Induction of Urokinase Activity by Retinoic Acid in Two Cell Lines of Neuronal Origin



Abstract
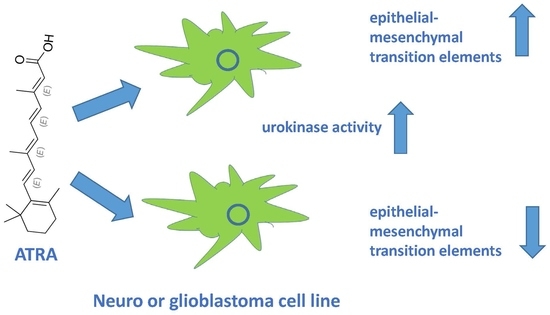
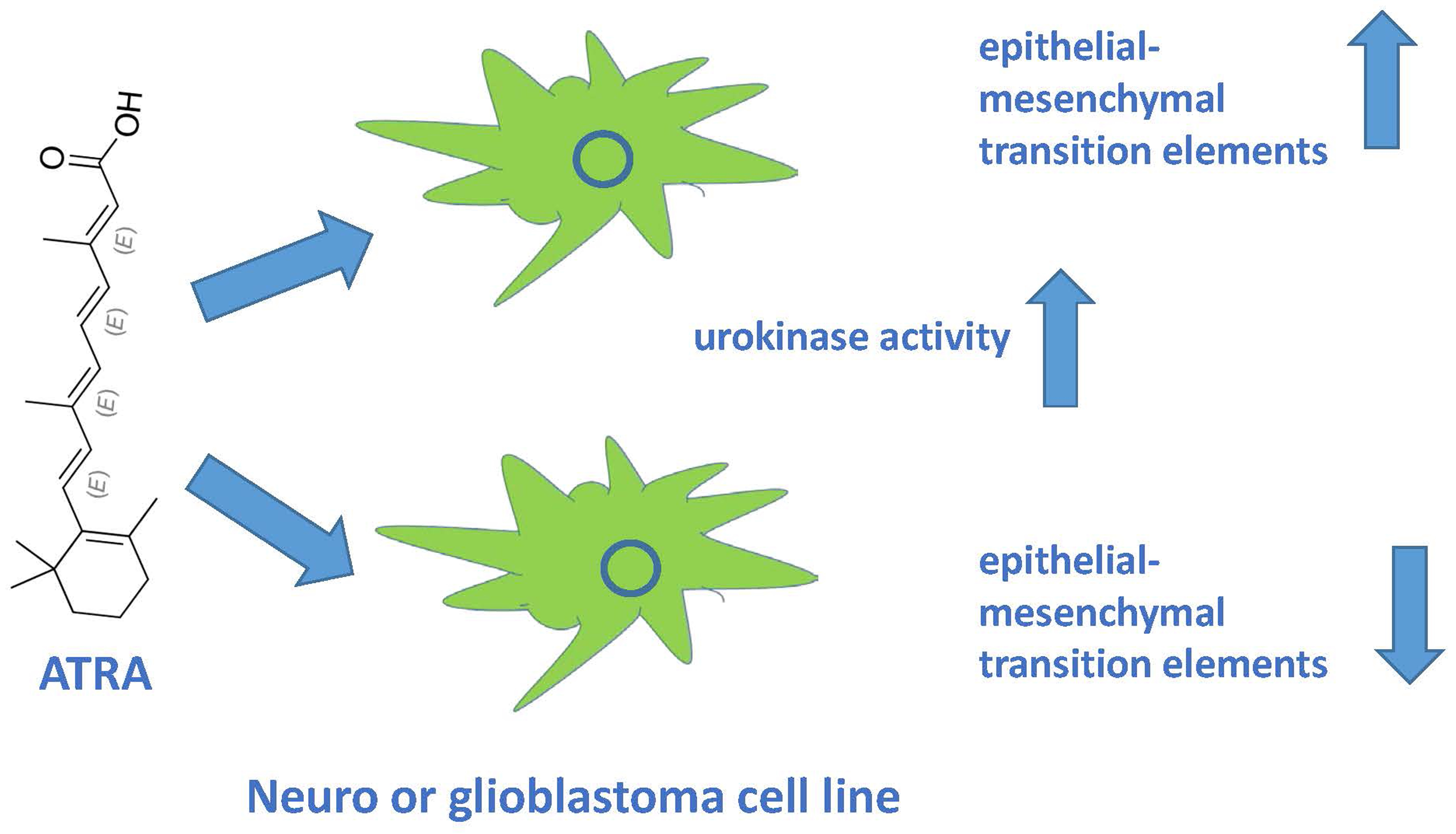{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Growth Assessment
2.2. Analysis of Enzymatic Activity
2.3. RNA Preparation, cDNA Synthesis, and Quantitative Real-Time PCR
2.4. SDS PAGE and Western Blot
2.5. Migration Assays
2.6. Statistical Analysis
3. Results
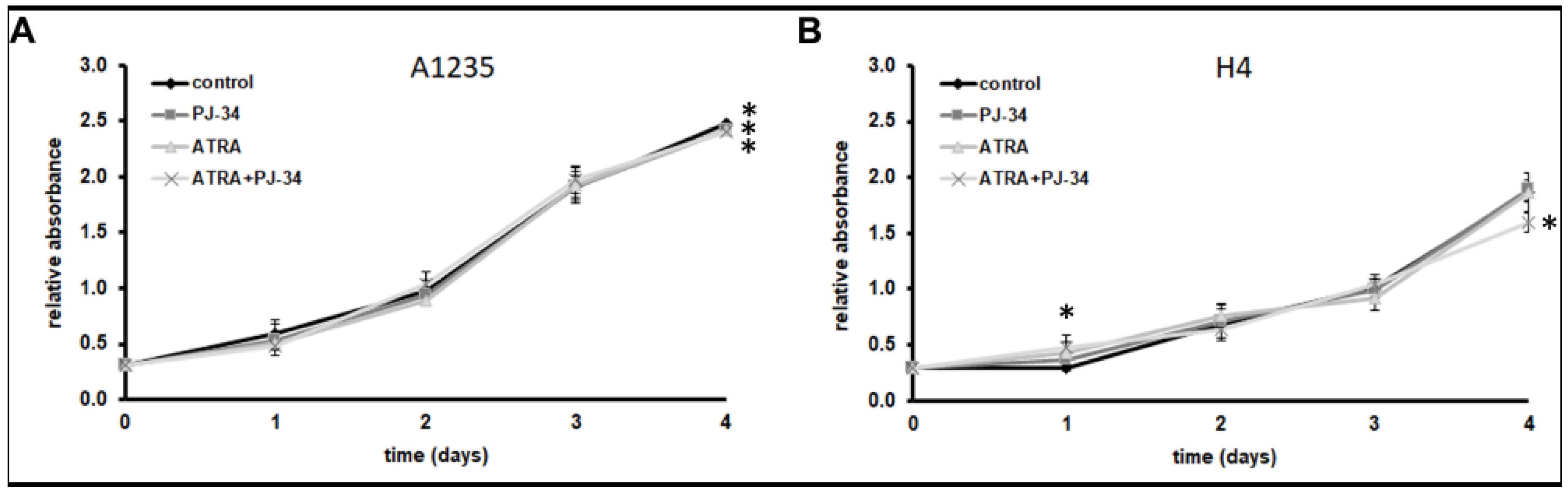
3.1. The Effect of ATRA and PARP Inhibition on Neuroglioblastoma Cell Growth and Morphology
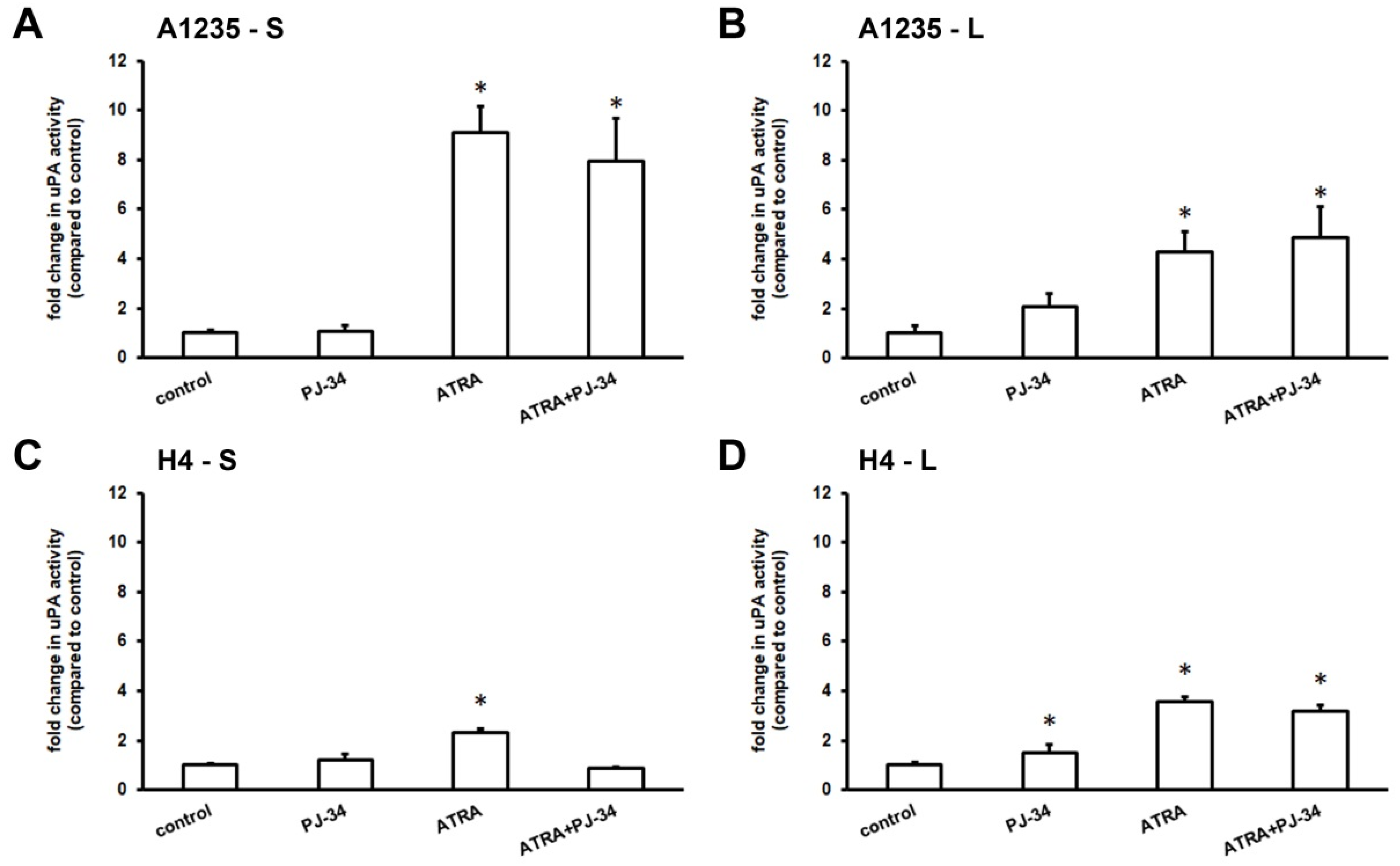
3.2. The Effect of ATRA and PARP Inhibition on Urokinase Activity
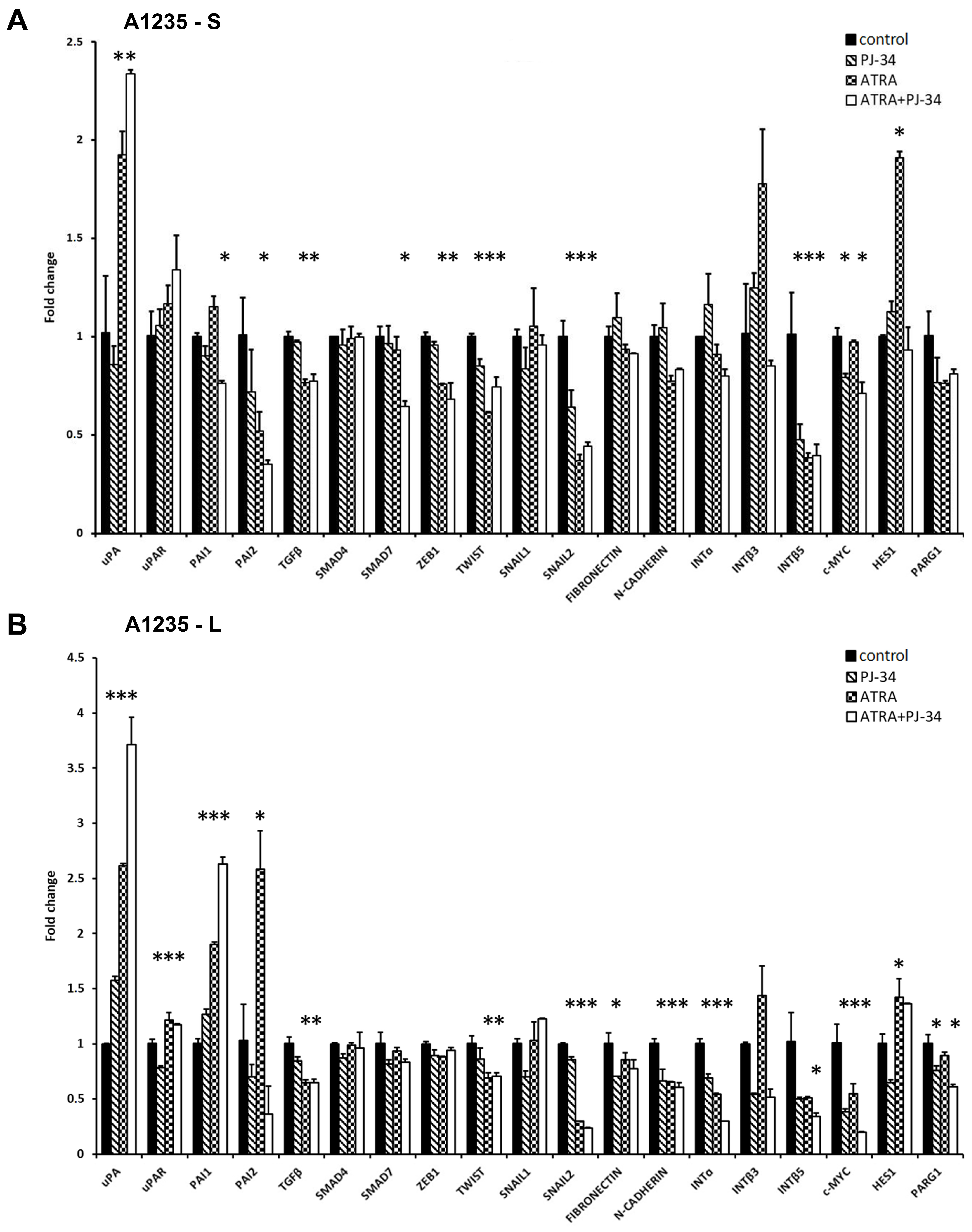
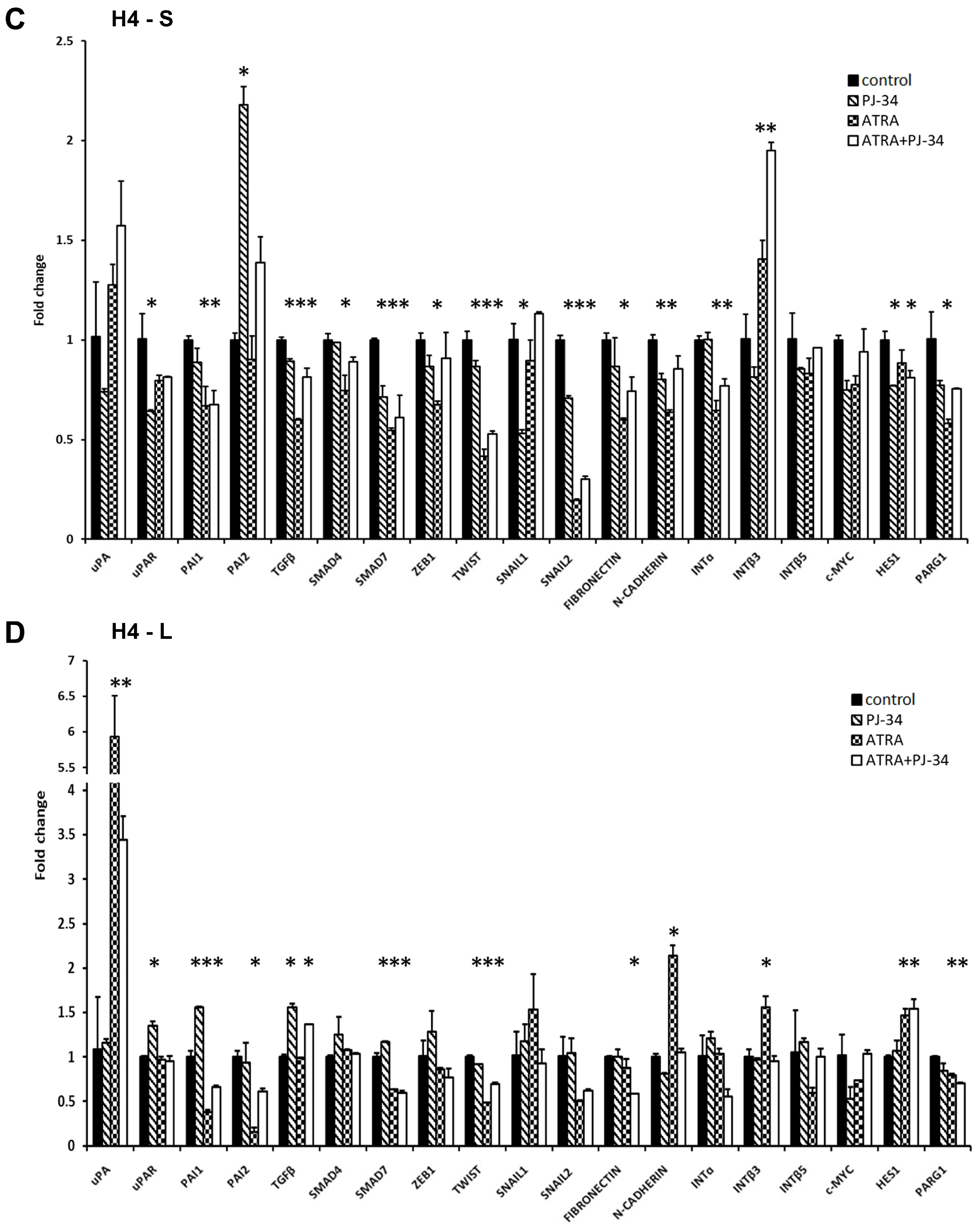
3.3. The Effect of ATRA and PARP Inhibition on EMT Gene Expression
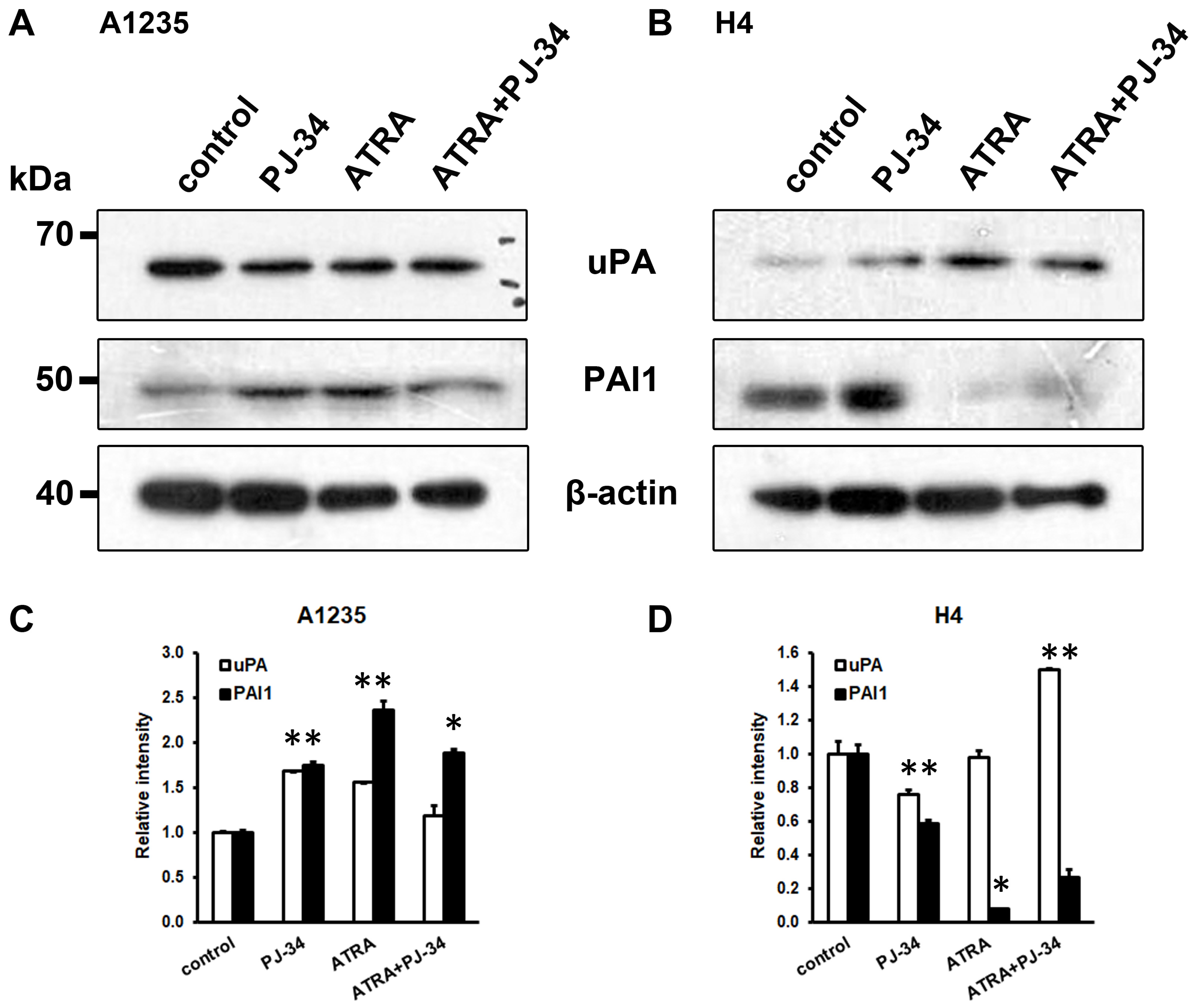
3.4. The Effect of ATRA and PARP Inhibition on the Plasminogen Activation System Proteins
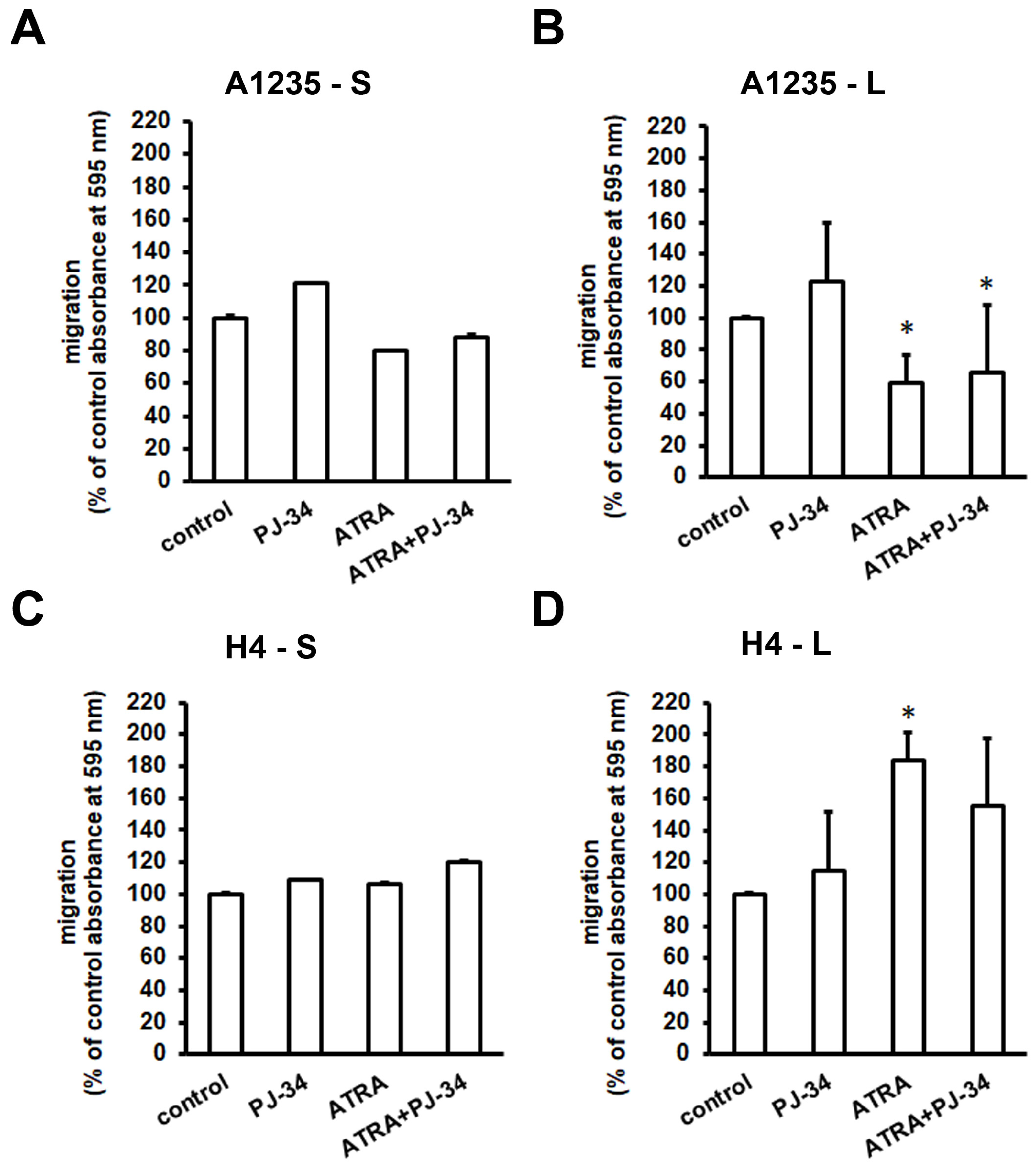
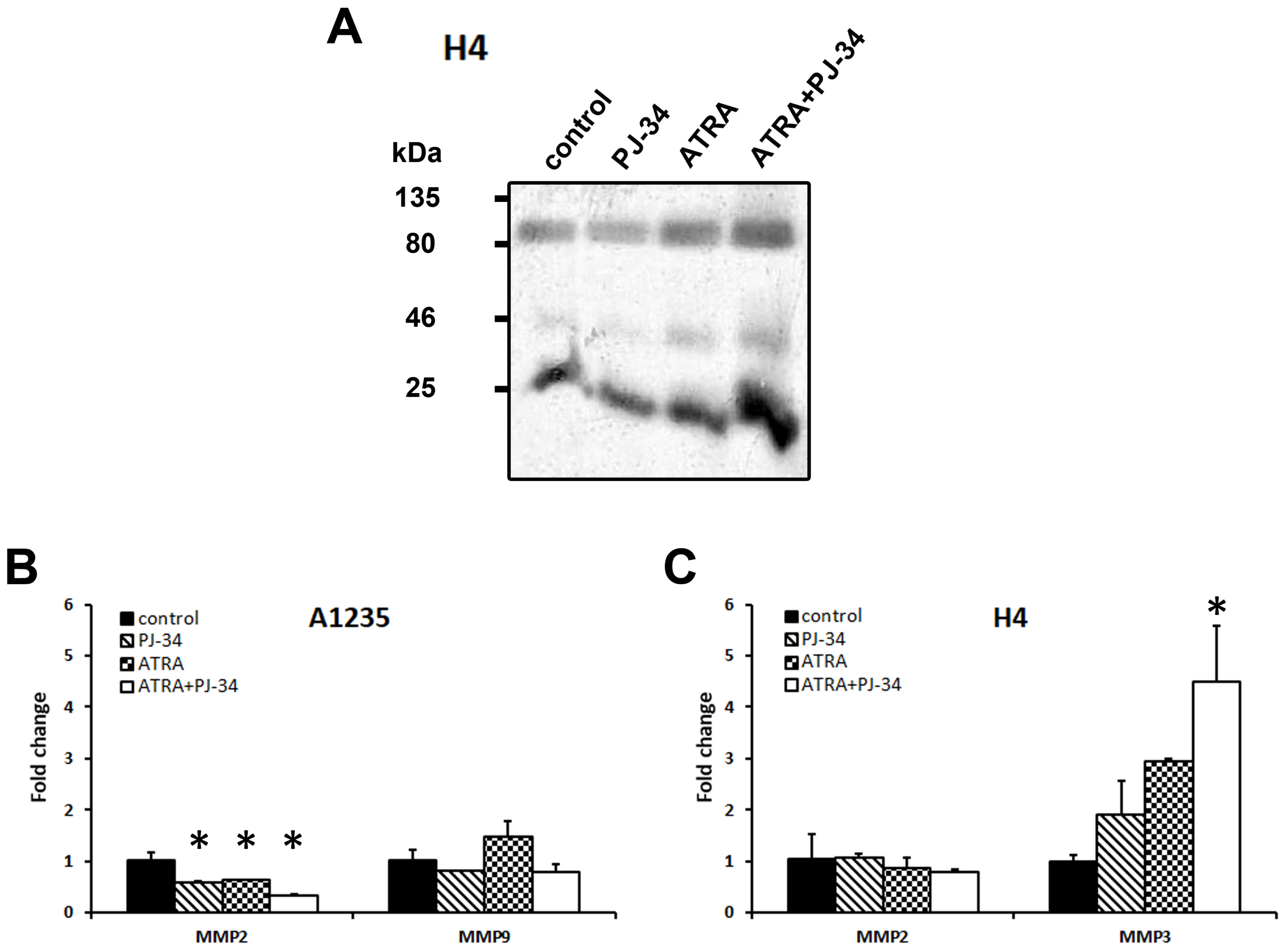
3.5. The Effect of ATRA and PARP Inhibition on Cell Migration and Matrix Metalloprotease Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Breitman, T.R.; Selonick, S.E.; Collins, S.J. Induction of differentiation of the human promyelocytic leukemia cell line (HL-60) by retinoic acid. Proc. Natl. Acad. Sci. USA 1980, 77, 2936–2940. [Google Scholar] [CrossRef] [PubMed]
