Interplay between Endoplasmic Reticulum Stress and Large Extracellular Vesicles (Microparticles) in Endothelial Cell Dysfunction



Abstract
1. Introduction
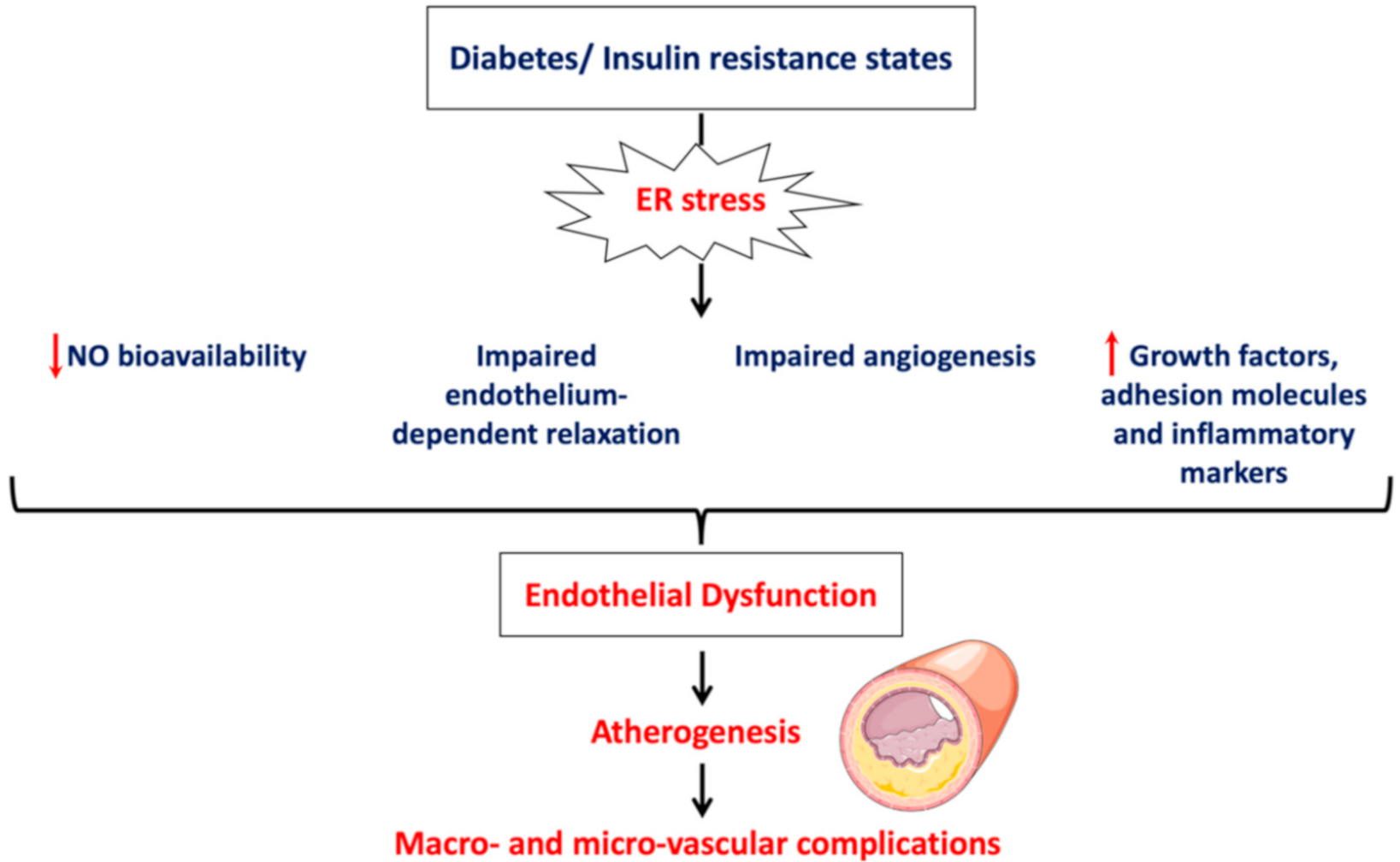
2. Physiology of Endothelium and Endothelial Cell Dysfunction
- Impairment of vascular function indicated by reduced NO bioavailability, impaired endothelium-depended smooth muscle relaxation and perturbated angiogenesis;
- Induction of vascular inflammation marked by increased production of inflammatory mediators and adhesion molecules;
- Activation of a prothrombotic state by increasing production of procoagulant factors and enhancing platelet aggregation.
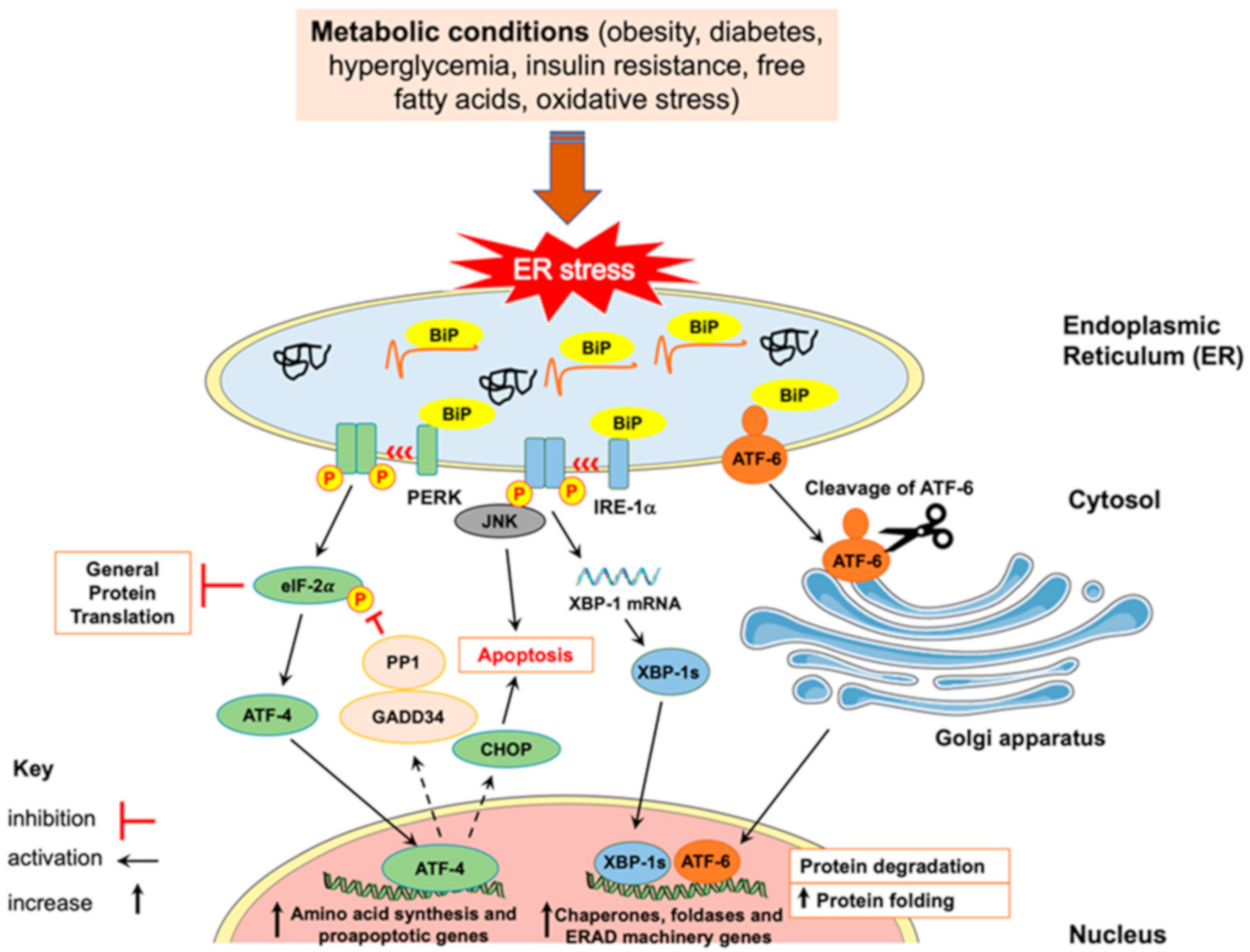
3. Endoplasmic Reticulum (ER) Stress
- a.
