The Links between Parkinson’s Disease and Cancer

 , , , , , and
, , , , , and 
Abstract
1. Introduction
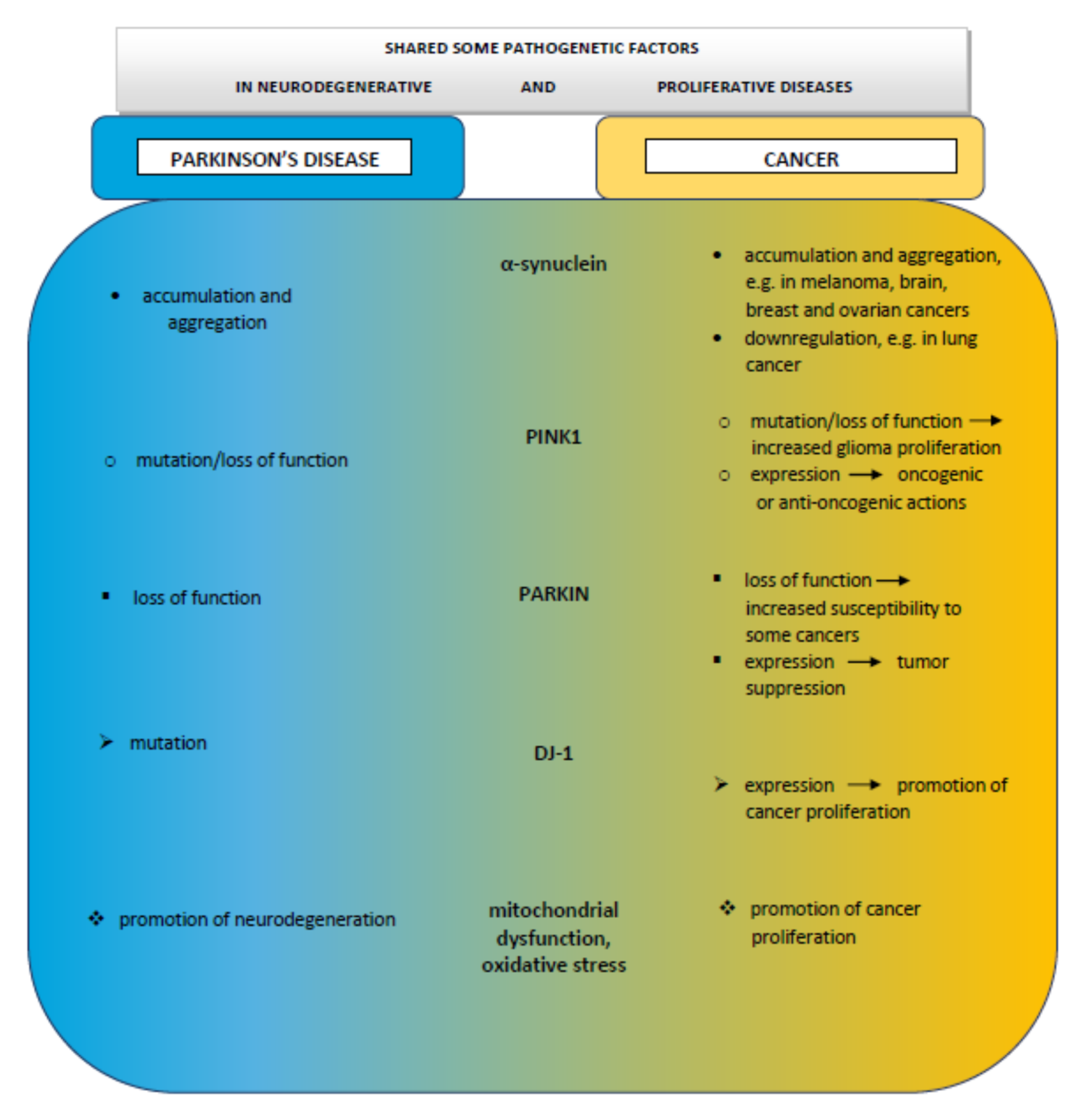
2. Epidemiologic Links between Parkinson’s Disease and Cancer
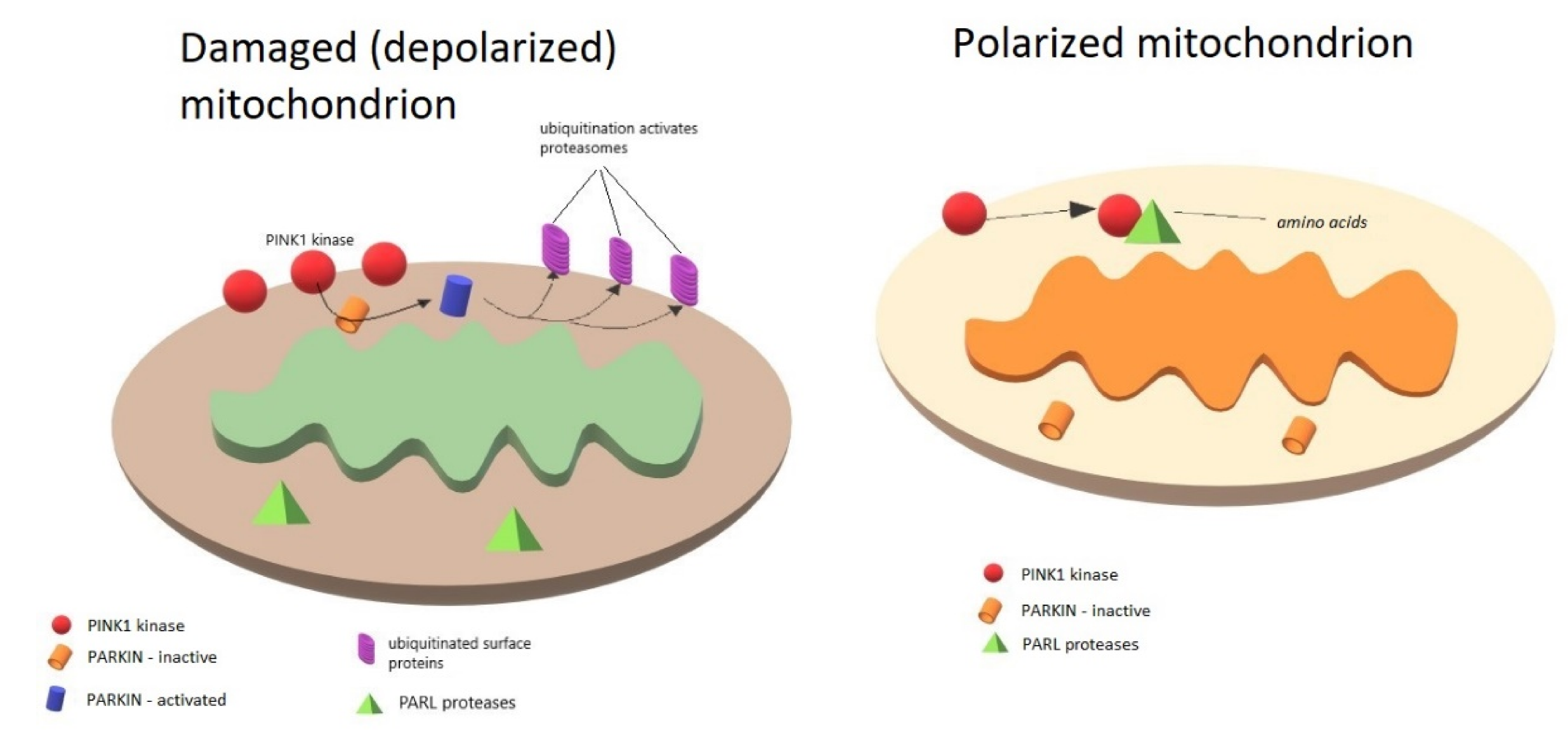
3. Role of PINK1, PARKIN, and DJ-1 in Mitochondria
4. PINK1, Parkinson’s Disease, and Cancer
5. PARKIN, Parkinson’s Disease, and Cancer
6. DJ-1, Parkinson’s Disease, and Cancer
7. Cancer and α-Synuclein
8. Mitochondrial Impairment in Parkinson’s Disease and Cancer
9. Oxidative Stress, Parkinson’s Disease, and Cancer
9.1. Oxidative Stress in Parkinson’s Disease
9.2. Oxidative Stress in Cancer
10. Comments
Author Contributions
Funding
Conflicts of Interest
References
- Parkinson, J. An essay on the shaking palsy. 1817. J. Neuropsychiatry Clin. Neurosci. 2002, 14, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Van der Vlag, M.; Havekes, R.; Heckman, P.R.A. The contribution of Parkin, PINK1 and DJ-1 genes to selective neuronal degeneration in Parkinson’s disease. Eur. J. Neurosci. 2020, 52, 3256–3268. [Google Scholar] [CrossRef] [PubMed]
- Post, M.R.; Lieberman, O.J.; Mosharov, E.V. Can Interactions Between α-Synuclein, Dopamine and Calcium Explain Selective Neurodegeneration in Parkinson’s Disease? Front. Neurosci. 2018, 12, 161. [Google Scholar] [CrossRef] [PubMed]
- Airavaara, M.; Parkkinen, I.; Konovalova, J.; Albert, K.; Chmielarz, P.; Domanskyi, A. Back and to the Future: From Neurotoxin-Induced to Human Parkinson’s Disease Models. Curr. Protoc. Neurosci. 2020, 91, e88. [Google Scholar] [CrossRef]
- Barazzuol, L.; Giamogante, F.; Brini, M.; Calì, T. PINK1/Parkin Mediated Mitophagy, Ca2+ Signalling, and ER-Mitochondria Contacts in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 1772. [Google Scholar] [CrossRef]
- Tabarés-Seisdedos, R.; Rubenstein, J.L. Inverse cancer comorbidity: A serendipitous opportunity to gain insight into CNS disorders. Nat. Rev. Neurosci. 2013, 14, 293–304. [Google Scholar] [CrossRef]
- Tansey, M.G.; Goldberg, M.S. Neuroinflammation in Parkinson’s disease: Its role in neuronal death and implications for therapeutic intervention. Neurobiol. Dis. 2010, 37, 510–518. [Google Scholar] [CrossRef]
- Kawashima, M.; Suzuki, S.O.; Doh-ura, K.; Iwaki, T. Alpha-Synuclein is expressed in a variety of brain tumors showing neuronal differentiation. Acta Neuropathol. 2000, 99, 154–160. [Google Scholar] [CrossRef]
- Dube, U.; Ibanez, L.; Budde, J.P.; Benitez, B.A.; Davis, A.A.; Harari, O.; Iles, M.M.; Law, M.H.; Brown, K.M.; 23andMe Research Team; et al. Overlapping genetic architecture between Parkinson disease and melanoma. Acta Neuropathol. 2020, 139, 347–364. [Google Scholar] [CrossRef]
- Agalliu, I.; Ortega, R.A.; Luciano, M.S.; Mirelman, A.; Pont-Sunyer, C.; Brockmann, K.; Vilas, D.; Tolosa, E.; Berg, D.; Warø, B.; et al. Cancer outcomes among Parkinson’s disease patients with leucine rich repeat kinase 2 mutations, idiopathic Parkinson’s disease patients, and nonaffected controls. Mov. Disord. 2019, 34, 1392–1398. [Google Scholar] [CrossRef]
- Xie, X.; Luo, X.; Xie, M. Association between Parkinson’s disease and risk of colorectal cancer. Parkinsonism Relat. Disord. 2017, 35, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Disse, M.; Reich, H.; Lee, P.K.; Schram, S.S. A review of the association between Parkinson disease and malignant melanoma. Dermatol. Surg. 2016, 42, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.D.; Cai, W.; Chen, X. The associations between Parkinson’s disease and cancer: The plot thickens. Transl. Neurodegener. 2015, 4, 20. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.C.; Xu, L.; Chen, Y.; Yan, H.; Hazawa, M.; Doan, N.; Said, J.W.; Ding, L.-W.; Liu, L.-Z.; Yang, H.; et al. Genomic and functional analysis of the E3 Ligase PARK2 in glioma. Cancer Res. 2015, 75, 1815–1827. [Google Scholar] [CrossRef] [PubMed]
- Devine, M.J.; Plun-Favreau, H.; Wood, N.W. Parkinson’s disease and cancer: Two wars, one front. Nat. Rev. Cancer 2011, 11, 812–823. [Google Scholar] [CrossRef]
- Bajaj, A.; Driver, J.A.; Schernhammer, E.S. Parkinson’s disease and cancer risk: A systematic review and meta-analysis. Cancer Causes Control 2010, 21, 697–707. [Google Scholar] [CrossRef]
- Fois, A.F.; Wotton, C.J.; Yeates, D.; Turner, M.R.; Goldacre, M.J. Cancer in patients with motor neuron disease, multiple sclerosis and Parkinson’s disease: Record linkage studies. J. Neurol. Neurosurg. Psychiatry 2010, 81, 215–221. [Google Scholar] [CrossRef]
- Ferreira, J.J.; Neutel, D.; Mestre, T.; Coelho, M.; Rosa, M.M.; Rascol, O.; Sampaio, C. Skin cancer and Parkinson’s disease. Mov. Disord. 2010, 25, 139–148. [Google Scholar] [CrossRef]
- Ong, E.L.; Goldacre, R.; Goldacre, M. Differential risks of cancer types in people with Parkinson’s disease: A national record-linkage study. Eur. J. Cancer 2014, 50, 2456–2462. [Google Scholar] [CrossRef]
- Peretz, C.; Gurel, R.; Rozani, V.; Gurevich, T.; El-Ad, B.; Tsamir, J.; Giladi, N. Cancer incidence among Parkinson‘s disease patients in a 10-yrs time-window around disease onset: A large-scale cohort study. Parkinsonism Relat. Disord. 2016, 28, 68–72. [Google Scholar] [CrossRef]
- Ajdacic-Gross, V.; Rodgers, S.; Aleksandrowicz, A.; Mutsch, M.; Steinemann, N.; von Wyl, V.; von Känel, R.; Bopp, M. Cancer co-occurrence patterns in Parkinson‘s disease and multiple sclerosis-Do they mirror immune system imbalances? Cancer Epidemiol. 2016, 44, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Wirdefeldt, K.; Weibull, C.E.; Chen, H.; Kamel, F.; Lundholm, C.; Fang, F.; Ye, W. Parkinson‘s disease and cancer: A register-based family study. Am. J. Epidemiol. 2014, 179, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Catalá-López, F.; Suárez-Pinilla, M.; Suárez-Pinilla, P.; Valderas, J.M.; Gómez-Beneyto, M.; Martinez, S.; Balanzá-Martínez, V.; Climent, J.; Valencia, A.; McGrath, J.; et al. Inverse and direct cancer comorbidity in people with central nervous system disorders: A meta-analysis of cancer incidence in 577,013 participants of 50 observational studies. Psychother. Psychosom. 2014, 83, 89–105. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.H.; Friis, S.; Frederiksen, K.; McLaughlin, J.K.; Mellemkjaer, L.; Møller, H. Atypical cancer pattern in patients with Parkinson’s disease. Br. J. Cancer 2005, 92, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.Y.; Chang, S.N.; Hsiao, T.H.; Huang, B.T.; Lin, C.H.; Yang, P.C. Association between Parkinson disease and risk of cancer in taiwan. Jama Oncol. 2015, 1, 633–640. [Google Scholar] [CrossRef]
- Wang, T. The link between Parkinson‘s disease and breast and prostate cancers: A meta-analysis. Int. J. Neurosci. 2015, 125, 895–903. [Google Scholar] [CrossRef]
- Ye, R.; Shen, T.; Jiang, Y.; Xu, L.; Si, X.; Zhang, B. The Relationship between Parkinson Disease and Brain Tumor: A Meta-Analysis. PLoS ONE 2016, 11, e0164388. [Google Scholar] [CrossRef]