- de Thé, H. Differentiation therapy revisited. Nat. Rev. Cancer 2017, 18, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, C.P.; Matthay, K.K.; Villablanca, J.G.; Maurer, B.J. Retinoid therapy of high-risk neuroblastoma. Cancer Lett. 2003, 197, 185–192. [Google Scholar] [CrossRef]
- Janesick, A.; Wu, S.C.; Blumberg, B. Retinoic acid signalling and neuronal differentiation. Cell Mol. Life Sci. 2015, 72, 1559–1576. [Google Scholar] [CrossRef] [PubMed]
- Gearing, K.L.; Göttlicher, M.; Teboul, M.; Widmark, E.; Gustafsson, J.A. Interaction of the peroxisome-proliferator-activated receptor and retinoid X receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 1440–1444. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, T.J.; Duester, G. Mechanisms of retinoic acid signalling and its roles in organ and limb development. Nat. Rev. Mol. Cell Biol. 2015, 16, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Chuang, J.H.; Tung, L.C.; Lin, Y. Neural differentiation from embryonic stem cells in vitro: An overview of the signalling pathways. World J. Stem Cells 2015, 7, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Krishnakumar, R.; Kraus, W.L. The PARP side of the nucleus: Molecular actions, physiological outcomes, and clinical targets. Mol. Cell 2010, 39, 8–24. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 78–96. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Heldin, C.H. Mechanisms of TGFβ-Induced Epithelial-Mesenchymal Transition. J. Clin. Med. 2016, 5, 63. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Tilló, E.; de Barrios, O.; Siles, L.; Amendola, P.G.; Darling, D.S.; Cuatrecasas, M.; Castells, A.; Postigo, A. ZEB1 Promotes invasiveness of colorectal carcinoma cells through the opposing regulation of uPA and PAI-1. Clin. Cancer Res. 2013, 19, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Giard, D.J.; Aaronson, S.A.; Todaro, G.J.; Arnstein, P.; Kersey, J.H.; Dosik, H.; Parks, W.P. In vitro cultivation of human tumours: Establishment of cell lines derived from a series of solid tumours. J. Natl. Cancer Inst. 1973, 51, 1417–1423. [Google Scholar] [CrossRef] [PubMed]
- Shipley, M.M.; Mangold, C.C.; Szpara, M.L. Differentiation of the SH-SY5Y Human Neuroblastoma Cell Line. J. Vis. Exp. 2016, 108, e53193. [Google Scholar] [CrossRef] [PubMed]
- Carbone, M.; Rossi, M.N.; Cavaldesi, M.; Notari, A.; Amati, P.; Maione, R. Poly(ADP-ribosyl)ation is implicated in the G0-G1 transition of resting cells. Oncogene 2008, 27, 6083–6092. [Google Scholar] [CrossRef][Green Version]
- Saotome, K.; Morita, H.; Umeda, M. Cytotoxicity test with simplified crystal violet staining method using microtitre plates and its application to injection drugs. Toxicol. Vitr. 1989, 3, 317–321. [Google Scholar] [CrossRef]
- Matulić, M.; Brdar, B. Urokinase-type Plasminogen Activator and Plasminogen Activator Inhibitor Induction by Etoposide in a Glioblastoma Cell Strain. Food Tech. Biotech. 2002, 40, 1–7. [Google Scholar]