- After its dissociation from BiP, PERK undergoes dimerization and autophosphorylation, then it phosphorylates eukaryotic initiation factor (eIF)-2α at the alpha subunit. Phosphorylated eIF-2α reduces the general translation of mRNA in order to alleviate the load on the ER; however, it allows, selectively, the translation of ATF-4 mRNA. ATF-4, as a transcription factor, stimulates the expression of genes encoding proteins involved in amino acid biosynthesis and cell death such as tribbles homolog 3 (TRIB3) and CCAAT/enhancer binding proteins (C/EBP) homologous protein (CHOP), respectively. ATF-4 also controls the negative feedback of PERK activation by enhancing the expression of growth arrest and DNA damage inducible protein (GADD34), which leads to the dephosphorylation of eIF-2α by protein phosphatase 1 (PP1).
- b.
- IRE-1α endoribonuclease activity is mediated by its dimerization and autophosphorylation. Activated IRE-1α splices X box-binding protein 1 (XBP-1) mRNA to form active spliced form (XBP-1s), which then translocates into the nucleus and promotes the transcription of genes encoding ER-associated degradation (ERAD) proteins and ER chaperones. Upon prolonged ER stress, IRE-1α can activate inflammatory and apoptotic pathways. For example, IRE-1α can interact with tumor necrosis factor (TNF) receptor-associated factor (TRAF)-2 to promote activation of nuclear factor κB (NF-κB), which is involved in the upregulation of inflammatory genes. Additionally, IRE-1α/TRAF-2 can activate c-Jun-NH2-terminal kinase (JNK), which was shown to induce insulin resistance, inflammation and apoptosis. Additionally, TRAF-2 stimulates apoptosis-signaling-kinase 1 (ASK-1), which can enhance the activity of p38 mitogen activated protein kinase (MAPK), which can result in the activation of proapoptotic CHOP.
- c.
- After release from BiP, ATF-6 translocates into the Golgi apparatus to be cleaved by proteases, site-1 protease (S1P) and site-2 protease (S2P). Then, the transcriptionally active cytosolic domain of ATF-6 is released and translocates into the nucleus to trigger the transcription of genes encoding for proteins involved in degradation of irreversibly misfolded proteins (ERAD machinery proteins) and folding of proteins (ER chaperones such as BiP and GRP94).
3.1. ER Stress-Mediated Inflammation and Apoptosis
3.2. ER Stress-Mediated Oxidative Stress
3.3. ER Stress-Mediated Autophagy
3.4. ER Stress-Mediated Insulin Resistance
4. ER Stress and Endothelial Dysfunction
5. Extracellular Vesicles (EVs)
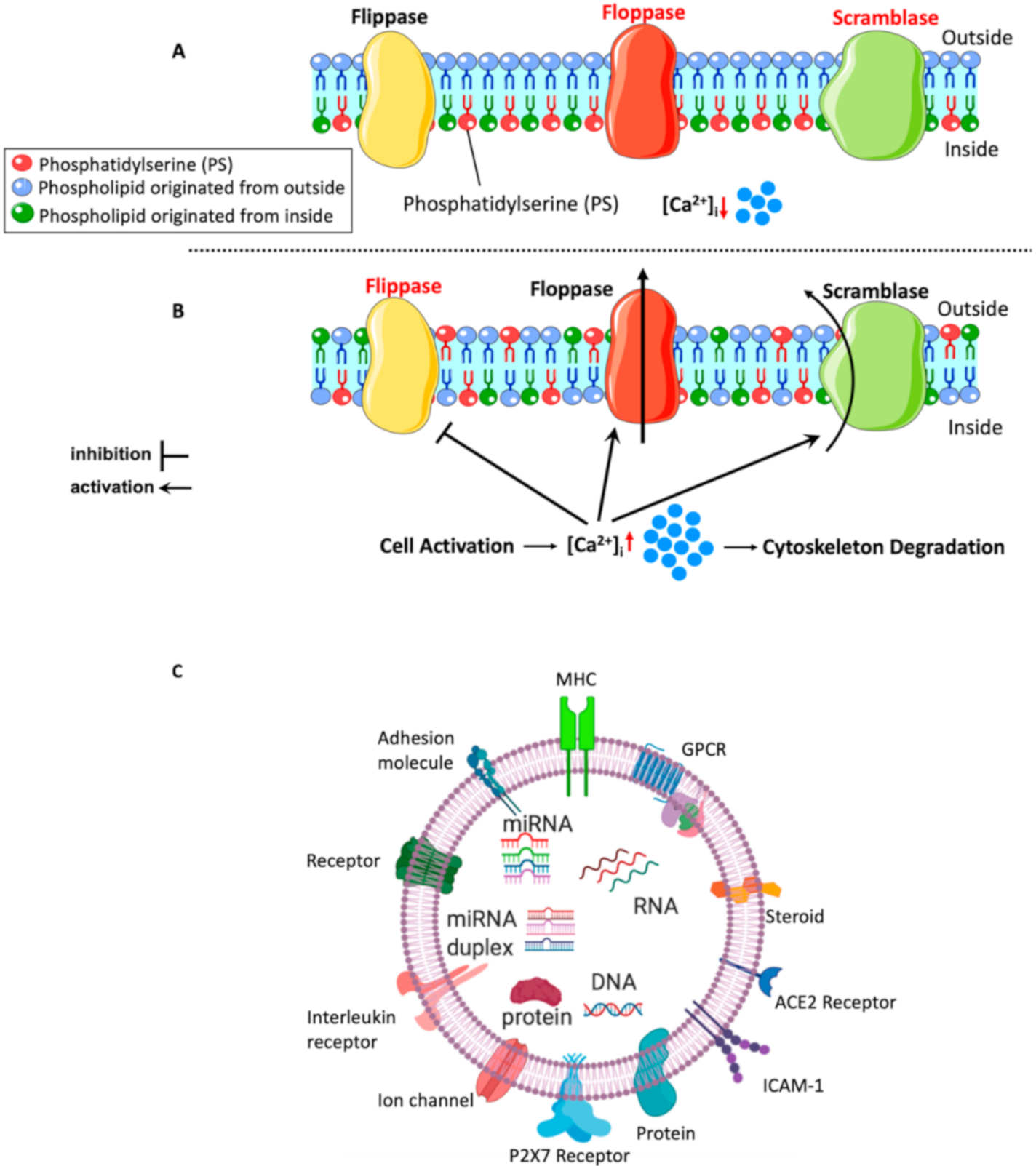
5.1. Mechanisms of lEVs (MPs) Formation
5.2. Content of lEVs
5.3. Mechanisms of lEVs (MPs) Interaction with Target Cells
6. lEVs (MPs) and Endothelial Dysfunction
7. Crosstalk between ER Stress and lEVs (MPs) in Endothelial Dysfunction
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Heijnen, H.F.G.; Schiel, A.E.; Fijnheer, R.; Geuze, H.J.; Sixma, J.J. Activated Platelets Release Two Types of Membrane Vesicles: Microvesicles by Surface Shedding and Exosomes Derived From Exocytosis of Multivesicular Bodies and alpha-Granules. Blood 1999, 94, 3791. [Google Scholar] [CrossRef]
- Théry, C.; Boussac, M.; Véron, P.; Ricciardi-Castagnoli, P.; Raposo, G.; Garin, J.; Amigorena, S. Proteomic Analysis of Dendritic Cell-Derived Exosomes: A Secreted Subcellular Compartment Distinct from Apoptotic Vesicles. J. Immunol. 2001, 166, 7309. [Google Scholar] [CrossRef] [PubMed]
- Zaborowski, M.P.; Balaj, L.; Breakefield, X.O.; Lai, C.P. Extracellular Vesicles: Composition, Biological Relevance, and Methods of Study. Bioscience 2015, 65, 783–797. [Google Scholar] [CrossRef] [PubMed]
- Flamment, M.; Hajduch, E.; Ferré, P.; Foufelle, F. New insights into ER stress-induced insulin resistance. Trends Endocrinol. Metab. 2012, 23, 381–390. [Google Scholar] [CrossRef]
- Maamoun, H.; Abdelsalam, S.S.; Zeidan, A.; Korashy, H.M.; Agouni, A. Endoplasmic Reticulum Stress: A Critical Molecular Driver of Endothelial Dysfunction and Cardiovascular Disturbances Associated with Diabetes. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef]
- Maamoun, H.; Benameur, T.; Pintus, G.; Munusamy, S.; Agouni, A. Crosstalk Between Oxidative Stress and Endoplasmic Reticulum (ER) Stress in Endothelial Dysfunction and Aberrant Angiogenesis Associated With Diabetes: A Focus on the Protective Roles of Heme Oxygenase (HO)-1. Front. Physiol. 2019, 10, 70. [Google Scholar] [CrossRef]
- Lenna, S.; Han, R.; Trojanowska, M. Endoplasmic reticulum stress and endothelial dysfunction. IUBMB Life 2014, 66, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Agouni, A.; Andriantsitohaina, R.; Martinez, M.C. Microparticles as biomarkers of vascular dysfunction in metabolic syndrome and its individual components. Curr Vasc Pharm. 2014, 12, 483–492. [Google Scholar] [CrossRef]
- El-Gamal, H.; Parray, A.S.; Mir, F.A.; Shuaib, A.; Agouni, A. Circulating microparticles as biomarkers of stroke: A focus on the value of endothelial- and platelet-derived microparticles. J. Cell Physiol. 2019, 234, 16739–16754. [Google Scholar] [CrossRef]