- Olsen, J.H.; Friis, S.; Frederiksen, K. Malignant melanoma and other types of cancer preceding Parkinson disease. Epidemiology 2006, 17, 582–587. [Google Scholar] [CrossRef]
- D’Amelio, M.; Ragonese, P.; Morgante, L.; Epifanio, A.; Callari, G.; Salemi, G.; Savettieri, G. Tumor diagnosis preceding Parkinson’s disease: A case-control study. Mov. Disord. 2004, 19, 807–811. [Google Scholar] [CrossRef]
- Elbaz, A.; Peterson, B.J.; Yang, P.; Van Gerpen, J.A.; Bower, J.H.; Maraganore, D.M.; McDonnell, S.K.; Ahlskog, J.E.; Rocca, W.A. Nonfatal cancer preceding Parkinson’s disease: A case-control study. Epidemiology 2002, 13, 157–164. [Google Scholar] [CrossRef]
- Driver, J.A.; Logroscino, G.; Buring, J.E.; Gaziano, J.M.; Kurth, T. A prospective cohort study of cancer incidence following the diagnosis of Parkinson‘s disease. Cancer Epidemiol. Biomark. Prev. 2007, 16, 1260–1265. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.M.; Liang, J.A.; Chang, S.N.; Sung, F.C.; Muo, C.H.; Kao, C.H. Analysis of Parkinson‘s disease and subsequent cancer risk in Taiwan: A nationwide population-based cohort study. Neuroepidemiology 2011, 37, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Bougea, A.; Spantideas, N.; Katoulis, A.; Stefanis, L. Levodopa-induced skin disorders in patients with Parkinson disease: A systematic literature review approach. Acta Neurol. Belg. 2019, 119, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Inzelberg, R.; Flash, S.; Friedman, E.; Azizi, E. Cutaneous malignant melanoma and Parkinson disease: Common pathways? Ann. Neurol. 2016, 80, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Zanetti, R.; Loria, D.; Rosso, S. Melanoma, Parkinson’s disease and levodopa: Causal or spurious link? A review of the literature. Melanoma Res. 2006, 16, 201–206. [Google Scholar] [CrossRef]
- Liu, R.; Gao, X.; Lu, Y.; Chen, H. Meta-analysis of the relationship between Parkinson disease and melanoma. Neurology 2011, 76, 2002–2009. [Google Scholar] [CrossRef]
- Dalvin, L.A.; Damento, G.M.; Yawn, B.P.; Abbott, B.A.; Hodge, D.O.; Pulido, J.S. Parkinson disease and melanoma: Confirming and reexamining an association. Mayo Clin. Proc. 2017, 92, 1070–1079. [Google Scholar] [CrossRef]
- Baade, P.D.; Fritschi, L.; Freedman, D.M. Mortality due to amyotrophic lateral sclerosis and Parkinson’s disease among melanoma patients. Neuroepidemiology 2007, 28, 16–20. [Google Scholar] [CrossRef]
- Kareus, S.A.; Figueroa, K.P.; Cannon-Albright, L.A.; Pulst, S.M. Shared predispositions of parkinsonism and cancer: A population-based pedigree-linked study. Arch. Neurol. 2012, 69, 1572–1577. [Google Scholar] [CrossRef]
- Freedman, D.M.; Sigurdson, A.; Doody, M.M.; Rao, R.; Linet, M.S. Risk of melanoma in relation to smoking, alcohol intake, and other factors in a large occupational cohort. Cancer Causes Control 2003, 14, 847–857. [Google Scholar] [CrossRef]
- DeLancey, J.O.; Hannan, L.M.; Gapstur, S.M.; Thun, M.J. Cigarette smoking and the risk of incident and fatal melanoma in a large prospective cohort study. Cancer Causes Control 2011, 22, 937–942. [Google Scholar] [CrossRef] [PubMed]
- Kenborg, L.; Lassen, C.F.; Ritz, B.; Andersen, K.K.; Christensen, J.; Schernhammer, E.S.; Hansen, J.; Wermuth, L.; Rod, N.H.; Olsen, J.H. Lifestyle, family history, and risk of idiopathic Parkinson disease: A large Danish case–control study. Am. J. Epidemiol. 2015, 181, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Song, F.; Qureshi, A.A.; Gao, X.; Li, T.; Han, J. Smoking and risk of skin cancer: A prospective analysis and a meta-analysis. Int. J. Epidemiol. 2012, 41, 1694–1705. [Google Scholar] [CrossRef] [PubMed]
- Van der Mark, M.; Nijssen, P.C.G.; Vlaanderen, J.; Huss, A.; Mulleners, W.M.; Sas, A.M.G.; van Laar, T.; Kromhout, H.; Vermeulen, R. A case–control study of the protective effect of alcohol, coffee, and cigarette consumption on Parkinson disease risk: Time-since-cessation modifies the effect of tobacco smoking. PLoS ONE 2014, 9, e95297. [Google Scholar] [CrossRef]
- Gao, X.; Simon, K.C.; Han, J.; Schwarzschild, M.A.; Ascherio, A. Family history of melanoma and Parkinson disease risk. Neurology 2009, 73, 1286–1291. [Google Scholar] [CrossRef]
- Gao, X.; Simon, K.C.; Han, J.; Schwarzschild, M.A.; Ascherio, A. Genetic determinants of hair color and Parkinson’s disease risk. Ann. Neurol. 2009, 65, 76–82. [Google Scholar] [CrossRef]
- Ruiz-Martínez, J.; de la Riva, P.; Rodríguez-Oroz, M.C.; Rezola, E.M.; Bergareche, A.; Gorostidi, A.; Gago, B.; Estanga, A.; Larranaga, N.; Sarasqueta, C.; et al. Prevalence of cancer in Parkinson‘s disease related to R1441G and G2019S mutations in LRRK2. Mov. Disord. 2014, 29, 750–755. [Google Scholar] [CrossRef]
- Kestenbaum, M.; Alcalay, R.N. Clinical Features of LRRK2 Carriers with Parkinson’s Disease. Adv. Neurobiol. 2017, 14, 31–48. [Google Scholar] [CrossRef]
- Agalliu, I.; San Luciano, M.; Mirelman, A.; Giladi, N.; Waro, B.; Aasly, J.; Inzelberg, R.; Hassin-Baer, S.; Friedman, E.; Ruiz-Martinez, J.; et al. Higher frequency of certain cancers in LRRK2 G2019S mutation carriers with Parkinson disease: A pooled analysis. JAMA Neurol. 2015, 72, 58–65. [Google Scholar] [CrossRef]
- Fung, K.-M.; Rorke, L.B.; Giasson, B.; Lee, V.M.-Y.; Trojanowski, J.Q. Expression of alpha-, beta-, and gamma-synuclein in glial tumors and medulloblastomas. Acta Neuropathol. 2003, 106, 167–175. [Google Scholar] [CrossRef]
- Bethge, N.; Lothe, R.A.; Honne, H.; Andresen, K.; Trøen, G.; Eknæs, M.; Liestøl, K.; Holte, H.; Delabie, J.; Smeland, E.B.; et al. Colorectal cancer DNA methylation marker panel validated with high performance in Non-Hodgkin lymphoma. Epigenetics 2014, 9, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Israeli, E.; Yakunin, E.; Zarbiv, Y.; Hacohen-Solovich, A.; Kisos, H.; Loeb, V.; Lichtenstein, M.; Ben-Gedalya, T.; Sabag, O.; Pikarsky, E.; et al. α-Synuclein expression selectively affects tumorigenesis in mice modeling Parkinson’s disease. PLoS ONE 2011, 6, e19622. [Google Scholar] [CrossRef] [PubMed]
- Li, W.H.; Zhang, H.; Guo, Q.; Wu, X.D.; Xu, Z.S.; Dang, C.X.; Xia, P.; Song, Y.C. Detection of SNCA and FBN1 methylation in the stool as a biomarker for colorectal cancer. Dis. Markers 2015, 2015, 657570. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.H.; Kannengiesser, C.; Lesage, S.; Andre, J.; Mourah, S.; Michel, L.; Descamps, V.; Basset-Seguin, N.; Bagot, M.; Bensussan, A.; et al. PARKIN inactivation links Parkinson’s disease to melanoma. J. Natl. Cancer Inst. 2015, 108. [Google Scholar] [CrossRef]
- Duan, H.; Lei, Z.; Xu, F.; Pan, T.; Lu, D.; Ding, P.; Zhu, C.; Pan, C.; Zhang, S. PARK2 Suppresses Proliferation and Tumorigenicity in Non-small Cell Lung Cancer. Front. Oncol. 2019, 9, 790. [Google Scholar] [CrossRef] [PubMed]
- Mehdi, S.J.; Ali, A.; Rizvi, M.M. Parkin gene alteration in ovarian carcinoma from northern Indian population. Pathol. Oncol. Res. 2011, 17, 579–586. [Google Scholar] [CrossRef]
