Ultrastructure in Transthyretin Amyloidosis: From Pathophysiology to Therapeutic Insights


Abstract
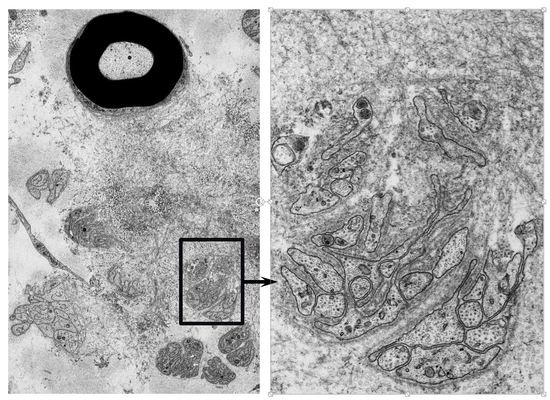
:
1. Introduction
2. Mechanisms of Amyloid Deposition
3. Diversity of Clinical Features
4. Pathological Findings Corresponding to Clinical Characteristics
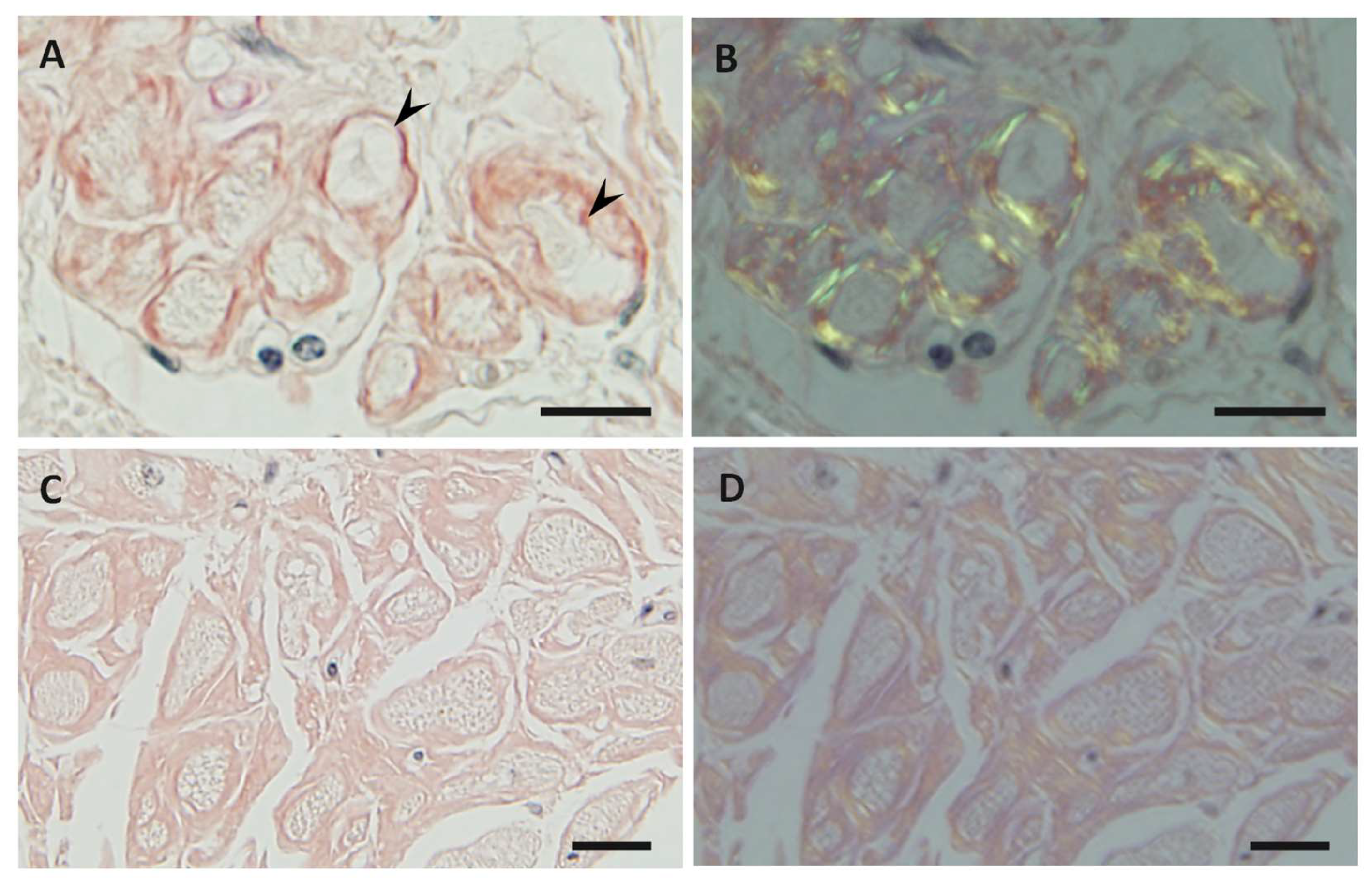
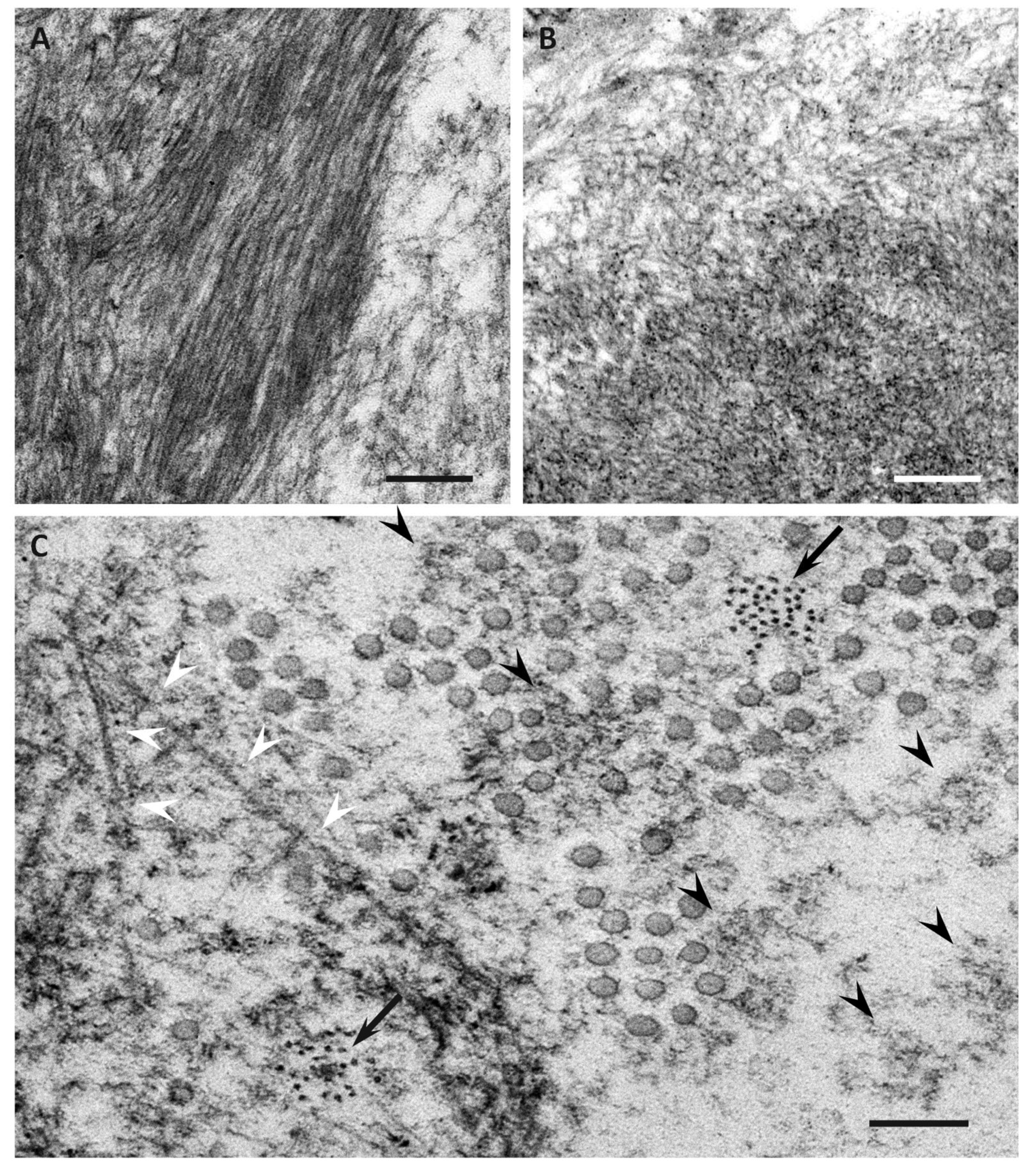
5. Characteristics of Amyloid Fibrils Determining the Clinicopathological Features
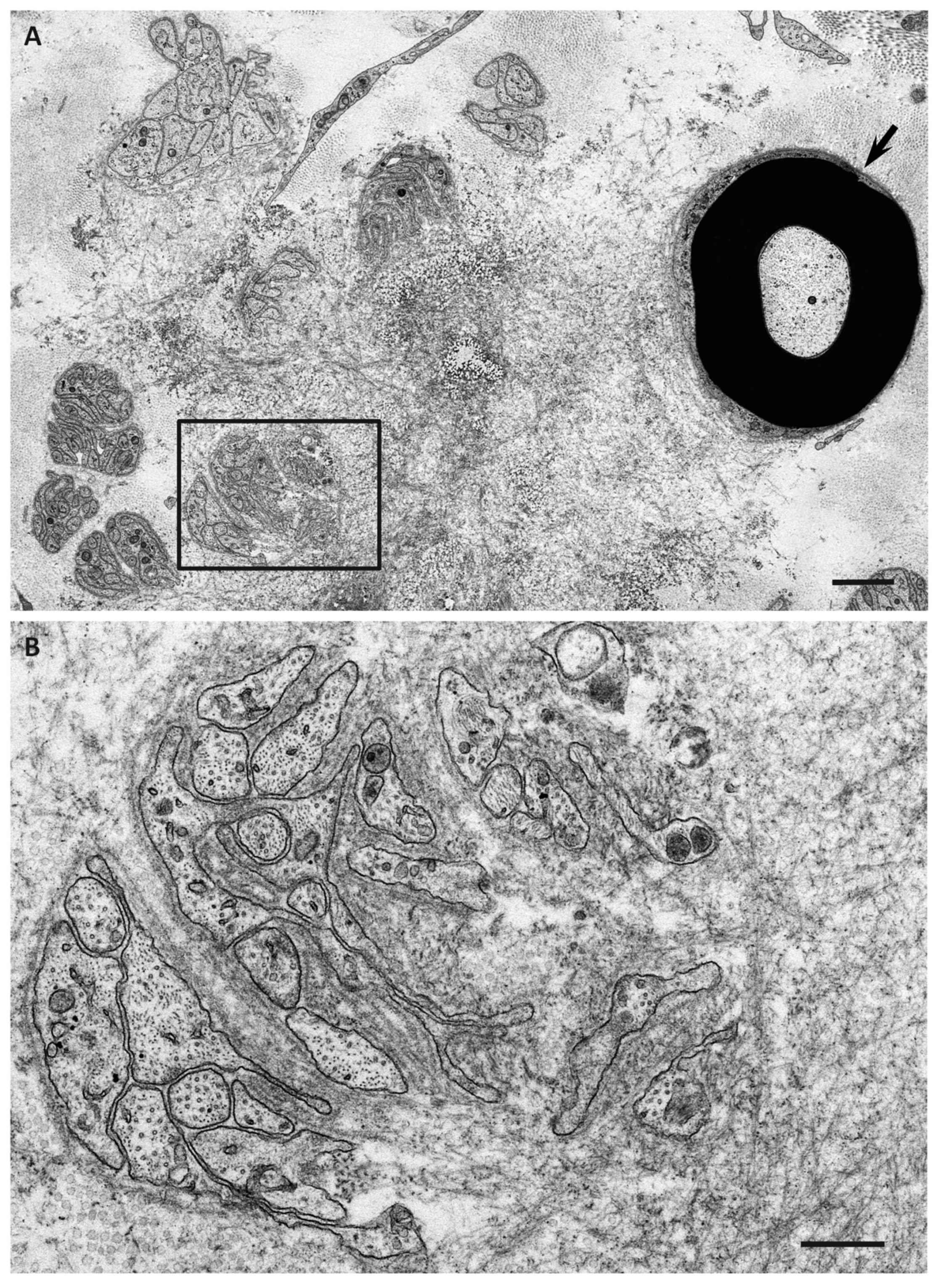
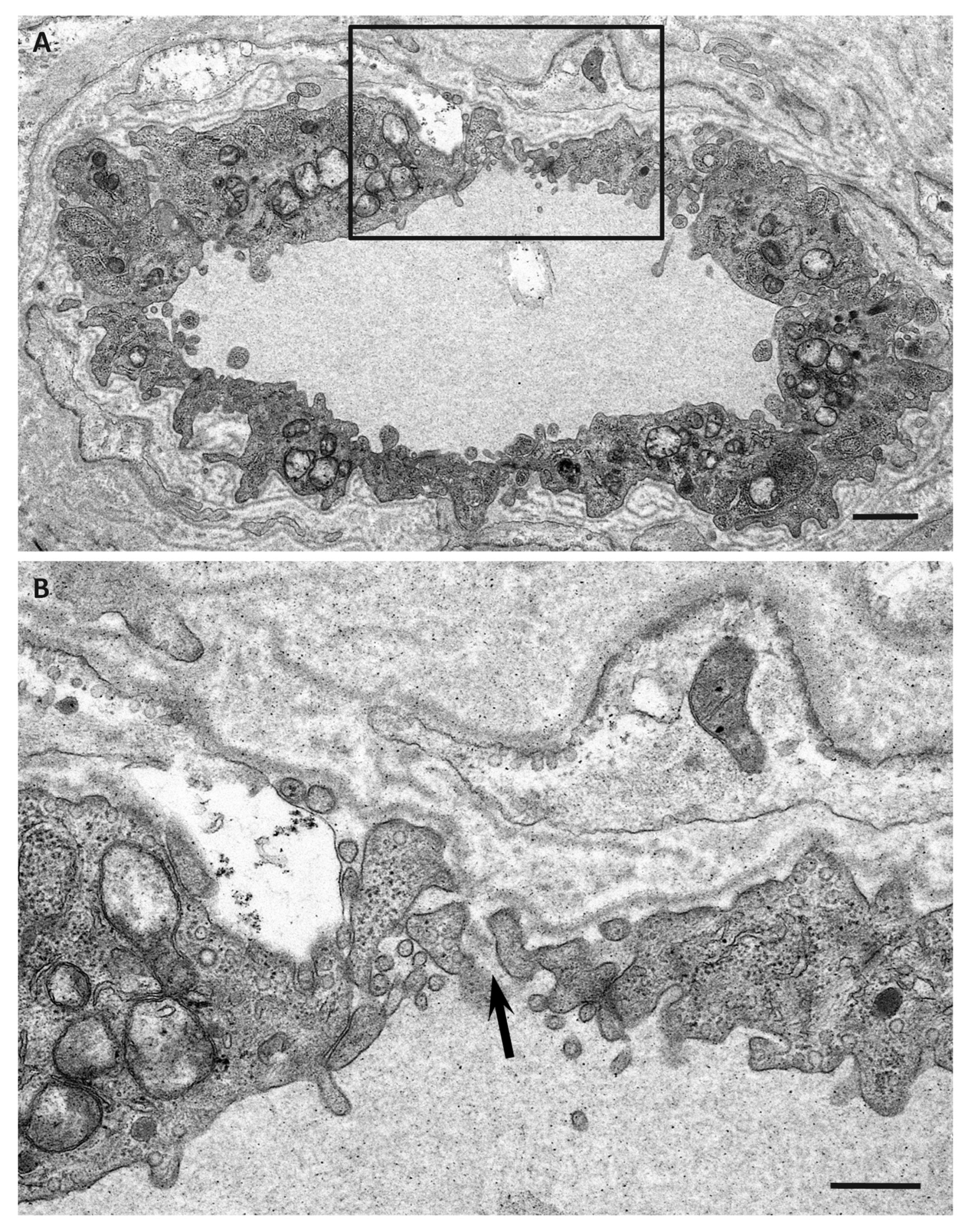
6. Impact of Amyloid Fibril Formation on Neighboring Tissues
7. Toxicity of Nonfibrillar TTR
8. Angiopathy Enhancing the Leakage of Circulating TTR
9. Therapeutic Insights from Pathology
Acknowledgments
Conflicts of Interest
References
- Pitkänen, P.; Westermark, P.; Cornwell, G.G., 3rd. Senile systemic amyloidosis. Am. J. Pathol. 1984, 117, 391–399. [Google Scholar]
- Koike, H.; Misu, K.; Ikeda, S.; Ando, Y.; Nakazato, M.; Ando, E.; Yamamoto, M.; Hattori, N.; Sobue, G.; Study Group for Hereditary Neuropathy in Japan. Type I (transthyretin Met30) familial amyloid polyneuropathy in Japan: Early- vs. late-onset form. Arch. Neurol. 2002, 59, 1771–1776. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.D.; Kincaid, J.C. The molecular biology and clinical features of amyloid neuropathy. Muscle Nerve 2007, 36, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Planté-Bordeneuve, V.; Said, G. Familial amyloid polyneuropathy. Lancet Neurol. 2011, 10, 1086–1097. [Google Scholar] [CrossRef]
- Adams, D.; Cauquil, C.; Labeyrie, C. Familial amyloid polyneuropathy. Curr. Opin. Neurol. 2017, 30, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Andrade, C. A peculiar form of peripheral neuropathy; familiar atypical generalized amyloidosis with special involvement of the peripheral nerves. Brain 1952, 75, 408–427. [Google Scholar] [CrossRef] [PubMed]
- Araki, S.; Mawatari, S.; Ohta, M.; Nakajima, A.; Kuroiwa, Y. Polyneuritic amyloidosis in a Japanese family. Arch. Neurol. 1968, 18, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Andersson, R. Hereditary amyloidosis with polyneuropathy. Acta Med. Scand. 1970, 1–2, 85–94. [Google Scholar] [CrossRef]
- Ando, Y.; Nakamura, M.; Araki, S. Transthyretin-related familial amyloidotic polyneuropathy. Arch. Neurol. 2005, 62, 1057–1062. [Google Scholar] [CrossRef] [PubMed]
- Koike, H.; Tanaka, F.; Hashimoto, R.; Tomita, M.; Kawagashira, Y.; Iijima, M.; Fujitake, J.; Kawanami, T.; Kato, T.; Yamamoto, M.; et al. Natural history of transthyretin Val30Met familial amyloid polyneuropathy: Analysis of late-onset cases from non-endemic areas. J. Neurol. Neurosurg. Psychiatry 2012, 83, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Parman, Y.; Adams, D.; Obici, L.; Galán, L.; Guergueltcheva, V.; Suhr, O.B.; Coelho, T. European Network for TTR-FAP (ATTReuNET). Sixty years of transthyretin familial amyloid polyneuropathy (TTR-FAP) in Europe: Where are we now? A European network approach to defining the epidemiology and management patterns for TTR-FAP. Curr. Opin. Neurol. 2016, 29 (Suppl. 1), S3–S13. [Google Scholar] [CrossRef] [PubMed]