- Doyle, A.; Griffiths, J.B.; Newell, D.G. Cell and Tissue Culture: Laboratory Procedures; John Wiley and Sons: Hoboken, NJ, USA, 1995; Available online: https://archive.org/stream/Cell_And_Tissue_Culture_Laboratory_Procedures_In_Biotechnology/Cell_And_Tissue_Culture_Laboratory_Procedures_In_Biotechnology_djvu.txt (accessed on 11 September 2019).
- Horvat, L.; Antica, M.; Matulić, M. Effect of Notch and PARP Pathways’ Inhibition in Leukemic Cells. Cells 2018, 7, 58. [Google Scholar] [CrossRef]
- Horvat, L.; Antica, M.; Matulić, M. Inhibition of PARP activity does not affect the differentiation processes caused by retinoic acid in SH-SY5Y cells. Mol. Exp. Biol. Med. 2019, 1, 38–43. [Google Scholar]
- Sambrook, J.F.; Russell, D.W. Molecular Cloning: A Laboratory Manual, 3rd ed.; ColdSpring Harbor Laboratory Press: New York, NY, USA, 2001. [Google Scholar]
- Nagakawa, O.; Ogasawara, M.; Murata, J.; Fuse, H.; Saiki, I. Effect of prostatic neuropeptides on migration of prostate cancer cell lines. Int. J. Urol. 2001, 8, 65–70. [Google Scholar] [CrossRef]
- Madunić, J.; Antica, M.; Cvjetko, P.; Požgaj, L.; Matulić, M. Modulation of urokinase plasminogen activator system by poly(ADP-ribose)polymerase-1 inhibition. Cytotechnology 2016, 68, 783–794. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Matulić, M.; Brdar, B. Induction of urokinase-type plasminogen activator by sodium salicylate in a glioblastoma cell strain. Food Technol. Biotechnol. 2001, 39, 5–11. [Google Scholar]