- Benameur, T.; Osman, A.; Parray, A.; Ait Hssain, A.; Munusamy, S.; Agouni, A. Molecular Mechanisms Underpinning Microparticle-Mediated Cellular Injury in Cardiovascular Complications Associated with Diabetes. Oxidative Med. Cellular Longev. 2019, 2019. [Google Scholar] [CrossRef]
- Safiedeen, Z.; Rodríguez-Gómez, I.; Vergori, L.; Soleti, R.; Vaithilingam, D.; Douma, I.; Agouni, A.; Leiber, D.; Dubois, S.; Simard, G.; et al. Temporal Cross Talk Between Endoplasmic Reticulum and Mitochondria Regulates Oxidative Stress and Mediates Microparticle-Induced Endothelial Dysfunction. Antioxid. Redox Signal. 2016, 26, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Agouni, A.; Osman, A.; El Gamal, H.; Pasha, M. Endoplasmic Reticulum (ER) stress-generated microparticles self-perpetuate ER stress and mediate endothelial cell dysfunction independently of cell survival. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Galley, H.F.; Webster, N.R. Physiology of the endothelium. Br. J. Anaesth. 2004, 93, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Palmer, R.M.J.; Ashton, D.S.; Moncada, S. Vascular endothelial cells synthesize nitric oxide from L-arginine. Nature 1988, 333, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Lincoln, T.M.; Komalavilas, P.; Cornwell, T.L. Pleiotropic regulation of vascular smooth muscle tone by cyclic GMP-dependent protein kinase. Hypertension 1994, 23, 1141–1147. [Google Scholar] [CrossRef]
- Mendelsohn, M.E.; O’Neill, S.; George, D.; Loscalzo, J. Inhibition of fibrinogen binding to human platelets by S-nitroso-N-acetylcysteine. J. Biol. Chem. 1990, 265, 19028–19034. [Google Scholar]
- Behrendt, D.; Ganz, P. Endothelial function: From vascular biology to clinical applications. Am. J. Cardiol. 2002, 90, L40–L48. [Google Scholar] [CrossRef]
- Marks, D.S.; Vita, J.A.; Folts, J.D.; Keaney, J.F., Jr.; Welch, G.N.; Loscalzo, J. Inhibition of neointimal proliferation in rabbits after vascular injury by a single treatment with a protein adduct of nitric oxide. J. Clin. Investig. 1995, 96, 2630–2638. [Google Scholar] [CrossRef]
- Garg, U.C.; Hassid, A. Nitric oxide-generating vasodilators and 8-bromo-cyclic guanosine monophosphate inhibit mitogenesis and proliferation of cultured rat vascular smooth muscle cells. J. Clin. Investig. 1989, 83, 1774–1777. [Google Scholar] [CrossRef]
- Kota, S.K.; Meher, L.K.; Jammula, S.; Kota, S.K.; Krishna, S.V.S.; Modi, K.D. Aberrant angiogenesis: The gateway to diabetic complications. Indian J. Endocrinol. Metab. 2012, 16, 918–930. [Google Scholar] [CrossRef]
- Cade, W.T. Diabetes-related microvascular and macrovascular diseases in the physical therapy setting. Phys. Ther. 2008, 88, 1322–1335. [Google Scholar] [CrossRef] [PubMed]
- Sheikh-Ali, M.; Sultan, S.; Alamir, A.-R.; Haas, M.J.; Mooradian, A.D. Hyperglycemia-induced endoplasmic reticulum stress in endothelial cells. Nutrition 2010, 26, 1146–1150. [Google Scholar] [CrossRef] [PubMed]
- Battson, M.L.; Lee, D.M.; Gentile, C.L. Endoplasmic reticulum stress and the development of endothelial dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H355–H367. [Google Scholar] [CrossRef] [PubMed]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of Stress in the ER to Activation of JNK Protein Kinases by Transmembrane Protein Kinase IRE1. Science 2000, 287, 664. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; Brenner, D.A.; Karin, M. A liver full of JNK: Signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology 2012, 143, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Tan, M.; Hu, Y.; Wang, J.-L.; Scheuner, D.; Kaufman, R.J. Ultraviolet Light Activates NFκB through Translational Inhibition of IκBα Synthesis. J. Biol. Chem. 2004, 279, 34898–34902. [Google Scholar] [CrossRef] [PubMed]
- Pahl, H.L.; Baeuerle, P.A. A novel signal transduction pathway from the endoplasmic reticulum to the nucleus is mediated by transcription factor NF-kappa B. EMBO J. 1995, 14, 2580–2588. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Hiramatsu, N.; Hayakawa, K.; Tagawa, Y.; Okamura, M.; Ogata, R.; Huang, T.; Nakajima, S.; Yao, J.; Paton, A.W.; et al. Activation of the Akt-NF-kappaB pathway by subtilase cytotoxin through the ATF6 branch of the unfolded protein response. J. Immunol. 2009, 183, 1480–1487. [Google Scholar] [CrossRef]
- Maamoun, H.; Zachariah, M.; McVey, J.H.; Green, F.R.; Agouni, A. Heme oxygenase (HO)-1 induction prevents Endoplasmic Reticulum stress-mediated endothelial cell death and impaired angiogenic capacity. Biochem. Pharmacol. 2017, 127, 46–59. [Google Scholar] [CrossRef]
- Kamiya, T.; Hara, H.; Adachi, T. Effect of endoplasmic reticulum (ER) stress inducer thapsigargin on the expression of extracellular-superoxide dismutase in mouse 3T3-L1 adipocytes. J. Clin. Biochem. Nutr. 2013, 52, 101–105. [Google Scholar] [CrossRef]
- Landmesser, U.; Spiekermann, S.; Dikalov, S.; Tatge, H.; Wilke, R.; Kohler, C.; Harrison David, G.; Hornig, B.; Drexler, H. Vascular Oxidative Stress and Endothelial Dysfunction in Patients With Chronic Heart Failure. Circulation 2002, 106, 3073–3078. [Google Scholar] [CrossRef] [PubMed]