- Sekijima, Y.; Ueda, M.; Koike, H.; Misawa, S.; Ishii, T.; Ando, Y. Diagnosis and management of transthyretin familial amyloid polyneuropathy in Japan: Red-flag symptom clusters and treatment algorithm. Orphanet J. Rare Dis. 2018, 13, 6. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.C.; Huang, C.M.; Chiang, H.H.; Luo, K.R.; Kan, H.W.; Yang, N.C.; Chiang, H.; Lin, W.M.; Lai, S.M.; Lee, M.J.; et al. Sudomotor innervation in transthyretin amyloid neuropathy: Pathology and functional correlates. Ann. Neurol. 2015, 78, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.S.; Pelayo-Negro, A.L.; Evans, M.R.; Laurà, M.; Blake, J.; Stancanelli, C.; Iodice, V.; Wechalekar, A.D.; Whelan, C.J.; Gillmore, J.D.; et al. A study of the neuropathy associated with transthyretin amyloidosis (ATTR) in the UK. J. Neurol. Neurosurg. Psychiatry 2016, 87, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Durmuş-Tekçe, H.; Matur, Z.; Mert Atmaca, M.; Poda, M.; Çakar, A.; Hıdır Ulaş, Ü.; Oflazer-Serdaroğlu, P.; Deymeer, F.; Parman, Y.G. Genotypic and phenotypic presentation of transthyretin-related familial amyloid polyneuropathy (TTR-FAP) in Turkey. Neuromuscul. Disord. 2016, 26, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, G.; Steen, L.; Ekstedt, J.; Groth, C.G.; Ericzon, B.G.; Eriksson, S.; Andersen, O.; Karlberg, I.; Nordén, G.; Nakazato, M.; et al. Biochemical effect of liver transplantation in two Swedish patients with familial amyloidotic polyneuropathy (FAP-met30). Clin. Genet. 1991, 40, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Ando, Y.; Okamoto, S.; Misumi, Y.; Hirahara, T.; Ueda, M.; Obayashi, K.; Nakamura, M.; Jono, H.; Shono, M.; et al. Long-term survival after liver transplantation in patients with familial amyloid polyneuropathy. Neurology 2012, 78, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Coelho, T.; Maia, L.F.; Martins da Silva, A.; Waddington Cruz, M.; Planté-Bordeneuve, V.; Lozeron, P.; Suhr, O.B.; Campistol, J.M.; Conceição, I.M.; Schmidt, H.H.; et al. Tafamidis for transthyretin familial amyloid polyneuropathy: A randomized, controlled trial. Neurology 2012, 79, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Berk, J.L.; Suhr, O.B.; Obici, L.; Sekijima, Y.; Zeldenrust, S.R.; Yamashita, T.; Heneghan, M.A.; Gorevic, P.D.; Litchy, W.J.; Wiesman, J.F.; et al. Repurposing diflunisal for familial amyloid polyneuropathy: A randomized clinical trial. JAMA 2013, 310, 2658–2667. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.S.; Schwartz, J.H.; Gundapaneni, B.; Elliott, P.M.; Merlini, G.; Waddington-Cruz, M.; Kristen, A.V.; Grogan, M.; Witteles, R.; Damy, T.; et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N. Engl. J. Med. 2018, 379, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.D.; Waddington-Cruz, M.; Berk, J.L.; Polydefkis, M.; Dyck, P.J.; Wang, A.K.; Planté-Bordeneuve, V.; Barroso, F.A.; Merlini, G.; Obici, L.; et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018, 379, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Blake, C.C.F.; Geisow, M.J.; Swan, I.D.A.; Rerat, C.; Rerat, B. Structure of human plasma prealbumin at 2–5 A resolution. a preliminary report on the polypeptide chain conformation, quaternary structure and thyroxine binding. J. Mol. Biol. 1974, 88, 1–12. [Google Scholar] [CrossRef]
- Dickson, P.W.; Aldred, A.R.; Marley, P.D.; Tu, G.F.; Howlett, G.J.; Schreiber, G. High prealbumin and transferrin mRNA levels in the choroid plexus of rat brain. Biochem. Biophys. Res. Commun. 1985, 127, 890–895. [Google Scholar] [CrossRef]
- Soprano, D.R.; Herbert, J.; Soprano, K.J.; Schon, E.A.; Goodman, D.S. Demonstration of transthyretin mRNA in the brain and other extrahepatic tissues in the rat. J. Biol. Chem. 1985, 260, 11793–11798. [Google Scholar] [PubMed]
- Uemichi, T.; Uitti, R.J.; Koeppen, A.H.; Donat, J.R.; Benson, M.D. Oculoleptomeningeal amyloidosis associated with a new transthyretin variant Ser64. Arch. Neurol. 1999, 56, 1152–1155. [Google Scholar] [CrossRef] [PubMed]
- Ingbar, S.H. Pre-albumin: A thyroxinebinding protein of human plasma. Endocrinology 1958, 63, 256–259. [Google Scholar] [PubMed]
- Raz, A.; Goodman, D.S. The interaction of thyroxine with human plasma prealbumin and with the prealbumin-retinol-binding protein complex. J. Biol. Chem. 1969, 244, 3230–3237. [Google Scholar] [PubMed]
- Kelly, J.W. Amyloid fibril formation and protein misassembly: A structural quest for insights into amyloid and prion diseases. Structure 1997, 5, 595–600. [Google Scholar] [CrossRef]
- Sekijima, Y.; Wiseman, R.L.; Matteson, J.; Hammarström, P.; Miller, S.R.; Sawkar, A.R.; Balch, W.E.; Kelly, J.W. The biological and chemical basis for tissue-selective amyloid disease. Cell 2005, 121, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Westermark, P.; Sletten, K.; Johansson, B.; Cornwell, G.G., 3rd. Fibril in senile systemic amyloidosis is derived from normal transthyretin. Proc. Natl. Acad. Sci. USA 1990, 87, 2843–2845. [Google Scholar] [CrossRef] [PubMed]
- Suhr, O.B.; Lundgren, E.; Westermark, P. One mutation, two distinct disease variants: Unravelling the impact of transthyretin amyloid fibril composition. J. Intern. Med. 2017, 281, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Mangione, P.P.; Verona, G.; Corazza, A.; Marcoux, J.; Canetti, D.; Giorgetti, S.; Raimondi, S.; Stoppini, M.; Esposito, M.; Relini, A.; et al. Plasminogen activation triggers transthyretin amyloidogenesis in vitro. J. Biol. Chem. 2018, 293, 14192–14199. [Google Scholar] [CrossRef] [PubMed]