- Snoek-van Beurden, P.A.; Von den Hoff, J.W. Zymographic techniques for the analysis of matrix metalloproteinases and their inhibitors. Biotechniques 2005, 38, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Carén, H.; Stricker, S.H.; Bulstrode, H.; Gagrica, S.; Johnstone, E.; Bartlett, T.E.; Feber, A.; Wilson, G.; Teschendorff, A.E.; Bertone, P.; et al. Glioblastoma Stem Cells Respond to Differentiation Cues but Fail to Undergo Commitment and Terminal Cell-Cycle Arrest. Stem Cell Rep. 2015, 5, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Duffy, D.J.; Krstic, A.; Halasz, M.; Schwarzl, T.; Konietzny, A.; Iljin, K.; Higgins, D.G.; Kolch, W. Retinoic acid and TGF-β signalling cooperate to overcome MYCN-induced retinoid resistance. Genome Med. 2017, 9, 15. [Google Scholar] [CrossRef] [PubMed]
- Irigoyen, J.P.; Muñoz-Cánoves, P.; Montero, L.; Koziczak, M.; Nagamine, Y. The plasminogen activator system: Biology and regulation. Cell Mol. Life Sci. 1999, 56, 104–132. [Google Scholar] [CrossRef] [PubMed]
- Savagner, P.; Kusewitt, D.F.; Carver, E.A.; Magnino, F.; Choi, C.; Gridley, T.; Hudson, L.G. Developmental transcription factor slug is required for effective re-epithelialization by adult keratinocytes. J. Cell Physiol. 2005, 202, 858–866. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Boareto, M.; Huang, B.; Jia, D.; Lu, M.; Ben-Jacob, E.; Onuchic, J.N.; Levine, H. Implications of the Hybrid Epithelial/Mesenchymal Phenotype in Metastasis. Front. Oncol. 2015, 5, 155. [Google Scholar] [CrossRef] [PubMed]
- Barrallo-Gimeno, A.; Nieto, M.A. The Snail genes as inducers of cell movement and survival: Implications in development and cancer. Development 2005, 132, 3151–3161. [Google Scholar] [CrossRef]