- Ogata, M.; Hino, S.-i.; Saito, A.; Morikawa, K.; Kondo, S.; Kanemoto, S.; Murakami, T.; Taniguchi, M.; Tanii, I.; Yoshinaga, K.; et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Molecular Cellular Biol. 2006, 26, 9220–9231. [Google Scholar] [CrossRef] [PubMed]
- Hosoi, T.; Nomura, J.; Tanaka, K.; Ozawa, K.; Nishi, A.; Nomura, Y. Link between endoplasmic reticulum stress and autophagy in neurodegenerative diseases. Endoplasmic Reticulum Stress Dis. 2017, 4, 37. [Google Scholar] [CrossRef]
- Yang, J.; Yao, S. JNK-Bcl-2/Bcl-xL-Bax/Bak Pathway Mediates the Crosstalk between Matrine-Induced Autophagy and Apoptosis via Interplay with Beclin 1. Int. J. Mol. Sci. 2015, 16, 25744–25758. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Farrokhi, F.R.; Butler, A.E.; Sahebkar, A. Insulin resistance: Review of the underlying molecular mechanisms. J. Cell. Physiol. 2019, 234, 8152–8161. [Google Scholar] [CrossRef] [PubMed]
- Abdelsalam, S.S.; Korashy, H.M.; Zeidan, A.; Agouni, A. The Role of Protein Tyrosine Phosphatase (PTP)-1B in Cardiovascular Disease and Its Interplay with Insulin Resistance. Biomolecules 2019, 9. [Google Scholar] [CrossRef]
- Potenza, M.A.; Gagliardi, S.; Nacci, C.; Carratu, M.R.; Montagnani, M. Endothelial dysfunction in diabetes: From mechanisms to therapeutic targets. Curr. Med. Chem. 2009, 16, 94–112. [Google Scholar] [CrossRef]
- Okon, E.B.; Chung, A.W.; Rauniyar, P.; Padilla, E.; Tejerina, T.; McManus, B.M.; Luo, H.; van Breemen, C. Compromised arterial function in human type 2 diabetic patients. Diabetes 2005, 54, 2415–2423. [Google Scholar] [CrossRef]
- Duncan, E.R.; Crossey, P.A.; Walker, S.; Anilkumar, N.; Poston, L.; Douglas, G.; Ezzat, V.A.; Wheatcroft, S.B.; Shah, A.M.; Kearney, M.T. Effect of endothelium-specific insulin resistance on endothelial function in vivo. Diabetes 2008, 57, 3307–3314. [Google Scholar] [CrossRef]
- Zhou, J.; Lhoták, Š.; Hilditch Brooke, A.; Austin Richard, C. Activation of the Unfolded Protein Response Occurs at All Stages of Atherosclerotic Lesion Development in Apolipoprotein E–Deficient Mice. Circulation 2005, 111, 1814–1821. [Google Scholar] [CrossRef]
- Villalobos-Labra, R.; Sáez, P.J.; Subiabre, M.; Silva, L.; Toledo, F.; Westermeier, F.; Pardo, F.; Farías, M.; Sobrevia, L. Pre-pregnancy maternal obesity associates with endoplasmic reticulum stress in human umbilical vein endothelium. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2018, 1864, 3195–3210. [Google Scholar] [CrossRef]
- Di Pietro, N.; Marcovecchio, M.L.; Di Silvestre, S.; de Giorgis, T.; Cordone, V.G.P.; Lanuti, P.; Chiarelli, F.; Bologna, G.; Mohn, A.; Pandolfi, A. Plasma from pre-pubertal obese children impairs insulin stimulated Nitric Oxide (NO) bioavailability in endothelial cells: Role of ER stress. Mol. Cell. Endocrinol. 2017, 443, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Seals, D.R.; Chung, E.; Kaplon, R.E.; Cox-York, K.; Reese, L.; Gentile, C.L. Activation of the Unfolded Protein Response in Vascular Endothelial Cells of Nondiabetic Obese Adults. J. Clin. Endocrinol. Metab. 2013, 98, E1505–E1509. [Google Scholar] [CrossRef]
- Bhatta, M.; Ma, J.H.; Wang, J.J.; Sakowski, J.; Zhang, S.X. Enhanced endoplasmic reticulum stress in bone marrow angiogenic progenitor cells in a mouse model of long-term experimental type 2 diabetes. Diabetologia 2015, 58, 2181–2190. [Google Scholar] [CrossRef] [PubMed]
- Suganya, N.; Dornadula, S.; Chatterjee, S.; Mohanram, R.K. Quercetin improves endothelial function in diabetic rats through inhibition of endoplasmic reticulum stress-mediated oxidative stress. Eur. J. Pharmacol. 2018, 819, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Liu, X.; Feng, L.; Yang, H.; Yu, W.; Feng, T.; Wang, S.; Wang, J.; Liu, N. Glycation of paraoxonase 1 by high glucose instigates endoplasmic reticulum stress to induce endothelial dysfunction in vivo. Sci. Rep. 2017, 7, 45827. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-K.; Lim, M.; Yeon, S.-I.; Lee, Y.-H. Inhibition of endoplasmic reticulum stress improves coronary artery function in type 2 diabetic mice. Exp. Physiol. 2016, 101, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Sarvani, C.; Sireesh, D.; Ramkumar, K.M. Unraveling the role of ER stress inhibitors in the context of metabolic diseases. Pharm. Res. 2017, 119, 412–421. [Google Scholar] [CrossRef]
- Yang, Y.; Zhou, Q.; Gao, A.; Chen, L.; Li, L. Endoplasmic reticulum stress and focused drug discovery in cardiovascular disease. Clin. Chim. Acta 2020, 504, 125–137. [Google Scholar] [CrossRef]
- Zhang, Z.; Tong, N.; Gong, Y.; Qiu, Q.; Yin, L.; Lv, X.; Wu, X. Valproate protects the retina from endoplasmic reticulum stress-induced apoptosis after ischemia-reperfusion injury. Neurosci. Lett. 2011, 504, 88–92. [Google Scholar] [CrossRef]