- Koike, H.; Misu, K.; Sugiura, M.; Iijima, M.; Mori, K.; Yamamoto, M.; Hattori, N.; Mukai, E.; Ando, Y.; Ikeda, S.; et al. Pathology of early- vs. late-onset TTR Met30 familial amyloid polyneuropathy. Neurology 2004, 63, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Cornwell, G.G., 3rd; Murdoch, W.L.; Kyle, R.A.; Westermark, P.; Pitkänen, P. Frequency and distribution of senile cardiovascular amyloid. A clinicopathologic correlation. Am. J. Med. 1983, 75, 618–623. [Google Scholar] [CrossRef]
- Tanskanen, M.; Peuralinna, T.; Polvikoski, T.; Notkola, I.L.; Sulkava, R.; Hardy, J.; Singleton, A.; Kiuru-Enari, S.; Paetau, A.; Tienari, P.J.; et al. Senile systemic amyloidosis affects 25% of the very aged and associates with genetic variation in α2-macroglobulin and tau: A population-based autopsy study. Ann. Med. 2008, 40, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Ueda, M.; Horibata, Y.; Shono, M.; Misumi, Y.; Oshima, T.; Su, Y.; Tasaki, M.; Shinriki, S.; Kawahara, S.; Jono, H.; et al. Clinicopathological features of senile systemic amyloidosis: An ante- and post-mortem study. Mod. Pathol. 2011, 24, 1533–1544. [Google Scholar] [CrossRef] [PubMed]
- Sekijima, Y.; Yazaki, M.; Ueda, M.; Koike, H.; Yamada, M.; Ando, Y. First nationwide survey on systemic wild-type ATTR amyloidosis in Japan. Amyloid 2018, 25, 8–10. [Google Scholar] [CrossRef] [PubMed]
- Grogan, M.; Scott, C.G.; Kyle, R.A.; Zeldenrust, S.R.; Gertz, M.A.; Lin, G.; Klarich, K.W.; Miller, W.L.; Maleszewski, J.J.; Dispenzieri, A. Natural history of wild-type transthyretin cardiac amyloidosis and risk stratification using a novel staging system. J. Am. Coll. Cardiol. 2016, 68, 1014–1020. [Google Scholar] [CrossRef] [PubMed]
- Westermark, P.; Westermark, G.T.; Suhr, O.B.; Berg, S. Transthyretin-derived amyloidosis: Probably a common cause of lumbar spinal stenosis. Ups J. Med. Sci. 2014, 119, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, A.; Ueda, M.; Sueyoshi, T.; Okada, T.; Fujimoto, T.; Ogi, Y.; Kitagawa, K.; Tasaki, M.; Misumi, Y.; Oshima, T.; et al. Amyloid deposits derived from transthyretin in the ligamentum flavum as related to lumbar spinal canal stenosis. Mod. Pathol. 2015, 28, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Koike, H.; Kawagashira, Y.; Iijima, M.; Yamamoto, M.; Hattori, N.; Tanaka, F.; Hirayama, M.; Ando, Y.; Ikeda, S.; Sobue, G. Electrophysiological features of late-onset transthyretin Met30 familial amyloid polyneuropathy unrelated to endemic foci. J. Neurol. 2008, 255, 1526–1533. [Google Scholar] [CrossRef] [PubMed]