- Mladinich, M.; Ruan, D.; Chan, C.H. Tackling Cancer Stem Cells via Inhibition of EMT Transcription Factors. Stem Cells Int. 2016, 2016, 5285892. [Google Scholar] [CrossRef]
- Lönn, P.; van der Heide, L.P.; Dahl, M.; Hellman, U.; Heldin, C.H.; Moustakas, A. PARP-1 attenuates Smad-mediated transcription. Mol. Cell 2010, 40, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Dahl, M.; Maturi, V.; Lönn, P.; Papoutsoglou, P.; Zieba, A.; Vanlandewijck, M.; van der Heide, L.P.; Watanabe, Y.; Söderberg, O.; Hottiger, M.O.; et al. Fine-tuning of Smad protein function by poly(ADP-ribose) polymerases and poly(ADP-ribose) glycohydrolase during transforming growth factor β signalling. PLoS ONE 2014, 9, e103651. [Google Scholar] [CrossRef] [PubMed]
- Schacke, M.; Kumar, J.; Colwell, N.; Hermanson, K.; Folle, G.A.; Nechaev, S.; Dhasarathy, A.; Lafon-Hughes, L. PARP-1/2 Inhibitor Olaparib Prevents or Partially Reverts EMT Induced by TGF-β in NMuMG Cells. Int. J. Mol. Sci. 2019, 20, 518. [Google Scholar] [CrossRef] [PubMed]
- Hollestelle, A.; Peeters, J.K.; Smid, M.; Timmermans, M.; Verhoog, L.C.; Westenend, P.J.; Heine, A.A.J.; Chan, A.; Sieuwerts, A.M.; Wiemer, E.A.C.; et al. Loss of E-cadherin is not a necessity for epithelial to mesenchymal transition in human breast cancer. Breast Cancer Res. Treat. 