- O’Flanagan, C.; Morais, V.; O’Neill, C. PINK1, cancer and neurodegeneration. Oncoscience 2016, 3, 1–2. [Google Scholar] [CrossRef]
- Li, Y.; Cui, J.; Zhang, C.H.; Yang, D.J.; Chen, J.H.; Zan, W.H.; Li, B.; Li, Z.; He, Y.L. High-expression of DJ-1 and loss of PTEN associated with tumor metastasis and correlated with poor prognosis of gastric carcinoma. Int. J. Med. Sci. 2013, 10, 1689–1697. [Google Scholar] [CrossRef]
- Kawate, T.; Iwaya, K.; Kikuchi, R.; Kaise, H.; Oda, M.; Sato, E.; Hiroi, S.; Matsubara, O.; Kohno, N. DJ1 protein expression as a predictor of pathological complete remission after neoadjuvant chemotherapy in breast cancer patients. Breast Cancer Res. Treat. 2013, 139, 51–59. [Google Scholar] [CrossRef]
- Park, K.R.; Yun, J.S.; Park, M.H.; Jung, Y.Y.; Yeo, I.J.; Nam, K.T.; Kim, H.D.; Song, J.K.; Choi, D.Y.; Park, P.H.; et al. Loss of parkin reduces lung tumor development by blocking p21 degradation. PLoS ONE 2019, 14, e0217037. [Google Scholar] [CrossRef]
- Wahabi, K.; Perwez, A.; Kamarudheen, S.; Bhat, Z.I.; Mehta, A.; Rizvi, M.M.A. Parkin gene mutations are not common, but its epigenetic inactivation is a frequent event and predicts poor survival in advanced breast cancer patients. BMC Cancer 2019, 19, 820. [Google Scholar] [CrossRef] [PubMed]
- Hernan, M.A.; Takkouche, B.; Caamano-Isorna, F.; Gestal-Otero, J.J. A meta-analysis of coffee drinking, cigarette smoking, and the risk of Parkinson’s disease. Ann. Neurol. 2002, 52, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Welinder, C.; Jönsson, G.B.; Ingvar, C.; Lundgren, L.; Baldetorp, B.; Olsson, H.; Breslin, T.; Rezeli, M.; Jansson, B.; Fehniger, T.E.; et al. Analysis of alpha-synuclein in malignant melanoma—development of a SRM quantification assay. PLoS ONE 2014, 9, e110804. [Google Scholar] [CrossRef] [PubMed]
- Mu, J.; Huang, W.; Tan, Z.; Li, M.; Zhang, L.; Ding, Q.; Wu, X.; Lu, J.; Liu, Y.; Dong, Q.; et al. Dopamine receptor D2 is correlated with gastric cancer prognosis. Oncol. Lett. 2017, 13, 1223–1227. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Z.B.; Luo, C.; Mao, X.Y.; Li, X.; Yin, J.Y.; Zhang, W.; Zhou, H.H.; Liu, Z.Q. The Prospective Value of Dopamine Receptors on Bio-Behavior of Tumor. J. Cancer 2019, 10, 1622–1632. [Google Scholar] [CrossRef]
- Grossrubatscher, E.; Veronese, S.; Ciaramella, P.D.; Pugliese, R.; Boniardi, M.; De Carlis, L.; Torre, M.; Ravini, M.; Gambacorta, M.; Loli, P. High expression of dopamine receptor subtype 2 in a large series of neuroendocrine tumors. Cancer Biol. Ther. 2008, 7, 1970–1978. [Google Scholar] [CrossRef][Green Version]
- Li, J.; Zhu, S.; Kozono, D.; Ng, K.; Futalan, D.; Shen, Y.; Akers, J.C.; Steed, T.; Kushwaha, D.; Schlabach, M.; et al. Genome-wide shRNA screen revealed integrated mitogenic signaling between dopamine receptor D2 (DRD2) and epidermal growth factor receptor (EGFR) in glioblastoma. Oncotarget 2014, 5, 882–893. [Google Scholar] [CrossRef]
- Akbari, M.E.; Kashani, F.L.; Ahangari, G.; Pornour, M.; Hejazi, H.; Nooshinfar, E.; Kabiri, M.; Hosseini, L. The effects of spiritual intervention and changes in dopamine receptor gene expression in breast cancer patients. Breast Cancer 2016, 23, 893–900. [Google Scholar] [CrossRef]
- Weissenrieder, J.S.; Neighbors, J.D.; Mailman, R.B.; Hohl, R.J. Cancer and the dopamine D2 receptor: A pharmacological perspective. J. Pharmacol. Exp. Ther. 2019, 370, 111–126. [Google Scholar] [CrossRef]
- Dolma, S.; Selvadurai, H.J.; Lan, X.; Lee, L.; Kushida, M.; Voisin, V.; Whetstone, H.; So, M.; Aviv, T.; Park, N.; et al. Inhibition of Dopamine Receptor D4 Impedes Autophagic Flux, Proliferation, and Survival of Glioblastoma Stem Cells. Cancer Cell 2016, 29, 859–873. [Google Scholar] [CrossRef]
- Salazar, C.; Ruiz-Hincapie, P.; Ruiz, L.M. The interplay among PINK1/PARKIN/Dj-1 network during mitochondrial quality control in cancer biology: Protein interaction analysis. Cells 2018, 7, 154. [Google Scholar] [CrossRef] [PubMed]
- Vasseur, S.; Afzal, S.; Tardivel-Lacombe, J.; Park, D.S.; Iovanna, J.L.; Mak, T.W. Dj-1/park7 is an important mediator of hypoxia-induced cellular responses. Proc. Natl. Acad. Sci. USA 2009, 106, 1111–1116. [Google Scholar] [CrossRef] [PubMed]
- Poole, A.C.; Thomas, R.E.; Andrews, L.A.; McBride, H.M.; Whitworth, A.J.; Pallanck, L.J. The pink1/parkin pathway regulates mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2008, 105, 1638–1643. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Dorn, G.W. Pink1-phosphorylated mitofusin 2 is a parkin receptor for culling damaged mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Strobbe, D.; Robinson, A.A.; Harvey, K.; Rossi, L.; Ferraina, C.; de Biase, V.; Rodolfo, C.; Harvey, R.J.; Campanella, M. Distinct Mechanisms of Pathogenic DJ-1 Mutations in Mitochondrial Quality Control. Front. Mol. Neurosci. 2018, 11, 68. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef]
- Birsa, N.; Norkett, R.; Wauer, T.; Mevissen, T.E.T.; Wu, H.-C.; Foltynie, T.; Bhatia, K.; Hirst, W.D.; Komander, D.; Plun-Favreau, H.; et al. Lysine 27 ubiquitination of the mitochondrial transport protein Miro is dependent on serine 65 of the Parkin ubiquitin ligase. J. Biol. Chem. 2014, 289, 14569–14582. [Google Scholar] [CrossRef]
- Lovas, J.R.; Wang, X. The meaning of mitochondrial movement to a neuron’s life. Biochim. Biophys. Acta 2013, 1833, 184–194. [Google Scholar] [CrossRef]
- Liu, S.; Sawada, T.; Lee, S.; Yu, W.; Silverio, G.; Alapatt, P.; Millan, I.; Shen, A.; Saxton, W.; Kanao, T.; et al. Parkinson’s disease-associated kinase PINK1 regulates Miro protein level and axonal transport of mitochondria. PLoS Genet. 2012, 8, e1002537. [Google Scholar] [CrossRef]
- Wang, X.; Winter, D.; Ashrafi, G.; Schlehe, J.; Wong, Y.L.; Selkoe, D.; Rice, S.; Steen, J.; LaVoie, M.J.; Schwarz, T.L. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 2011, 147, 893–906. [Google Scholar] [CrossRef]
- Imai, Y. PINK1-Parkin signaling in Parkinson’s disease: Lessons from Drosophila. Neurosci. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Chu, C. Tickled PINK1 mitochondrial homeostasis and autophagy in recessive Parkinsonism. Biochim. Biophys. Acta 2010, 1802, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Arena, G.; Gelmetti, V.; Torosantucci, L.; Vignone, D.; Lamorte, G.; De Rosa, P.; Cilia, E.; Jonas, E.A.; Valente, E.M. PINK1 protects against cell death induced by mitochondrial depolarization, by phosphorylating Bcl-xL and impairing its pro-apoptotic cleavage. Cell Death Differ. 2013, 20, 920–930. [Google Scholar] [CrossRef] [PubMed]
- Arena, G.; Valente, E.M. PINK1 in the limelight: Multiple functions of an eclectic protein in human health and disease. J. Pathol. 2017, 241, 251–263. [Google Scholar] [CrossRef]
- Hernández, C.J.; Báez-Becerra, C.; Contreras-Zárate, M.J.; Arboleda, H.; Arboleda, G. PINK1 Silencing Modifies Dendritic Spine Dynamics of Mouse Hippocampal Neurons. J. Mol. Neurosci. 2019, 69, 570–579. [Google Scholar] [CrossRef]