- Koike, H.; Morozumi, S.; Kawagashira, Y.; Iijima, M.; Yamamoto, M.; Hattori, N.; Tanaka, F.; Nakamura, T.; Hirayama, M.; Ando, Y.; et al. The significance of carpal tunnel syndrome in transthyretin Val30Met familial amyloid polyneuropathy. Amyloid 2009, 16, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Ueda, M.; Misumi, Y.; Masuda, T.; Nomura, T.; Tasaki, M.; Takamatsu, K.; Sasada, K.; Obayashi, K.; Matsui, H.; et al. Genetic and clinical characteristics of hereditary transthyretin amyloidosis in endemic and non-endemic areas: Experience from a single-referral center in Japan. J. Neurol. 2018, 265, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Koike, H.; Sobue, G. Diagnosis of familial amyloid polyneuropathy: Wide-ranged clinicopathological features. Expert Opin. Med. Diagn. 2010, 4, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Koike, H.; Sobue, G. Late-onset familial amyloid polyneuropathy in Japan. Amyloid 2012, 19 (Suppl. 1), 55–57. [Google Scholar] [CrossRef] [PubMed]
- Lemos, C.; Coelho, T.; Alves-Ferreira, M.; Martins-da-Silva, A.; Sequeiros, J.; Mendonça, D.; Sousa, A. Overcoming artefact: Anticipation in 284 Portuguese kindreds with familial amyloid polyneuropathy (FAP) ATTRV30M. J. Neurol. Neurosurg. Psychiatry 2014, 85, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Koike, H.; Hashimoto, R.; Tomita, M.; Kawagashira, Y.; Iijima, M.; Tanaka, F.; Sobue, G. Diagnosis of sporadic transthyretin Val30Met familial amyloid polyneuropathy: A practical analysis. Amyloid 2011, 18, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Misu, K.; Hattori, N.; Nagamatsu, M.; Ikeda, S.; Ando, Y.; Nakazato, M.; Takei, Y.; Hanyu, N.; Usui, Y.; Tanaka, F.; et al. Late-onset familial amyloid polyneuropathy type I (transthyretin Met30-associated familial amyloid polyneuropathy) unrelated to endemic focus in Japan. Clinicopathological and genetic features. Brain 1999, 122, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Misu, K.; Hattori, N.; Ando, Y.; Ikeda, S.; Sobue, G. Anticipation in early- but not late-onset familial amyloid polyneuropathy (TTR met 30) in Japan. Neurology 2000, 55, 451–452. [Google Scholar] [CrossRef] [PubMed]
- Koike, H.; Ikeda, S.; Takahashi, M.; Kawagashira, Y.; Iijima, M.; Misumi, Y.; Ando, Y.; Ikeda, S.I.; Katsuno, M.; Sobue, G. Schwann cell and endothelial cell damage in transthyretin familial amyloid polyneuropathy. Neurology 2016, 87, 2220–2229. [Google Scholar] [CrossRef] [PubMed]
- González-López, E.; Gallego-Delgado, M.; Guzzo-Merello, G.; de Haro-Del Moral, F.J.; Cobo-Marcos, M.; Robles, C.; Bornstein, B.; Salas, C.; Lara-Pezzi, E.; Alonso-Pulpon, L.; et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur. Heart J. 2015, 36, 2585–2594. [Google Scholar] [CrossRef] [PubMed]