2013, 138, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.R.; Kang, Y.; Massagué, J. Defective repression of c-myc in breast cancer cells: A loss at the core of the transforming growth factor beta growth arrest program. Proc. Natl. Acad. Sci. USA 2001, 98, 992–999. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, J.; Lázcoz, P.; Inda, M.M.; Nistal, M.; Pestaña, A.; Encío, I.J.; Castresana, J.S. Homozygous deletion and expression of PTEN and DMBT1 in human primary neuroblastoma and cell lines. Int. J. Cancer 2004, 109, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Heiple, J.M.; Ossowski, L. Human neutrophil plasminogen activator is localized in specific granules and is translocated to the cell surface by exocytosis. J. Exp. Med. 1986, 164, 826–840. [Google Scholar] [CrossRef] [PubMed]
- Ishigami, S.; Sandkvist, M.; Tsui, F.; Moore, E.; Coleman, T.A.; Lawrence, D.A. Identification of a novel targeting sequence for regulated secretion in the serine protease inhibitor neuroserpin. Biochem. J. 2007, 402, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Knop, M.; Gerke, V. Ca2+ -regulated secretion of tissue-type plasminogen activator and von Willebrand factor in human endothelial cells. Biochim. Biophys. Acta 2002, 1600, 162–167. [Google Scholar] [CrossRef]
- Wei, L.; Lun, Y.; Zhou, X.; He, S.; Gao, L.; Liu, Y.; He, Z.; Li, B.; Wang, C. Novel urokinase-plasminogen activator inhibitor SPINK13 inhibits growth and metastasis of hepatocellular carcinoma in vivo. Pharmacol. Res. 2019, 143, 73–85. [Google Scholar] [CrossRef]
