
| In-focus dots | White streaks | |
| Concentric structures |
| Sonidegib 200 mg Daily Central Review RECIST-Like 18-Month Follow-up | Vismodegib 150 mg Daily Central Review RECIST 21-Month Follow-up | |
|---|---|---|
| Overall response rate n (%); 95% CI | 40 (60.6); 47.8–72.4 | 30 (47.6); 35.5–60.6 |
| Complete response n (%) | 14 (21.2%) | 14 (22.2%) |
| Partial response n (%) | 26 (39.4%) | 16 (25.4%) |
| Stable disease n (%) | 20 (30.3%) | 22 (34.9%) |
| Progressive disease n (%) | 1 (1.5%) | 8 (12.7%) |
| Unknown n (%) | 5 (7.6%) | 3 (4.8%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fania, L.; Didona, D.; Morese, R.; Campana, I.; Coco, V.; Di Pietro, F.R.; Ricci, F.; Pallotta, S.; Candi, E.; Abeni, D.; et al. Basal Cell Carcinoma: From Pathophysiology to Novel Therapeutic Approaches. Biomedicines 2020, 8, 449. https://doi.org/10.3390/biomedicines8110449
Fania L, Didona D, Morese R, Campana I, Coco V, Di Pietro FR, Ricci F, Pallotta S, Candi E, Abeni D, et al. Basal Cell Carcinoma: From Pathophysiology to Novel Therapeutic Approaches. Biomedicines. 2020; 8(11):449. https://doi.org/10.3390/biomedicines8110449
Chicago/Turabian StyleFania, Luca, Dario Didona, Roberto Morese, Irene Campana, Valeria Coco, Francesca Romana Di Pietro, Francesca Ricci, Sabatino Pallotta, Eleonora Candi, Damiano Abeni, and et al. 2020. "Basal Cell Carcinoma: From Pathophysiology to Novel Therapeutic Approaches" Biomedicines 8, no. 11: 449. https://doi.org/10.3390/biomedicines8110449
APA StyleFania, L., Didona, D., Morese, R., Campana, I., Coco, V., Di Pietro, F. R., Ricci, F., Pallotta, S., Candi, E., Abeni, D., & Dellambra, E. (2020). Basal Cell Carcinoma: From Pathophysiology to Novel Therapeutic Approaches. Biomedicines, 8(11), 449. https://doi.org/10.3390/biomedicines8110449