- Eldeeb, M.A.; Ragheb, M.A. N-degron-mediated degradation and regulation of mitochondrial PINK1 kinase. Curr. Genet. 2020, 66, 693–701. [Google Scholar] [CrossRef]
- Gandhi, S.; Wood-Kaczmar, A.; Yao, Z.; Plun-Favreau, H.; Deas, E.; Klupsch, K.; Downward, J.; Latchman, D.S.; Tabrizi, S.J.; Wood, N.W.; et al. PINK1-Associated Parkinson’s Disease Is Caused by Neuronal Vulnerability to Calcium-Induced Cell Death. Mol. Cell 2009, 33, 627–638. [Google Scholar] [CrossRef]
- Bueno, M.; Lai, Y.C.; Romero, Y.; Brands, J.; St Croix, C.M.; Kamga, C.; Corey, C.; Herazo-Maya, J.D.; Sembrat, J.; Lee, J.S.; et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J. Clin. Investig. 2015, 125, 521–538. [Google Scholar] [CrossRef]
- Zhang, R.; Gu, J.; Chen, J.; Ni, J.; Hung, J.; Wang, Z.; Zhang, X.; Feng, J.; Ji, L. High expression of PINK1 promotes proliferation and chemoresistance of NSCLC. Oncol. Rep. 2017, 37, 2137–2146. [Google Scholar] [CrossRef]
- Dai, K.; Radin, D.P.; Leonardi, D. PINK1 depletion sensitizes non-small cell lung cancer to glycolytic inhibitor 3-bromopyruvate: Involvement of ROS and mitophagy. Pharmacol. Rep. 2019, 71, 1184–1189. [Google Scholar] [CrossRef]
- Chang, G.; Zhang, W.; Ma, Y.; Wen, Q. PINK1 Expression Is Associated with Poor Prognosis in Lung Adenocarcinoma. Tohokuj. Exp. Med. 2018, 245, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Agnihotri, S.; Golbourn, B.; Huang, X.; Remke, M.; Younger, S.; Cairns, R.A.; Chalil, A.; Smith, C.A.; Krumholtz, S.-L.; Mackenzie, D.; et al. PINK1 Is a Negative Regulator of Growth and the Warburg Effect in Glioblastoma. Cancer Res. 2016, 76, 4708–4719. [Google Scholar] [CrossRef] [PubMed]
- Li, G.B.; Fu, R.Q.; Shen, H.M.; Zhou, J.; Hu, X.Y.; Liu, X.Y.; Li, Y.N.; Zhang, H.W.; Liu, X.; Zhang, Y.H.; et al. Polyphyllin I induces mitophagic and apoptotic cell death in human breast cancer cells by increasing mitochondrial PINK1 levels. Oncotarget 2017, 8, 10359–10374. [Google Scholar] [CrossRef]
- Chew, K.C.; Matsuda, N.; Saisho, K.; Lim, G.G.Y.; Chai, C.; Tan, H.M.; Tanaka, K.; Lim, K.L. Parkin mediates apparent E2-independent monoubiquitination in vitro and contains an intrinsic activity that catalyzes polyubiquitination. PLoS ONE 2011, 6, e19720. [Google Scholar] [CrossRef] [PubMed]
- Durcan, T.M.; Tang, M.Y.; Pérusse, J.R.; Dashti, E.A.; Aguileta, M.A.; McLelland, G.L.; Gros, P.; Shaler, T.A.; Faubert, D.; Coulombe, B.; et al. USP8 regulates mitophagy by removing K6-linked ubiquitin conjugates from parkin. Embo J. 2014, 33, 2473–2491. [Google Scholar] [CrossRef]
- Charan, R.A.; Johnson, B.N.; Zaganelli, S.; Nardozzi, J.D.; LaVoie, M.J. Inhibition of apoptotic Bax translocation to the mitochondria is a central function of parkin. Cell Death Dis. 2014, 5, e1313. [Google Scholar] [CrossRef]
- Carroll, R.G.; Hollville, E.; Martin, S.J. Parkin sensitizes toward apoptosis induced by mitochondrial depolarization through promoting degradation of Mcl-1. Cell Rep. 2014, 9, 1538–1553. [Google Scholar] [CrossRef]
- Miklya, I.; Göltl, P.; Hafenscher, F.; Pencz, N. The role of parkin in Parkinson’s disease. Neuropsychopharmacol. Hung. 2014, 16, 67–76. [Google Scholar]
- Pinto, M.; Nissanka, N.; Moraes, C.T. Lack of Parkin Anticipates the Phenotype and Affects Mitochondrial Morphology and mtDNA Levels in a Mouse Model of Parkinson’s Disease. J. Neurosci. 2018, 38, 1042–1053. [Google Scholar] [CrossRef]
- Dawson, T.M.; Dawson, V.L. Parkin plays a role in sporadic Parkinson’s disease. Neurodegener. Dis. 2014, 13, 69–71. [Google Scholar] [CrossRef]
- Cha, S.J.; Choi, H.J.; Kim, H.J.; Choi, E.J.; Song, K.H.; Im, D.S.; Kim, K. Parkin expression reverses mitochondrial dysfunction in fused in sarcoma-induced amyotrophic lateral sclerosis. Insect. Mol. Biol. 2020, 29, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Castellazzi, M.; Patergnani, S.; Donadio, M.; Giorgi, C.; Bonora, M.; Fainardi, E.; Casetta, I.; Granieri, E.; Pugliatti, M.; Pinton, P. Correlation between auto/mitophagic processes and magnetic resonance imaging activity in multiple sclerosis patients. J. NeuroInflamm. 2019, 16, 131. [Google Scholar] [CrossRef] [PubMed]
- Alexiou, A.; Nizami, B.; Khan, F.I.; Soursou, G.; Vairaktarakis, C.; Chatzichronis, S.; Tsiamis, V.; Manztavinos, V.; Yarla, N.S.; Ashraf, G.M. Mitochondrial Dynamics and Proteins Related to Neurodegenerative Diseases. Curr. Protein. Pept. Sci. 2018, 19, 850–857. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, U.; Banerjee, A.; Chakrabarti, S.S.; Kaur, U.; Sen, O.; Cappai, R.; Chakrabarti, S. Interaction of α-synuclein and Parkin in iron toxicity on SH-SY5Y cells: Implications in the pathogenesis of Parkinson’s disease. Biochem. J. 2020, 477, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- González-Barbosa, E.; Mejía-García, A.; Bautista, E.; Gonzalez, F.J.; Segovia, J.; Elizondo, G. TCDD induces UbcH7 expression and synphilin-1 protein degradation in the mouse ventral midbrain. J. Biochem. Mol. Toxicol. 2017, 31. [Google Scholar] [CrossRef]
- Lee, S.B.; Kim, J.J.; Han, S.A.; Fan, Y.; Guo, L.S.; Aziz, K.; Nowsheen, S.; Kim, S.S.; Park, S.Y.; Luo, O.; et al. The AMPK-Parkin axis negatively regulates necroptosis and tumorigenesis by inhibiting the necrosome. Nat. Cell Biol. 2019, 21, 940–951. [Google Scholar] [CrossRef]
- Park, M.H.; Lee, H.J.; Lee, H.L.; Son, D.J.; Ju, J.H.; Hyun, B.K.; Jung, S.H.; Song, J.K.; Lee, D.H.; Hwang, C.J.; et al. Parkin Knockout Inhibits Neuronal Development via Regulation of Proteasomal Degradation of p21. Theranostics 2017, 7, 2033–2045. [Google Scholar] [CrossRef]
- Lee, S.B.; Kim, J.J.; Nam, H.J.; Gao, B.; Yin, P.; Qin, B.; Yi, S.Y.; Ham, H.; Evans, D.; Kim, S.H.; et al. Parkin Regulates Mitosis and Genomic Stability through Cdc20/Cdh1. Mol. Cell. 2015, 60, 21–34. [Google Scholar] [CrossRef]
- Li, Z.; Wang, Y.; Wu, L.; Dong, Y.; Zhang, J.; Chen, F.; Xie, W.; Huang, J.; Lu, N. Apurinic endonuclease 1 promotes the cisplatin resistance of lung cancer cells by inducing Parkin-mediated mitophagy. Oncol. Rep. 2019, 42, 2245–2254. [Google Scholar] [CrossRef]
- Veeriah, S.; Taylor, B.S.; Meng, S.; Fang, F.; Yilmaz, E.; Vivanco, I.; Janakiraman, M.; Schultz, N.; Hanrahan, A.J.; Pao, W.; et al. Somatic mutations of the Parkinson’s disease-associated gene PARK2 in glioblastoma and other human malignancies. Nat. Genet. 2010, 42, 77–82. [Google Scholar] [CrossRef]
- Viotti, J.; Duplan, E.; Caillava, C.; Condat, J.; Goiran, T.; Giordano, C.; Marie, Y.; Idbaih, A.; Delattre, J.Y.; Honnorat, J.; et al. Glioma tumor grade correlates with parkin depletion in mutant p53-linked tumors and results from loss of function of p53 transcriptional activity. Oncogene 2014, 33, 1764–1775. [Google Scholar] [CrossRef] [PubMed]