- Cui, J.; Gong, M.; He, Y.; Li, Q.; He, T.; Bi, Y. All-trans retinoic acid inhibits proliferation, migration, invasion and induces differentiation of hepa1-6 cells through reversing EMT in vitro. Int. J. Oncol. 2016, 48, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Zanetti, A.; Affatato, R.; Centritto, F.; Fratelli, M.; Kurosaki, M.; Barzago, M.M.; Bolis, M.; Terao, M.; Garattini, E.; Paroni, G. All-trans-retinoic Acid Modulates the Plasticity and Inhibits the Motility of Breast Cancer Cells: ROLE OF NOTCH1 AND TRANSFORMING GROWTH FACTOR (TGFβ). J. Biol. Chem. 2015, 290, 17690–17709. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Horvat, L.; Madunić, J.; Grubar, M.; Antica, M.; Matulić, M. Induction of Urokinase Activity by Retinoic Acid in Two Cell Lines of Neuronal Origin. Biomedicines 2019, 7, 70. https://doi.org/10.3390/biomedicines7030070
Horvat L, Madunić J, Grubar M, Antica M, Matulić M. Induction of Urokinase Activity by Retinoic Acid in Two Cell Lines of Neuronal Origin. Biomedicines. 2019; 7(3):70. https://doi.org/10.3390/biomedicines7030070
Chicago/Turabian StyleHorvat, Luka, Josip Madunić, Martina Grubar, Mariastefania Antica, and Maja Matulić. 2019. "Induction of Urokinase Activity by Retinoic Acid in Two Cell Lines of Neuronal Origin" Biomedicines 7, no. 3: 70. https://doi.org/10.3390/biomedicines7030070
APA StyleHorvat, L., Madunić, J., Grubar, M., Antica, M., & Matulić, M. (2019). Induction of Urokinase Activity by Retinoic Acid in Two Cell Lines of Neuronal Origin. Biomedicines, 7(3), 70. https://doi.org/10.3390/biomedicines7030070