- Ihse, E.; Ybo, A.; Suhr, O.; Lindqvist, P.; Backman, C.; Westermark, P. Amyloid fibril composition is related to the phenotype of hereditary transthyretin V30M amyloidosis. J. Pathol. 2008, 216, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Ihse, E.; Rapezzi, C.; Merlini, G.; Benson, M.D.; Ando, Y.; Suhr, O.B.; Ikeda, S.; Lavatelli, F.; Obici, L.; Quarta, C.C.; et al. Amyloid fibrils containing fragmented ATTR may be the standard fibril composition in ATTR amyloidosis. Amyloid 2013, 20, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Koike, H.; Ando, Y.; Ueda, M.; Kawagashira, Y.; Iijima, M.; Fujitake, J.; Hayashi, M.; Yamamoto, M.; Mukai, E.; Nakamura, T.; et al. Distinct characteristics of amyloid deposits in early- and late-onset transthyretin Val30Met familial amyloid polyneuropathy. J. Neurol. Sci. 2009, 287, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Koike, H.; Nishi, R.; Ikeda, S.; Kawagashira, Y.; Iijima, M.; Sakurai, T.; Shimohata, T.; Katsuno, M.; Sobue, G. The morphology of amyloid fibrils and their impact on tissue damage in hereditary transthyretin amyloidosis: An ultrastructural study. J. Neurol. Sci. 2018, 394, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Bergström, J.; Gustavsson, A.; Hellman, U.; Sletten, K.; Murphy, C.L.; Weiss, D.T.; Solomon, A.; Olofsson, B.O.; Westermark, P. Amyloid deposits in transthyretin-derived amyloidosis: Cleaved transthyretin is associated with distinct amyloid morphology. J. Pathol. 2005, 206, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Yazaki, M.; Mitsuhashi, S.; Tokuda, T.; Kametani, F.; Takei, Y.I.; Koyama, J.; Kawamorita, A.; Kanno, H.; Ikeda, S.I. Progressive wild-type transthyretin deposition after liver transplantation preferentially occurs onto myocardium in FAP patients. Am. J. Transplant. 2007, 7, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, S.; Wixner, J.; Obayashi, K.; Ando, Y.; Ericzon, B.G.; Friman, S.; Uchino, M.; Suhr, O.B. Liver transplantation for familial amyloidotic polyneuropathy: Impact on Swedish patients’ survival. Liver Transplant. 2009, 15, 1229–1235. [Google Scholar] [CrossRef] [PubMed]
- Oshima, T.; Kawahara, S.; Ueda, M.; Kawakami, Y.; Tanaka, R.; Okazaki, T.; Misumi, Y.; Obayashi, K.; Yamashita, T.; Ohya, Y.; et al. Changes in pathological and biochemical findings of systemic tissue sites in familial amyloid polyneuropathy more than 10 years after liver transplantation. J. Neurol. Neurosurg. Psychiatry 2014, 85, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Marcoux, J.; Mangione, P.P.; Porcari, R.; Degiacomi, M.T.; Verona, G.; Taylor, G.W.; Giorgetti, S.; Raimondi, S.; Sanglier-Cianférani, S.; Benesch, J.L.; et al. A novel mechano-enzymatic cleavage mechanism underlies transthyretin amyloidogenesis. EMBO Mol. Med. 2015, 7, 1337–1349. [Google Scholar] [CrossRef] [PubMed]
- Sueyoshi, T.; Ueda, M.; Jono, H.; Irie, H.; Sei, A.; Ide, J.; Ando, Y.; Mizuta, H. Wild-type transthyretin-derived amyloidosis in various ligaments and tendons. Hum. Pathol. 2011, 42, 1259–1264. [Google Scholar] [CrossRef] [PubMed]