- Denison, S.R.; Wang, F.; Becker, N.A.; Schüle, B.; Kock, N.; Phillips, L.A.; Klein, C.; Smith, D.I. Alterations in the common fragile site gene Parkin in ovarian and other cancers. Oncogene 2003, 22, 8370–8378. [Google Scholar] [CrossRef]
- Picchio, M.C.; Martin, E.S.; Cesari, R.; Calin, G.A.; Yendamuri, S.; Kuroki, T.; Pentimalli, F.; Sarti, M.; Yoder, K.; Kaiser, L.R.; et al. Alterations of the tumor suppressor gene Parkin in non-small cell lung cancer. Clin. Cancer Res. 2004, 10, 2720–2724. [Google Scholar] [CrossRef]
- Wang, F.; Denison, S.; Lai, J.P.; Philips, L.A.; Montoya, D.; Kock, N.; Schüle, B.; Klein, C.; Shridhar, V.; Roberts, L.R.; et al. Parkin gene alterations in hepatocellular carcinoma. Genes Chromosomes Cancer 2004, 40, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, C.; Hu, W.; Feng, Z. Parkinson’s disease-associated protein Parkin: An unusual player in cancer. Cancer Commun. 2018, 38, 40. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, C.; Zhao, Y.; Yue, X.; Wu, H.; Huang, S.; Chen, J.; Tomsky, K.; Xie, H.; Khella, C.A.; et al. Parkin targets HIF-1alpha for ubiquitination and degradation to inhibit breast tumor progression. Nat. Commun. 2017, 8, 1823. [Google Scholar] [CrossRef] [PubMed]
- Toma, M.I.; Wuttig, D.; Kaiser, S.; Herr, A.; Weber, T.; Zastrow, S.; Koch, R.; Meinhardt, M.; Baretton, G.B.; Wirth, M.P.; et al. PARK2 and PACRG are commonly downregulated in clear-cell renal cell carcinoma and are associated with aggressive disease and poor clinical outcome. Genes Chromosomes Cancer 2013, 52, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lin, C.; Song, J.; Chen, H.; Chen, X.; Ren, L.; Zhou, Z.; Pan, J.; Yang, Z.; Bao, W.; et al. Parkin facilitates proteasome inhibitor-induced apoptosis via suppression of NF-κB activity in hepatocellular carcinoma. Cell Death Dis. 2019, 10, 719. [Google Scholar] [CrossRef]
- Agirre, X.; Román-Gómez, J.; Vázquez, I.; Jimenez-Velasco, A.; Garate, L.; Montiel-Duarte, C.; Artieda, P.; Cordeu, L.; Lahortiga, I.; Calasanz, M.J.; et al. Abnormal methylation of the common PARK2 and PACRG promoter is associated with downregulation of gene expression in acute lymphoblastic leukemia and chronic myeloid leukemia. Int. J. Cancer 2006, 118, 1945–1953. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, M.; Marusawa, H.; Wang, H.Q.; Iwai, A.; Ikeuchi, K.; Imai, Y.; Kataoka, A.; Nukina, N.; Takahashi, R.; Chiba, T. Parkin as a tumor suppressor gene for hepatocellular carcinoma. Oncogene 2008, 27, 6002–6011. [Google Scholar] [CrossRef] [PubMed]
- Tay, S.P.; Yeo, C.W.; Chai, C.; Chua, P.J.; Tan, H.M.; Ang, A.X.Y.; Yip, D.L.H.; Sung, J.X.; Tan, P.H.; Bay, B.H.; et al. Parkin enhances the expression of cyclin-dependent kinase 6 and negatively regulates the proliferation of breast cancer cells. J. Biol. Chem. 2010, 285, 29231–29238. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.; Zhou, Z.; Jiang, B.; Yuan, X.; Cao, X.; Huang, G.; Li, Y. Inactivation of parkin by promoter methylation correlated with lymph node metastasis and genomic instability in nasopharyngeal carcinoma. Tumour Biol. 2017, 39, 1010428317695025. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Liu, M.; Hao, J.; Li, D.; Luo, Y.; Wang, X.; Yang, Y.; Li, F.; Shui, W.; Chen, Q.; et al. Parkin deficiency contributes to pancreatic tumorigenesis by inducing spindle multipolarity and misorientation. Cell Cycle 2013, 12, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Cho, Y.; Jung, B.C.; Kim, S.H.; Kang, Y.W.; Pan, C.H.; Rhee, K.J.; Kim, Y.S. Parkin induces G2/M cell cycle arrest in TNF-α-treated HeLa cells. Biochem. Biophys. Res. Commun. 2015, 464, 63–69. [Google Scholar] [CrossRef]
- Sarraf, S.A.; Sideris, D.P.; Giagtzoglou, N.; Ni, L.; Kankel, M.W.; Sen, A.; Bochicchio, L.E.; Huang, C.H.; Nussenzweig, S.C.; Worley, S.H.; et al. PINK1/Parkin Influences Cell Cycle by Sequestering TBK1 at Damaged Mitochondria, Inhibiting Mitosis. Cell Rep. 2019, 29, 225–235. [Google Scholar] [CrossRef]
- Ding, D.; Ao, X.; Liu, Y.; Wang, Y.Y.; Fa, H.G.; Wang, M.Y.; He, Y.Q.; Wang, J.X. Post-translational modification of Parkin and its research progress in cancer. Cancer Commun. 2019, 39, 77. [Google Scholar] [CrossRef]
- Lee, K.; Lee, M.H.; Kang, Y.W.; Rhee, K.J.; Kim, T.U.; Kim, Y.S. Parkin induces apoptotic cell death in TNF-α-treated cervical cancer cells. BMB Rep. 2012, 45, 526–531. [Google Scholar] [CrossRef]
- Zhang, C.; Lin, M.; Wu, R.; Wang, X.; Yang, B.; Levine, A.J.; Hu, W.; Feng, Z. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc. Natl. Acad. Sci. USA 2011, 108, 16259–16264. [Google Scholar] [CrossRef]
- Cao, K.; Lait, S.W.G. Parkin inhibits necroptosis to prevent cancer. Nat. Cell Biol. 2019, 21, 915–916. [Google Scholar] [CrossRef]
- Galluzzi, L.; Keep, O.; Krautwald, S.; Kroemer, G.; Linkermann, A. Molecular mechanisms of regulated necrosis. Semin. Cell Dev. Biol. 2014, 35, 24–32. [Google Scholar] [CrossRef]
- Alameda, J.P.; Moreno-Maldonado, R.; Navarro, M.; Bravo, A.; Ramírez, A.; Page, A.; Jorcano, J.L.; Fernández-Aceñero, M.J.; Casanova, M.L. An inactivating CYLD mutation promotes skin tumor progression by conferring enhanced proliferative, survival and angiogenic properties to epidermal cancer cells. Oncogene 2010, 29, 6522–6532. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Xu, B.; Shen, W.; Zhu, H.; Wu, W.; Fu, Y.; Chen, H.; Dong, H.; Zhu, Y.; Miao, K.; et al. Dysregulation of TNFα-induced necroptotic signaling in chronic lymphocytic leukemia: Suppression of CYLD gene by LEF1. Leukemia 2012, 26, 1293–1300. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Liu, P.; Li, J. Necroptosis: An emerging form of programmed cell death. Crit. Rev. Oncol. Hematol. 2012, 82, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, K.; Tsumoto, H.; Miura, Y.; Yamaguchi, J.; Iguchi-Ariga, S.M.M.; Sakuma, T.; Yamamoto, T.; Uchiyama, Y. DJ-1 is indispensable for the S-nitrosylation of Parkin, which maintains function of mitochondria. Sci. Rep. 2020, 10, 4377. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Zhou, Y.; Jiang, N.; Wang, T.; Zhu, J.; Chen, Y.; Li, L.; Zhang, J.; Yu, S.; Zhao, Y. DJ-1 exerts anti-inflammatory effects and regulates NLRX1-TRAF6 via SHP-1 in stroke. J. Neuroinflamm. 2020, 17, 81. [Google Scholar] [CrossRef] [PubMed]
- Van der Merwe, C.; Jalali Sefid Dashti, Z.; Christoffels, A.; Loos, B.; Bardien, S. Evidence for a common biological pathway linking three Parkinson’s disease-causing genes: Parkin, PINK1 and DJ-1. Eur. J. Neurosci. 2015, 41, 1113–1125. [Google Scholar] [CrossRef]
- Zhang, X.; Yuan, D.; Sun, Q.; Xu, L.; Lee, E.; Lewis, A.J.; Zuckerbraun, B.S.; Rosengart, M.R. Calcium/calmodulin-dependent protein kinase regulates the PINK1/Parkin and DJ-1 pathways of mitophagy during sepsis. FASEB J. 2017, 31, 4382–4395. [Google Scholar] [CrossRef]
- Kamp, F.; Exner, N.; Lutz, A.K.; Wender, N.; Hegermann, J.; Brunner, B.; Nuscher, B.; Bartels, T.; Giese, A.; Beyer, K.; et al. Inhibition of mitochondrial fusion by α-synuclein is rescued by pink1, parkin and dj-1. EMBO J. 2010, 29, 3571–3589. [Google Scholar] [CrossRef]