- Tong, Q.; Wu, L.; Jiang, T.; Ou, Z.; Zhang, Y.; Zhu, D. Inhibition of endoplasmic reticulum stress-activated IRE1alpha-TRAF2-caspase-12 apoptotic pathway is involved in the neuroprotective effects of telmisartan in the rotenone rat model of Parkinson’s disease. Eur. J. Pharmacol. 2016, 776, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Lakshmanan, A.P.; Thandavarayan, R.A.; Palaniyandi, S.S.; Sari, F.R.; Meilei, H.; Giridharan, V.V.; Soetikno, V.; Suzuki, K.; Kodama, M.; Watanabe, K. Modulation of AT-1R/CHOP-JNK-Caspase12 pathway by olmesartan treatment attenuates ER stress-induced renal apoptosis in streptozotocin-induced diabetic mice. Eur. J. Pharm. Sci. 2011, 44, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhao, L.; Zhang, Z.; Lu, X. Protective Effect of Enalapril against Methionine-Enriched Diet-Induced Hypertension: Role of Endoplasmic Reticulum and Oxidative Stress. Biomed. Res. Int. 2015, 2015, 724876. [Google Scholar] [CrossRef] [PubMed]
- Ai, W.; Wu, M.; Chen, L.; Jiang, B.; Mu, M.; Liu, L.; Yuan, Z. Ghrelin ameliorates atherosclerosis by inhibiting endoplasmic reticulum stress. Fundam. Clin. Pharmacol. 2017, 31, 147–154. [Google Scholar] [CrossRef]
- Ghosh, R.; Wang, L.; Wang, E.S.; Perera, B.G.; Igbaria, A.; Morita, S.; Prado, K.; Thamsen, M.; Caswell, D.; Macias, H.; et al. Allosteric inhibition of the IRE1alpha RNase preserves cell viability and function during endoplasmic reticulum stress. Cell 2014, 158, 534–548. [Google Scholar] [CrossRef]
- Lee, E.S.; Kim, H.M.; Kang, J.S.; Lee, E.Y.; Yadav, D.; Kwon, M.H.; Kim, Y.M.; Kim, H.S.; Chung, C.H. Oleanolic acid and N-acetylcysteine ameliorate diabetic nephropathy through reduction of oxidative stress and endoplasmic reticulum stress in a type 2 diabetic rat model. Nephrol Dial Transpl. 2016, 31, 391–400. [Google Scholar] [CrossRef]
- Natsume, Y.; Ito, S.; Satsu, H.; Shimizu, M. Protective effect of quercetin on ER stress caused by calcium dynamics dysregulation in intestinal epithelial cells. Toxicology 2009, 258, 164–175. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Surgucheva, I.; Sharov, V.S.; Surguchov, A. gamma-Synuclein: Seeding of alpha-synuclein aggregation and transmission between cells. Biochemistry 2012, 51, 4743–4754. [Google Scholar] [CrossRef]
- van der Pol, E.; Böing, A.N.; Gool, E.L.; Nieuwland, R. Recent developments in the nomenclature, presence, isolation, detection and clinical impact of extracellular vesicles. J. Thromb. Haemost. 2016, 14, 48–56. [Google Scholar] [CrossRef]
- van der Pol, E.; Coumans, F.A.W.; Grootemaat, A.E.; Gardiner, C.; Sargent, I.L.; Harrison, P.; Sturk, A.; van Leeuwen, T.G.; Nieuwland, R. Particle size distribution of exosomes and microvesicles determined by transmission electron microscopy, flow cytometry, nanoparticle tracking analysis, and resistive pulse sensing. J. Thromb. Haemost. 2014, 12, 1182–1192. [Google Scholar] [CrossRef] [PubMed]
- Henseleit, U.; Plasa, G.; Haest, C. Effects of divalent cations on lipid flip-flop in the human erythrocyte membrane. Biochim. Biophys. Acta (BBA)-Biomembr. 1990, 1029, 127–135. [Google Scholar] [CrossRef]
- Cauwenberghs, S.; Feijge, M.A.H.; Harper, A.G.S.; Sage, S.O.; Curvers, J.; Heemskerk, J.W.M. Shedding of procoagulant microparticles from unstimulated platelets by integrin-mediated destabilization of actin cytoskeleton. FEBS Lett. 2006, 580, 5313–5320. [Google Scholar] [CrossRef] [PubMed]
- Flaumenhaft, R.; Dilks, J.R.; Richardson, J.; Alden, E.; Patel-Hett, S.R.; Battinelli, E.; Klement, G.L.; Sola-Visner, M.; Italiano, J.E., Jr. Megakaryocyte-derived microparticles: Direct visualization and distinction from platelet-derived microparticles. Blood 2009, 113, 1112–1121. [Google Scholar] [CrossRef] [PubMed]
- Yano, Y.; Shiba, E.; Kambayashi, J.-i.; Sakon, M.; Kawasaki, T.; Fujitani, K.; Kang, J.; Mori, T. The effects of calpeptin (a calpain specific inhibitor) on agonist induced microparticle formation from the platelet plasma membrane. Thromb. Res. 1993, 71, 385–396. [Google Scholar] [CrossRef]
- Nolan, S.; Dixon, R.; Norman, K.; Hellewell, P.; Ridger, V. Nitric oxide regulates neutrophil migration through microparticle formation. Am. J. Pathol. 2008, 172, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Burger, D.; Montezano Augusto, C.; Nishigaki, N.; He, Y.; Carter, A.; Touyz Rhian, M. Endothelial Microparticle Formation by Angiotensin II Is Mediated via Ang II Receptor Type I/NADPH Oxidase/Rho Kinase Pathways Targeted to Lipid Rafts. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1898–1907. [Google Scholar] [CrossRef]
- van den Akker, J.; van Weert, A.; Afink, G.; Bakker, E.N.T.P.; van der Pol, E.; Böing, A.N.; Nieuwland, R.; VanBavel, E. Transglutaminase 2 is secreted from smooth muscle cells by transamidation-dependent microparticle formation. Amino acids 2012, 42, 961–973. [Google Scholar] [CrossRef][Green Version]
- Bevers, E.M.; Comfurius, P.; Dekkers, D.W.C.; Zwaal, R.F.A. Lipid translocation across the plasma membrane of mammalian cells. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 1999, 1439, 317–330. [Google Scholar] [CrossRef]
- Suzuki, J.; Umeda, M.; Sims, P.J.; Nagata, S. Calcium-dependent phospholipid scrambling by TMEM16F. Nature 2010, 468, 834. [Google Scholar] [CrossRef]