- Misumi, Y.; Ando, Y.; Ueda, M.; Obayashi, K.; Jono, H.; Su, Y.; Yamashita, T.; Uchino, M. Chain reaction of amyloid fibril formation with induction of basement membrane in familial amyloidotic polyneuropathy. J. Pathol. 2009, 219, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Richardson, S.J.; Aguilar, M.I.; Small, D.H. Binding of amyloidogenic transthyretin to the plasma membrane alters membrane fluidity and induces neurotoxicity. Biochemistry 2005, 44, 11618–11627. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.L.; Gandhi, S. Crucial role of protein oligomerization in the pathogenesis of Alzheimer’s and Parkinson’s diseases. FEBS J. 2018, 285, 3631–3644. [Google Scholar] [CrossRef] [PubMed]
- Madhivanan, K.; Greiner, E.R.; Alves-Ferreira, M.; Soriano-Castell, D.; Rouzbeh, N.; Aguirre, C.A.; Paulsson, J.F.; Chapman, J.; Jiang, X.; Ooi, F.K.; et al. Cellular clearance of circulating transthyretin decreases cell-nonautonomous proteotoxicity in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2018, 115, E7710–E7719. [Google Scholar] [CrossRef] [PubMed]
- Sousa, M.M.; Cardoso, I.; Fernandes, R.; Guimarães, A.; Saraiva, M.J. Deposition of transthyretin in early stages of familial amyloidotic polyneuropathy: Evidence for toxicity of nonfibrillar aggregates. Am. J. Pathol. 2001, 159, 1993–2000. [Google Scholar] [CrossRef]
- Monteiro, F.A.; Sousa, M.M.; Cardoso, I.; do Amaral, J.B.; Guimarães, A.; Saraiva, M.J. Activation of ERK1/2 MAP kinases in familial amyloidotic polyneuropathy. J. Neurochem. 2006, 97, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Fong, V.H.; Vieira, A. Pro-oxidative effects of aggregated transthyretin in human Schwannoma cells. Neurotoxicology 2013, 39, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Sousa, M.M.; Fernandes, R.; Palha, J.A.; Taboada, A.; Vieira, P.; Saraiva, M.J. Evidence for early cytotoxic aggregates in transgenic mice for human transthyretin Leu55Pro. Am. J. Pathol. 2002, 161, 1935–1948. [Google Scholar] [CrossRef]
- Ueda, M.; Ando, Y.; Hakamata, Y.; Nakamura, M.; Yamashita, T.; Obayashi, K.; Himeno, S.; Inoue, S.; Sato, Y.; Kaneko, T.; et al. A transgenic rat with the human ATTR V30M: A novel tool for analyses of ATTR metabolisms. Biochem. Biophys. Res. Commun. 2007, 352, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Naharro, A.; Treibel, T.A.; Abdel-Gadir, A.; Bulluck, H.; Zumbo, G.; Knight, D.S.; Kotecha, T.; Francis, R.; Hutt, D.F.; Rezk, T.; et al. Magnetic Resonance in Transthyretin Cardiac Amyloidosis. J. Am. Coll. Cardiol. 2017, 70, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, A.; Terrada, C.; Touhami, S.; Barreau, E.; Rothschild, P.R.; Valleix, S.; Benoudiba, F.; Errera, M.H.; Cauquil, C.; Guiochon-Mantel, A.; et al. Angiographic signatures of the predominant form of familial transthyretin amyloidosis (Val30Met Mutation). Am. J. Ophthalmol. 2018, 192, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yi, S.; Kimura, Y.; Araki, S. Familial amyloidotic polyneuropathy type 1 in Kumamoto, Japan: A clinicopathologic, histochemical, immunohistochemical, and ultrastructural study. Hum. Pathol. 1991, 22, 519–527. [Google Scholar] [CrossRef]