- Cao, J.; Chen, X.; Jiang, L.; Lu, B.; Yuan, M.; Zhu, D.; Zhu, H.; He, Q.; Yang, B.; Ying, M. DJ-1 suppresses ferroptosis through preserving the activity of S-adenosyl homocysteine hydrolase. Nat. Commun. 2020, 11, 1251. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by lipid peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic. Biol. Med. 2020, 152, 175–185. [Google Scholar] [CrossRef]
- Zondler, L.; Miller-Fleming, L.; Repici, M.; Gonçalves, S.; Tenreiro, S.; Rosado-Ramos, R.; Betzer, C.; Straatman, K.R.; Jensen, P.H.; Giorgini, F.; et al. DJ-1 interactions with α-synuclein attenuate aggregation and cellular toxicity in models of Parkinson’s disease. Cell Death Dis. 2014, 5, e1350. [Google Scholar] [CrossRef]
- Cao, J.; Lou, S.; Ying, M.; Yang, B. DJ-1 as a human oncogene and potential therapeutic target. Biochem. Pharmacol. 2015, 93, 241–250. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, H.; Zhang, L.; Liu, X.; Zhang, C.; Wang, Y.; He, Q.; Zhang, Y.; Li, Y.; Chen, Q.; et al. DJ-1 promotes colorectal cancer progression through activating PLAGL2/Wnt/BMP4 axis. Cell Death Dis. 2018, 9, 865. [Google Scholar] [CrossRef]
- Qin, X.; Lu, A.; Ke, M.; Zhu, W.; Ye, X.; Wang, G.; Weng, G. DJ-1 inhibits autophagy activity of prostate cancer cells by repressing JNK-Bcl2-Beclin1 signaling. Cell Biol. Int. 2020, 44, 937–946. [Google Scholar] [CrossRef]
- Zhu, X.; Luo, C.; Lin, K.; Bu, F.; Ye, F.; Huang, C.; Luo, H.; Huang, J.; Zhu, Z. Overexpression of DJ-1 enhances colorectal cancer cell proliferation through the cyclin-D1/MDM2-p53 signaling pathway. Biosci. Trends 2020, 14, 83–95. [Google Scholar] [CrossRef]
- Qiu, B.; Wang, J.; Yu, Y.; Zhen, C.; Gu, J.; Liu, W.; Wen, Y.; Chen, L.; Gao, Y.; Xia, Q.; et al. DJ-1 promotes development of DEN-induced hepatocellular carcinoma and proliferation of liver cancer cells. Oncotarget 2017, 8, 8499–8511. [Google Scholar] [CrossRef]
- Pei, X.; Wu, T.; Li, B.; Tian, X.; Li, Z.; Yang, Q. Increased expression of macrophage migration inhibitory factor and dj-1 contribute to cell invasion and metastasis of nasopharyngeal carcinoma. Int. J. Med. Sci. 2014, 11, 106–115. [Google Scholar] [CrossRef]
- Han, B.; Wang, J.; Gao, J.; Feng, S.; Zhu, Y.; Li, X.; Xiao, T.; Qi, J.; Cui, W. DJ-1 as a potential biomarker for the early diagnosis in lung cancer patients. Tumour Biol. 2017, 39, 1010428317714625. [Google Scholar] [CrossRef]
- Zhang, S.; Mukherjee, S.; Fan, X.; Salameh, A.; Mujoo, K.; Huang, Z.; Li, L.; To’a Salazar, G.; Zhang, N.; An, Z. Novel association of DJ-1 with HER3 potentiates HER3 activation and signaling in cancer. Oncotarget 2016, 7, 65758–65769. [Google Scholar] [CrossRef]
- Lin, J.P.; Pan, B.C.; Li, B.; Li, Y.; Tian, X.Y.; Li, Z. DJ-1 is activated in medulloblastoma and is associated with cell proliferation and differentiation. World J. Surg. Oncol. 2014, 12, 373. [Google Scholar] [CrossRef]
- Qiu, K.; Xie, Q.; Jiang, S.; Lin, T. Silencing of DJ-1 reduces proliferation, invasion, and migration of papillary thyroid cancer cells in vitro, probably by increase of PTEN expression. Int. J. Clin. Exp. Pathol. 2019, 12, 2046–2055. [Google Scholar]
- Zhang, J.; Xu, M.; Zhou, W.; Li, D.; Zhang, H.; Chen, Y.; Ning, L.; Zhang, Y.; Li, S.; Yu, M.; et al. Deficiency in the anti-apoptotic protein DJ-1 promotes intestinal epithelial cell apoptosis and aggravates inflammatory bowel disease via p53. J. Biol. Chem. 2020, 295, 4237–4251. [Google Scholar] [CrossRef]
- Mehra, S.; Sahay, S.; Maji, S.K. α-Synuclein misfolding and aggregation: Implications in Parkinson’s disease pathogenesis. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 890–908. [Google Scholar] [CrossRef]
- Diógenes, M.J.; Dias, R.B.; Rombo, D.M.; Miranda, H.V.; Maiolino, F.; Guerreiro, P.; Näsström, T.; Franquelim, H.G.; Oliveira, L.M.A.; Castanho, M.A.R.B.; et al. Extracellular alpha-synuclein oligomers modulate synaptic transmission and impair LTP via NMDA-receptor activation. J. Neurosci. 2012, 32, 11750–11762. [Google Scholar] [CrossRef]
- Rockenstein, E.; Number, S.; Overk, C.R.; Ubhi, K.; Mante, M.; Patrick, C.; Adame, A.; Trejo-Morales, M.; Gerez, J.; Picotti, P.; et al. Accumulation of oligomer-prone α-synuclein exacerbates synaptic and neuronal degeneration in vivo. Brain 2014, 137, 1496–1513. [Google Scholar] [CrossRef]
- Brundin, P.; Melki, R. Prying into the Prion Hypothesis for Parkinson’s Disease. J. Neurosci. 2017, 37, 9808–9818. [Google Scholar] [CrossRef]
- Courte, J.; Bousset, L.; Boxberg, Y.V.; Villard, C.; Melki, R.; Peyrin, J.M. The expression level of alpha-synuclein in different neuronal populations is the primary determinant of its prion-like seeding. Sci. Rep. 2020, 10, 4895. [Google Scholar] [CrossRef]
- Mezias, C.; Rey, N.; Brundin, P.; Raj, A. Neural connectivity predicts spreading of alpha-synuclein pathology in fibril-injected mouse models: Involvement of retrograde and anterograde axonal propagation. Neurobiol. Dis. 2020, 134, 104623. [Google Scholar] [CrossRef]
- Lee, B.R.; Matsuo, Y.; Cashikar, A.G.; Kamitani, T. Role of Ser129 phosphorylation of α-synuclein in melanoma cells. J. Cell Sci. 2013, 126, 696–704. [Google Scholar] [CrossRef]
- Bruening, W.; Giasson, B.I.; Klein-Szanto, A.J.; Lee, V.M.; Trojanowski, J.Q.; Godwin, A.K. Synucleins are expressed in the majority of breast and ovarian carcinomas and in preneoplastic lesions of the ovary. Cancer 2000, 88, 2154–2163. [Google Scholar] [CrossRef]
- Yan, Y.; Xu, Z.; Hu, X.; Qian, L.; Li, Z.; Zhou, Y.; Dai, S.; Zeng, S.; Gong, Z. SNCA Is a Functionally Low-Expressed Gene in Lung Adenocarcinoma. Genes 2018, 9, 16. [Google Scholar] [CrossRef]
- Rodríguez-Losada, N.; de la Rosa, J.; Larriva, M.; Wendelbo, R.; Aguirre, J.A.; Castresana, J.S.; Ballaz, S.J. Overexpression of alpha-synuclein promotes both cell proliferation and cell toxicity in human SH-SY5Y neuroblastoma cells. J. Adv. Res. 2020, 23, 37–45. [Google Scholar] [CrossRef]
- Li, Y.X.; Yu, Z.W.; Jiang, T.; Shao, L.W.; Liu, Y.; Li, N.; Wu, Y.F.; Zheng, C.; Wu, X.Y.; Zhang, M.; et al. SNCA, a novel biomarker for Group 4 medulloblastomas, can inhibit tumor invasion and induce apoptosis. Cancer Sci. 2018, 109, 1263–1275. [Google Scholar] [CrossRef]
- Plecitá-Hlavatá, L.; Ježek, P. Integration of superoxide formation and cristae morphology for mitochondrial redox signaling. Int. J. Biochem. Cell Biol. 2016, 80, 31–50. [Google Scholar] [CrossRef]
- Hamacher-Brady, A.; Brady, N. Mitophagy programs mechanisms and physiological implications of mitochondrial targeting by autophagy. Cell Mol. Life Sci. 2016, 73, 775–795. [Google Scholar] [CrossRef]
- Ge, P.; Dawson, V.L.; Dawson, T.M. PINK1 and Parkin mitochondrial quality control: A source of regional vulnerability in Parkinson’s disease. Mol. Neurodegener. 2020, 15, 20. [Google Scholar] [CrossRef]