- Connor, D.E.; Exner, T.; Ma, D.D.F.; Joseph, J.E. The majority of circulating platelet-derived microparticles fail to bind annexin V, lack phospholipid-dependent procoagulant activity and demonstrate greater expression of glycoprotein Ib. Thromb Haemost. 2010, 103, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Banfi, C.; Brioschi, M.; Wait, R.; Begum, S.; Gianazza, E.; Pirillo, A.; Mussoni, L.; Tremoli, E. Proteome of endothelial cell-derived procoagulant microparticles. Proteomics 2005, 5, 4443–4455. [Google Scholar] [CrossRef] [PubMed]
- Palmisano, G.; Jensen, S.S.; Le Bihan, M.-C.; Lainé, J.; McGuire, J.N.; Pociot, F.; Larsen, M.R. Characterization of membrane-shed microvesicles from cytokine-stimulated β-cells using proteomics strategies. Mol. Cell. Proteom. 2012, 11, 230–243. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.-D.; Wu, X.-Z.; Zhou, Y.; Xue, Y.; Zhang, K.-Q. Proteomic characteristics of circulating microparticles in patients with newly-diagnosed type 2 diabetes. Am. J. Transl. Res. 2016, 8, 209–220. [Google Scholar] [PubMed]
- Fan, G.-Q.; Qin, R.-R.; Li, Y.-H.; Song, D.-J.; Chen, T.-S.; Zhang, W.; Zhong, M.; Zhang, Y.; Xing, Y.-Q.; Wang, Z.-H. Endothelial cells microparticle-associated protein disulfide isomerase promotes platelet activation in metabolic syndrome. Oncotarget 2016, 7, 83231–83240. [Google Scholar] [CrossRef]
- Bicalho, B.; Holovati, J.L.; Acker, J.P. Phospholipidomics reveals differences in glycerophosphoserine profiles of hypothermically stored red blood cells and microvesicles. Biochim. Biophys. Acta (BBA)-Biomembr. 2013, 1828, 317–326. [Google Scholar] [CrossRef]
- Wolf, P. The Nature and Significance of Platelet Products in Human Plasma. Br. J. Haematol. 1967, 13, 269–288. [Google Scholar] [CrossRef]
- Sinauridze, E.I.; Kireev, D.A.; Popenko, N.Y.; Pichugin, A.V.; Panteleev, M.A.; Krymskaya, O.V.; Ataullakhanov, F.I. Platelet microparticle membranes have 50- to 100-fold higher specific procoagulant activity than activated platelets. Thromb Haemost. 2007, 97, 425–434. [Google Scholar]
- Diehl, P.; Fricke, A.; Sander, L.; Stamm, J.; Bassler, N.; Htun, N.; Ziemann, M.; Helbing, T.; El-Osta, A.; Jowett, J.B.M.; et al. Microparticles: Major transport vehicles for distinct microRNAs in circulation. Cardiovasc. Res. 2012, 93, 633–644. [Google Scholar] [CrossRef]
- Tual-Chalot, S.; Leonetti, D.; Andriantsitohaina, R.; Martínez, M.C. Microvesicles: Intercellular vectors of biological messages. Mol. Interv. 2011, 11, 88–94. [Google Scholar] [CrossRef]
- Panaro, M.A.; Benameur, T.; Porro, C. Extracellular Vesicles miRNA Cargo for Microglia Polarization in Traumatic Brain Injury. Biomolecules 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.-S.; Zhang, H.-G.; Zhang, Q.-J.; Xiu, R.-J. CD51+ endothelial microparticles as a biomarker of endothelial dysfunction in obese patients with hypertension. Endocrine 2015, 49, 283–285. [Google Scholar] [CrossRef] [PubMed]
- Gündüz, Z.; Dursun, İ; Tülpar, S.; Baştuğ, F.; Baykan, A.; Yıkılmaz, A.; Patıroğlu, T.; Poyrazoglu Hakan, M.; Akın, L.; Yel, S.; et al. Increased endothelial microparticles in obese and overweight children. J. Pediatric Endocrinol. Metab. 2012, 25, 1111. [Google Scholar]
- Schisano, B.; Giannetti, G.; Sardelli, L.; Ciotola, M.; Gualdiero, R.; Giugliano, D.; Esposito, K.; Misso, L. Endothelial Microparticles Correlate with Endothelial Dysfunction in Obese Women. J. Clin. Endocrinol. Metab. 2006, 91, 3676–3679. [Google Scholar] [CrossRef]
- Brodsky, S.V.; Zhang, F.; Nasjletti, A.; Goligorsky, M.S. Endothelium-derived microparticles impair endothelial function in vitro. Am. J. Physiol. -Heart Circ. Physiol. 2004, 286, H1910–H1915. [Google Scholar] [CrossRef]
- Martin, S.; Tesse, A.; Hugel, B.; Martínez, M.C.; Morel, O.; Freyssinet, J.-M.; Andriantsitohaina, R. Shed Membrane Particles From T Lymphocytes Impair Endothelial Function and Regulate Endothelial Protein Expression. Circulation 2004, 109, 1653–1659. [Google Scholar] [CrossRef]
- Feng, B.; Chen, Y.; Luo, Y.; Chen, M.; Li, X.; Ni, Y. Circulating level of microparticles and their correlation with arterial elasticity and endothelium-dependent dilation in patients with type 2 diabetes mellitus. Atherosclerosis 2010, 208, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Stork, C.J.; Xu, S.; Yuan, D.; Xia, X.; LaPenna, K.B.; Guo, G.; Sun, H.; Xu, L.-C.; Siedlecki, C.A.; et al. Increased circulating microparticles in streptozotocin-induced diabetes propagate inflammation contributing to microvascular dysfunction. J. Physiol. 2019, 597, 781–798. [Google Scholar] [CrossRef]
- Ishida, K.; Taguchi, K.; Hida, M.; Watanabe, S.; Kawano, K.; Matsumoto, T.; Hattori, Y.; Kobayashi, T. Circulating microparticles from diabetic rats impair endothelial function and regulate endothelial protein expression. Acta Physiol. 2016, 216, 211–220. [Google Scholar] [CrossRef]
- Jansen, F.; Yang, X.; Hoelscher, M.; Cattelan, A.; Schmitz, T.; Proebsting, S.; Wenzel, D.; Vosen, S.; Franklin Bernardo, S.; Fleischmann Bernd, K.; et al. Endothelial Microparticle–Mediated Transfer of MicroRNA-126 Promotes Vascular Endothelial Cell Repair via SPRED1 and Is Abrogated in Glucose-Damaged Endothelial Microparticles. Circulation 2013, 128, 2026–2038. [Google Scholar] [CrossRef]