- Sironi, L.; Restelli, L.M.; Tolnay, M.; Neutzner, A.; Frank, S. Dysregulated Interorganellar Crosstalk of Mitochondria in the Pathogenesis of Parkinson’s Disease. Cells 2020, 9, 233. [Google Scholar] [CrossRef]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef]
- Ravanan, P.; Srikumar, I.F.; Talwar, P. Autophagy: The spotlight for cellular stress responses. Life Sci. 2017, 188, 53–67. [Google Scholar] [CrossRef]
- Cuervo, A.M. Autophagy: Many paths to the same end. Mol. Cell Biochem. 2004, 263, 55–72. [Google Scholar] [CrossRef]
- Shefa, U.; Jeong, N.Y.; Song, I.O.; Chung, H.J.; Kim, D.; Jung, J.; Huh, Y. Mitophagy links oxidative stress conditions and neurodegenerative diseases. Neural Regen. Res. 2019, 14, 749–756. [Google Scholar] [CrossRef]
- Evans, C.S.; Holzbaur, E.L.F. Lysosomal degradation of depolarized mitochondria is rate-limiting in OPTN-dependent neuronal mitophagy. Autophagy 2020, 16, 962–964. [Google Scholar] [CrossRef]
- Rosenfeldt, M.T.; Ryan, K.M. The multiple roles of autophagy in cancer. Carcinogenesis 2011, 32, 955–963. [Google Scholar] [CrossRef]
- Rose, J.M.; Novoselov, S.S.; Robinson, P.A.; Cheetham, M.E. Molecular chaperone-mediated rescue of mitophagy by a parkin RING1 domain mutant. Hum. Mol. Genet. 2011, 20, 16–27. [Google Scholar] [CrossRef]
- Essick, E.E.; Sam, F. Oxidative stress and autophagy in cardiac disease, neurological disorders, aging and cancer. Oxid. Med. Cell Longev. 2010, 3, 168–177. [Google Scholar] [CrossRef]
- Choi, I.; Zhang, Y.; Seegobin, S.P.; Pruvost, M.; Wang, Q.; Purtell, K.; Zhang, B.; Yue, Z. Microglia clear neuron-released α-synuclein via selective autophagy and prevent neurodegeneration. Nat. Commun. 2020, 11, 1386. [Google Scholar] [CrossRef]
- Wu, X.; Zhou, Z.; Xu, S.; Liao, C.; Chen, X.; Li, B.; Peng, J.; Li, D.; Yang, L. Extracellular vesicle packaged LMP1-activated fibroblasts promote tumor progression via autophagy and stroma-tumor metabolism coupling. Cancer Lett. 2020, 478, 93–106. [Google Scholar] [CrossRef]
- Huang, F.; Wang, B.R.; Wang, Y.G. Role of autophagy in tumorigenesis, metastasis, targeted therapy and drug resistance of hepatocellular carcinoma. World J. Gastroenterol. 2018, 24, 4643–4651. [Google Scholar] [CrossRef]
- Kania, E.; Pająk, B.; Orzechowski, A. Calcium homeostasis and ER stress in control of autophagy in cancer cells. Biomed. Res. Int. 2015, 2015, 352794. [Google Scholar] [CrossRef]
- Chen, N.; Karantza-Wadsworth, V. Role and regulation of autophagy in cancer. Biochim. Biophis. Acta 2009, 1793, 1516–1523. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.M. p53 and autophagy in cancer: Guardian of the genome meets guardian of the proteome. Eur. J. Cancer 2011, 47, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Mrakovcic, M.; Fröhlich, L.F. p53-Mediated Molecular Control of Autophagy in Tumor Cells. Biomolecules 2018, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef]
- Hamurcu, Z.; Delibasi, N.; Gecene, S.; Şener, E.F.; Dönmez-Altuntaş, H.; Özkul, Y.; Canatan, H.; Ozpolat, B. Targeting LC3 and Beclin-1 autophagy genes suppresses proliferation, survival, migration and invasion by inhibition of Cyclin-D1 and uPAR/Integrin beta1/Src signaling in triple negative breast cancer cells. J. Cancer Res. Clin. Oncol. 2018, 144, 415–430. [Google Scholar] [CrossRef]
- Miracco, C.; Cevenini, G.; Franchi, A.; Luzi, P.; Cosci, E.; Mourmouras, V.; Monciatti, I.; Mannucci, S.; Biagioli, M.; Toscano, M.; et al. Beclin 1 and LC3 autophagic gene expression in cutaneous melanocytic lesions. Hum. Pathol. 2010, 41, 503–512. [Google Scholar] [CrossRef]
- Lazova, R.; Klump, V.; Pawelek, J. Autophagy in cutaneous malignant melanoma. J. Cutan. Pathol. 2010, 37, 256–268. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D. Oxidative Stress, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2007; pp. 45–48. [Google Scholar]
- Sies, H. Oxidative Stress; Academic Press: London, UK, 1985; pp. 1–507. [Google Scholar]
- Bellucci, A.; Mercuri, N.B.; Venneri, A.; Faustini, G.; Longhena, F.; Pizzi, M.; Missale, C.; Spano, P.F. Review: Parkinson’s disease: From synaptic loss to connectome dysfunction. Neuropathol. Appl. Neurobiol. 2016, 42, 77–94. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine; Oxford University Press: Oxford, UK, 2007. [Google Scholar]
- Valko, M.; Morris, H.; Cronin, M.T. Metals, toxicity and oxidative stress. Curr. Med. Chem. 2005, 12, 1161–1208. [Google Scholar] [CrossRef]
- Bosco, D.A.; Fowler, D.M.; Zhang, Q.; Nieva, J.; Powers, E.T.; Wentworth, P.; Lerner, R.A.; Kelly, J.W. Elevated levels of oxidized cholesterol metabolites in Lewy body disease brains accelerate alpha-synuclein fibrilization. Nat. Chem. Biol. 2006, 2, 249–253. [Google Scholar] [CrossRef]
- Nakabeppu, Y.; Tsuchimoto, D.; Yamaguchi, H.; Sakumi, K. Oxidative damage in nucleic acids and Parkinson’s disease. J. Neurosci. Res. 2007, 85, 919–934. [Google Scholar] [CrossRef] [PubMed]
- Floor, E.; Wetzel, M.G. Increased protein oxidation in human substantia nigra pars compacta in comparison with basal ganglia and prefrontal cortex measured with an improved dinitrophenylhydrazine assay. J. Neurochem. 1998, 70, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Farina, M.; Avila, D.S.; Teixeira da Rocha, J.B.; Aschner, M. Metals, oxidative stress and neurodegeneration: A focus on iron, manganese and mercury. Neurochem. Int. 2013, 62, 575–594. [Google Scholar] [CrossRef] [PubMed]
- Berg, D.; Youdim, M.B. Role of Iron in Neurodegenerative Disorders. Top. Magn. Reson. Imaging 2006, 17, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Zecca, L.; Youdim, M.B.; Riederer, P.; Connor, J.R.; Crichton, R.R. Iron, brain ageing and neurodegenerative disorders. Nat. Rev. Neurosci. 2004, 5, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Sian-Hülsmann, J.; Mandel, S.; Youdim, M.B.; Riederer, P. The relevance of iron in the pathogenesis of Parkinson’s disease. J. Neurochem. 2011, 118, 939–957. [Google Scholar] [CrossRef]
- Sofic, E.; Riederer, P.; Heinsen, H.; Beckmann, H.; Reynolds, G.P.; Hebenstreit, G.; Youdim, M.B.H. Increased iron (III) and total iron content in post mortem substantia nigra of parkinsonian brain. J. Neural Transm. 1988, 74, 199–205. [Google Scholar] [CrossRef]
- Sulzer, D.; Bogulavsky, J.; Larsen, K.E.; Behr, G.; Karatekin, E.; Kleinman, M.H.; Turro, N.; Krantzi, D.; Edwardsi, R.H.; Greene, L.A.; et al. Neuromelanin biosynthesis is driven by excess cytosolic catecholamines not accumulated by synaptic vesicles. Proc. Natl. Acad. Sci. USA 2000, 97, 11869–11874. [Google Scholar] [CrossRef]
- Segura-Aguilar, J.; Paris, I.; Munoz, P.; Ferrari, E.; Zecca, L.; Zucca, F.A. Protective and toxic roles of dopamine in Parkinson’s disease. J. Neurochem. 2014, 129, 898–915. [Google Scholar] [CrossRef]
- Wypijewska, A.; Galazka-Friedman, J.; Bauminger, E.R.; Wszolek, Z.K.; Schweitzer, K.J.; Dickson, D.W.; Jaklewicz, A.; Elbaum, D.; Friedman, A. Iron and reactive oxygen species activity in parkinsonian substantia nigra. Parkinsonism Relat. Disord. 2010, 16, 329–333. [Google Scholar] [CrossRef]