- Giannella, A.; Radu, C.M.; Franco, L.; Campello, E.; Simioni, P.; Avogaro, A.; de Kreutzenberg, S.V.; Ceolotto, G. Circulating levels and characterization of microparticles in patients with different degrees of glucose tolerance. Cardiovasc. Diabetol. 2017, 16, 118. [Google Scholar] [CrossRef] [PubMed]
- Burger, D.; Turner, M.; Xiao, F.; Munkonda, M.N.; Akbari, S.; Burns, K.D. High glucose increases the formation and pro-oxidative activity of endothelial microparticles. Diabetologia 2017, 60, 1791–1800. [Google Scholar] [CrossRef]
- Saez, T.; Salsoso, R.; Leiva, A.; Toledo, F.; de Vos, P.; Faas, M.; Sobrevia, L. Human umbilical vein endothelium-derived exosomes play a role in foetoplacental endothelial dysfunction in gestational diabetes mellitus. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Jing, T.; Ya-Shu, K.; Xue-Jun, W.; Han-Jing, H.; Yan, L.; Yi-An, Y.; Fei, C.; Xue-Bo, L. Sirt6 mRNA-incorporated endothelial microparticles (EMPs) attenuates DM patient-derived EMP-induced endothelial dysfunction. Oncotarget 2017, 8, 114300–114313. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Alberti, K.G.M.M.; Eckel Robert, H.; Grundy Scott, M.; Zimmet Paul, Z.; Cleeman James, I.; Donato Karen, A.; Fruchart, J.-C.; James, W.P.T.; Loria Catherine, M.; Smith Sidney, C. Harmonizing the Metabolic Syndrome. Circulation 2009, 120, 1640–1645. [Google Scholar] [CrossRef]
- Arteaga, R.B.; Chirinos, J.A.; Soriano, A.O.; Jy, W.; Horstman, L.; Jimenez, J.J.; Mendez, A.; Ferreira, A.; de Marchena, E.; Ahn, Y.S. Endothelial Microparticles and Platelet and Leukocyte Activation in Patients With the Metabolic Syndrome. Am. J. Cardiol. 2006, 98, 70–74. [Google Scholar] [CrossRef]
- Agouni, A.; Lagrue-Lak-Hal, A.H.; Ducluzeau, P.H.; Mostefai, H.A.; Draunet-Busson, C.; Leftheriotis, G.; Heymes, C.; Martinez, M.C.; Andriantsitohaina, R. Endothelial dysfunction caused by circulating microparticles from patients with metabolic syndrome. Am. J. Pathol. 2008, 173, 1210–1219. [Google Scholar] [CrossRef]
- Zhang, X.; McGeoch, S.C.; Johnstone, A.M.; Holtrop, G.; Sneddon, A.A.; MacRury, S.M.; Megson, I.L.; Pearson, D.W.M.; Abraham, P.; De Roos, B.; et al. Platelet-derived microparticle count and surface molecule expression differ between subjects with and without type 2 diabetes, independently of obesity status. J. Thromb. Thrombolysis 2014, 37, 455–463. [Google Scholar] [CrossRef]
- Jia, L.-X.; Zhang, W.-M.; Li, T.-T.; Liu, Y.; Piao, C.-M.; Ma, Y.-C.; Lu, Y.; Wang, Y.; Liu, T.-T.; Qi, Y.-F.; et al. ER stress dependent microparticles derived from smooth muscle cells promote endothelial dysfunction during thoracic aortic aneurysm and dissection. Clin. Sci. 2017, 131, 1287–1299. [Google Scholar] [CrossRef]
- Chen, Y.; Li, G.; Liu, M.L. Microvesicles as Emerging Biomarkers and Therapeutic Targets in Cardiometabolic Diseases. Genom. Proteom. Bioinform. 2018, 16, 50–62. [Google Scholar] [CrossRef]
- Suades, R.; Padro, T.; Alonso, R.; Mata, P.; Badimon, L. Lipid-lowering therapy with statins reduces microparticle shedding from endothelium, platelets and inflammatory cells. Thromb Haemost 2013, 110, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Morel, O.; Jesel, L.; Hugel, B.; Douchet, M.P.; Zupan, M.; Chauvin, M.; Freyssinet, J.M.; Toti, F. Protective effects of vitamin C on endothelium damage and platelet activation during myocardial infarction in patients with sustained generation of circulating microparticles. J. Thromb Haemost 2003, 1, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Bulut, D.; Becker, V.; Mugge, A. Acetylsalicylate reduces endothelial and platelet-derived microparticles in patients with coronary artery disease. Can. J. Physiol. Pharmacol. 2011, 89, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Baron, M.; Boulanger, C.M.; Staels, B.; Tailleux, A. Cell-derived microparticles in atherosclerosis: Biomarkers and targets for pharmacological modulation? J. Cell Mol. Med. 2012, 16, 1365–1376. [Google Scholar] [CrossRef]
- Martinez, M.C.; Tual-Chalot, S.; Leonetti, D.; Andriantsitohaina, R. Microparticles: Targets and tools in cardiovascular disease. Trends Pharm. Sci. 2011, 32, 659–665. [Google Scholar] [CrossRef]
- Ranghino, A.; Cantaluppi, V.; Grange, C.; Vitillo, L.; Fop, F.; Biancone, L.; Deregibus, M.C.; Tetta, C.; Segoloni, G.P.; Camussi, G. Endothelial progenitor cell-derived microvesicles improve neovascularization in a murine model of hindlimb ischemia. Int. J. Immunopathol. Pharmacol. 2012, 25, 75–85. [Google Scholar] [CrossRef]
- Soleti, R.; Benameur, T.; Porro, C.; Panaro, M.A.; Andriantsitohaina, R.; Martinez, M.C. Microparticles harboring Sonic Hedgehog promote angiogenesis through the upregulation of adhesion proteins and proangiogenic factors. Carcinogenesis 2009, 30, 580–588. [Google Scholar] [CrossRef]
- Hugel, B.; Martinez, M.C.; Kunzelmann, C.; Freyssinet, J.M. Membrane microparticles: Two sides of the coin. Physiology 2005, 20, 22–27. [Google Scholar] [CrossRef]
- Blajchman, M.A. Substitutes and alternatives to platelet transfusions in thrombocytopenic patients. J. Thromb Haemost 2003, 1, 1637–1641. [Google Scholar] [CrossRef]
- Piccin, A.; Murphy, W.G.; Smith, O.P. Circulating microparticles: Pathophysiology and clinical implications. Blood Rev. 2007, 21, 157–171. [Google Scholar] [CrossRef]
- Berezin, A. The Clinical Utility of Circulating Microparticles’ Measurement in Heart Failure Patients. J. Vasc. Med. Surg 2016, 4, 2. [Google Scholar]
- Berezin, A.E.; Kremzer, A.A.; Martovitskaya, Y.V.; Samura, T.A.; Berezina, T.A. The predictive role of circulating microparticles in patients with chronic heart failure. BBA Clin. 2015, 3, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Berezin, A.E.; Kremzer, A.A.; Samura, T.A.; Berezina, T.A.; Kruzliak, P. Impaired immune phenotype of circulating endothelial-derived microparticles in patients with metabolic syndrome and diabetes mellitus. J. Endocrinol. Investig. 2015, 38, 865–874. [Google Scholar] [CrossRef] [PubMed]
- Esposito, K.; Maiorino, M.I.; Di Palo, C.; Gicchino, M.; Petrizzo, M.; Bellastella, G.; Saccomanno, F.; Giugliano, D. Effects of pioglitazone versus metformin on circulating endothelial microparticles and progenitor cells in patients with newly diagnosed type 2 diabetes—a randomized controlled trial. Diabetes Obes. Metab. 2011, 13, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Nomura, S.; Omoto, S.; Yokoi, T.; Fujita, S.; Ozasa, R.; Eguchi, N.; Shouzu, A. Effects of miglitol in platelet-derived microparticle, adiponectin, and selectin level in patients with type 2 diabetes mellitus. Int. J. Gen. Med. 2011, 4, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Shimazu, T.; Inami, N.; Satoh, D.; Kajiura, T.; Yamada, K.; Iwasaka, T.; Nomura, S. Effect of acarbose on platelet-derived microparticles, soluble selectins, and adiponectin in diabetic patients. J. Thromb. Thrombolysis 2009, 28, 429. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Relaxing Factors | Contracting Factors | Contracting Factors | Antithrombotic Factors | Growth Factors | Inflammatory Mediators |
|---|---|---|---|---|---|
|
|
|
|
|
|
| Origins of MPs and Markers Used for Detection | Human, Cell and Animal Models | Key Observations | References |
|---|---|---|---|
| OBESITY | |||
| Endothelial lEVs (CD51+, CD144+, and CD31+/CD42b−) | Obese patients with hypertension (HTN) |
| Hu et al. [82] |
| Endothelial lEVs (CD144+ and CD146+) | Overweight and obese children |
| Gündüz et al. [83] |
| Endothelial lEVs (CD31+/CD42b−) and platelet lEVs (CD31+/CD42b+) | Obese women |
| Esposito et al. [84] |
| Origins of MPs and Markers Used for Detection | Human, Cell and Animal Models | Key Observations | References |
|---|---|---|---|
| DIABETES | |||
| Platelet, endothelial and leukocyte lEVs | STZ-induced diabetic rats |
| Feng et al. [88] |
| Endothelial lEVs | Cultured HUVECs induced with high glucose for 24 h |
| Burger et al. [92] |
| Endothelial lEVs (CD31+/CD42−, CD62+/CD42−, CD42+) |
|
| Jing et al. [94] |
| Endothelial lEVs (CD62E+, Annexin V+/CD62E+, Annexin V+) | Prediabetic and diabetic patients |
| Giannella et al. [91] |
| Platelet lEVs (P-selectin (CD62P+) and CD61+) | STZ-induced male Wistar rats |
| Ishida et al. [89] |
| AnnexinV+ lEVs, endothelial lEVs (CD31+/CD42− and CD51+) | Patients with type-2 diabetes |
| Feng et al. [87] |
| Lymphocyte lEVs and circulating lEVs | Lymphocyte lEVs derived from:
|
| Martin et al. [86] |
| Platelet, leukocytes and monocytes lEVs | Patients with diabetes and obese patients |
| Zhang et al. [98] |
| Origins of MPs and Markers Used for Detection | Humans, Cell and Animal Models | Key Observations | References |
|---|---|---|---|
| METABOLIC SYNDROME | |||
| Procoagulant lEVs (annexin V+), platelet (CD41+), endothelial (CD146+), erythrocyte (CD235a+) and leukocyte (CD45+) lEVs | Metabolic syndrome patients |
| Safiedeen et al. [11] |
| Circulating lEVs, procoagulant (annexin V+), platelet (CD41+), endothelial (CD146+), and erythrocyte (CD235a+) lEVs | Metabolic syndrome patients |
| Agouni et al. [97] |
| Platelet (CD31+), endothelial (CD31+, CD62E+ and CD51+) lEVs | Metabolic syndrome patients |
| Arteaga et al. [96] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osman, A.; Benameur, T.; Korashy, H.M.; Zeidan, A.; Agouni, A. Interplay between Endoplasmic Reticulum Stress and Large Extracellular Vesicles (Microparticles) in Endothelial Cell Dysfunction. Biomedicines 2020, 8, 409. https://doi.org/10.3390/biomedicines8100409
Osman A, Benameur T, Korashy HM, Zeidan A, Agouni A. Interplay between Endoplasmic Reticulum Stress and Large Extracellular Vesicles (Microparticles) in Endothelial Cell Dysfunction. Biomedicines. 2020; 8(10):409. https://doi.org/10.3390/biomedicines8100409
Chicago/Turabian StyleOsman, Aisha, Tarek Benameur, Hesham M. Korashy, Asad Zeidan, and Abdelali Agouni. 2020. "Interplay between Endoplasmic Reticulum Stress and Large Extracellular Vesicles (Microparticles) in Endothelial Cell Dysfunction" Biomedicines 8, no. 10: 409. https://doi.org/10.3390/biomedicines8100409
APA StyleOsman, A., Benameur, T., Korashy, H. M., Zeidan, A., & Agouni, A. (2020). Interplay between Endoplasmic Reticulum Stress and Large Extracellular Vesicles (Microparticles) in Endothelial Cell Dysfunction. Biomedicines, 8(10), 409. https://doi.org/10.3390/biomedicines8100409