- Friedman, A.; Galazka-Friedman, J. The history of the research of iron in parkinsonian substantia nigra. J. Neural Transm. 2012, 119, 1507–1510. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, H.; Choong, C.J.; Baba, K. Parkinson’s disease and iron. J. Neural Transm. 2020, 127, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Saran, M.; Michel, C.; Stettmaier, K.; Bors, W. Arguments against the significance of the Fenton reaction contributing to signal pathways under in vivo conditions. Free Radic. Res. 2000, 33, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Knörle, R. Neuromelanin in Parkinson’s Disease: From Fenton Reaction to Calcium Signaling. Neurotox. Res. 2018, 33, 515–522. [Google Scholar] [CrossRef]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.A.; Ostaszewski, M.; Matsuoka, Y.; Ghosh, S.; Glaab, E.; Trefois, C.; Crespo, I.; Perumal, T.M.; Jurkowski, W.; Antony, P.M.A.; et al. Integrating pathways of Parkinson’s disease in a molecular interaction map. Mol. Neurobiol. 2014, 49, 88–102. [Google Scholar] [CrossRef]
- Chinta, S.J.; Woods, G.; Demaria, M.; Rane, A.; Zou, Y.; McQuade, A. Cellular senescence is induced by the environmental neurotoxin paraquat and contributes to neuropathology linked to Parkinson’s disease. Cell Rep. 2018, 22, 930–940. [Google Scholar] [CrossRef]
- Vlková, B.; Stanko, P.; Minárik, G.; Tóthová, L.; Szemes, T.; Baňasová, L.; Novotňáková, D.D.; Hodosy, J.; Celec, P. Salivary markers of oxidative stress in patients with oral premalignant lesions. Arch. Oral Biol. 2012, 57, 1651–1656. [Google Scholar] [CrossRef]
- Russo, P.; Cardinale, A.; Margaritora, S.; Cesario, A. Nicotinic receptor and tobacco-related cancer. Life Sci. 2012, 91, 1087–1092. [Google Scholar] [CrossRef]
- Klaunig, J.E. Oxidative Stress and Cancer. Curr. Pharm. Des. 2018, 24, 4771–4778. [Google Scholar] [CrossRef]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.H. Oxidative stress, DNA damage, and breast cancer. AACN Clin. Issues 2002, 13, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Francuz, T.; Czajka-Francuz, P.; Cisoń-Jurek, S.; Wojnar, J. The role of inflammation in colon cancer pathogenesis. Postepy Hig. Med. Dosw. 2016, 70, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Simone, V.; D’Avenia, M.; Argentiero, A.; Felici, C.; Rizzo, F.M.; De Pergola, G.; Silvestris, F. Obesity and Breast Cancer: Molecular Interconnections and Potential Clinical Applications. Oncologist 2016, 21, 404–417. [Google Scholar] [CrossRef] [PubMed]
- Perri, F.; Pisconti, S.; Della Vittoria Scarpati, G. P53 mutations and cancer: A tight linkage. Ann. Transl. Med. 2016, 4, 522. [Google Scholar] [CrossRef] [PubMed]
- Sajadimajd, S.; Khazaei, M. Oxidative Stress and Cancer: The Role of Nrf2. Curr. Cancer Drug Targets 2018, 18, 538–557. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate hypoxic signaling. Curr. Opin. Cell Biol. 2009, 21, 894–899. [Google Scholar] [CrossRef]
- Sporn, M.B.; Liby, K.T. NRF2 and cancer: The good, the bad and the importance of context. Nat. Rev. Cancer. 2012, 12, 564–571. [Google Scholar] [CrossRef]
- Mitsuishi, Y.; Motohashi, H.; Yamamoto, M. The Keap1-Nrf2 system in cancers: Stress response and anabolic metabolism. Front. Oncol. 2012, 2, 200. [Google Scholar] [CrossRef]
- Piskovatska, V.; Strilbytska, O.; Koliada, A.; Vaiserman, A.; Lushchak, O. Health Benefits of Anti-aging Drugs. Subcell Biochem. 2019, 91, 339–392. [Google Scholar] [CrossRef]
- Takaki, A.; Yamamoto, K. Control of oxidative stress in hepatocellular carcinoma: Helpful or harmful? World J. Hepatol. 2015, 7, 968–979. [Google Scholar] [CrossRef] [PubMed]
- Sena, P.; Mancini, S.; Benincasa, M.; Mariani, F.; Palumbo, C.; Roncucci, L. Metformin Induces Apoptosis and Alters Cellular Responses to Oxidative Stress in Ht29 Colon Cancer Cells: Preliminary Findings. Int. J. Mol. Sci. 2018, 19, 1478. [Google Scholar] [CrossRef] [PubMed]
- Queiroz, E.A.A.F.; Puukila, S.; Eichler, R.; Sampaio, S.C.; Forsyth, H.L.; Lees, S.J.; Barbosa, A.M.; Dekker, R.F.H.; Fortes, Z.B.; Khaper, N. Metformin induces apoptosis and cell cycle arrest mediated by oxidative stress, AMPK and FOXO3a in MCF-7 breast cancer cells. PLoS ONE 2014, 9, e98207. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.G.; Piskounova, E.; Morrison, S.J. Cancer, Oxidative Stress, and Metastasis. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 163–175. [Google Scholar] [CrossRef]
- Chikara, S.; Nagaprashantha, L.D.; Singhal, J.; Horne, D.; Awasthi, S.; Singhal, S.S. Oxidative stress and dietary phytochemicals: Role in cancer chemoprevention and treatment. Cancer Lett. 2018, 413, 122–134. [Google Scholar] [CrossRef]
- Prasad, S.; Gupta, S.C.; Pandey, M.K.; Tyagi, A.K.; Deb, L. Oxidative Stress and Cancer: Advances and Challenges. Oxid. Med. Cell Longev. 2016, 2016, 5010423. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |



{kind=link}
{kind=link}
| Mutation/Polymorphism | Type of Neoplasm |
|---|---|
| MC1R polymorphism p.R151C | melanoma |
| LRRK2 G2019S mutation | skin cancer, breast cancer |
| R1441C/G mutation | colon cancer, hematological malignancies |
| LRRK2-PD | non-skin cancer, hormone-related cancers, breast cancer, leukemia |
| PARK1, PARK4 | Lung, intestine, prostate, and ovarian cancers; melanoma; non-Hodgkin’s lymphoma |
| PARK2 | lung, ovary, kidney, and pancreatic cancers; glioma; melanoma |
| PARK6 | glioma, ovarian cancer |
| PARK7 | breast, lung, pancreatic, stomach, and prostate cancers |
| PARK1/4, PINK1, PARK9 (ATP13A2) | brain tumors |
| DR polymorphisms | gastric, colorectal, and non-small-cell lung cancers; |
| increased expression of dopamine D2 receptors | gastric cancer; breast cancer; neuroendocrine tumors; glioma |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ejma, M.; Madetko, N.; Brzecka, A.; Guranski, K.; Alster, P.; Misiuk-Hojło, M.; Somasundaram, S.G.; Kirkland, C.E.; Aliev, G. The Links between Parkinson’s Disease and Cancer. Biomedicines 2020, 8, 416. https://doi.org/10.3390/biomedicines8100416
Ejma M, Madetko N, Brzecka A, Guranski K, Alster P, Misiuk-Hojło M, Somasundaram SG, Kirkland CE, Aliev G. The Links between Parkinson’s Disease and Cancer. Biomedicines. 2020; 8(10):416. https://doi.org/10.3390/biomedicines8100416
Chicago/Turabian StyleEjma, Maria, Natalia Madetko, Anna Brzecka, Konstanty Guranski, Piotr Alster, Marta Misiuk-Hojło, Siva G. Somasundaram, Cecil E. Kirkland, and Gjumrakch Aliev. 2020. "The Links between Parkinson’s Disease and Cancer" Biomedicines 8, no. 10: 416. https://doi.org/10.3390/biomedicines8100416
APA StyleEjma, M., Madetko, N., Brzecka, A., Guranski, K., Alster, P., Misiuk-Hojło, M., Somasundaram, S. G., Kirkland, C. E., & Aliev, G. (2020). The Links between Parkinson’s Disease and Cancer. Biomedicines, 8(10), 416. https://doi.org/10.3390/biomedicines8100416