- Said, G.; Planté-Bordeneuve, V. Familial amyloid polyneuropathy: A clinico-pathologic study. J. Neurol. Sci. 2009, 284, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Kollmer, J.; Sahm, F.; Hegenbart, U.; Purrucker, J.C.; Kimmich, C.; Schönland, S.O.; Hund, E.; Heiland, S.; Hayes, J.M.; Kristen, A.V.; et al. Sural nerve injury in familial amyloid polyneuropathy: MR neurography vs. clinicopathologic tools. Neurology 2017, 89, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Nunes, R.J.; de Oliveira, P.; Lages, A.; Becker, J.D.; Marcelino, P.; Barroso, E.; Perdigoto, R.; Kelly, J.W.; Quintas, A.; Santos, S.C. Transthyretin proteins regulate angiogenesis by conferring different molecular identities to endothelial cells. J. Biol. Chem. 2013, 288, 31752–31760. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Yin, Y.; Yin, X.; Ji, L.; Xin, Y.; Zou, J.; Yao, Y. Transthyretin exerts pro-apoptotic effects in human retinal microvascular endothelial cells through a GRP78-dependent pathway in diabetic retinopathy. Cell Physiol. Biochem. 2017, 43, 788–800. [Google Scholar] [CrossRef] [PubMed]
- Du, M.; Martin, A.; Hays, F.; Johnson, J.; Farjo, R.A.; Farjo, K.M. Serum retinol-binding protein-induced endothelial inflammation is mediated through the activation of toll-like receptor 4. Mol. Vis. 2017, 23, 185–197. [Google Scholar] [PubMed]
- Hosoi, A.; Su, Y.; Torikai, M.; Jono, H.; Ishikawa, D.; Soejima, K.; Higuchi, H.; Guo, J.; Ueda, M.; Suenaga, G.; et al. Novel Antibody for the Treatment of Transthyretin Amyloidosis. J. Biol. Chem. 2016, 291, 25096–25105. [Google Scholar] [CrossRef] [PubMed]
- Liepnieks, J.J.; Zhang, L.Q.; Benson, M.D. Progression of transthyretin amyloid neuropathy after liver transplantation. Neurology 2010, 75, 324–327. [Google Scholar] [CrossRef] [PubMed]
- Koike, H.; Hashimoto, R.; Tomita, M.; Kawagashira, Y.; Iijima, M.; Nakamura, T.; Watanabe, H.; Kamei, H.; Kiuchi, T.; Sobue, G. Impact of aging on the progression of neuropathy after liver transplantation in transthyretin Val30Met amyloidosis. Muscle Nerve 2012, 46, 964–970. [Google Scholar] [CrossRef] [PubMed]
- aus dem Siepen, F.; Bauer, R.; Aurich, M.; Buss, S.J.; Steen, H.; Altland, K.; Katus, H.A.; Kristen, A.V. Green tea extract as a treatment for patients with wild-type transthyretin amyloidosis: An observational study. Drug Des. Dev. Ther. 2015, 9, 6319–6325. [Google Scholar] [CrossRef] [PubMed]
- Rapezzi, C.; Lorenzini, M.; Longhi, S.; Milandri, A.; Gagliardi, C.; Bartolomei, I.; Salvi, F.; Maurer, M.S. Cardiac amyloidosis: The great pretender. Heart Fail. Rev. 2015, 20, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Said, G.; Ropert, A.; Faux, N. Length-dependent degeneration of fibers in Portuguese amyloid polyneuropathy: A clinicopathologic study. Neurology 1984, 34, 1025–1032. [Google Scholar] [CrossRef] [PubMed]
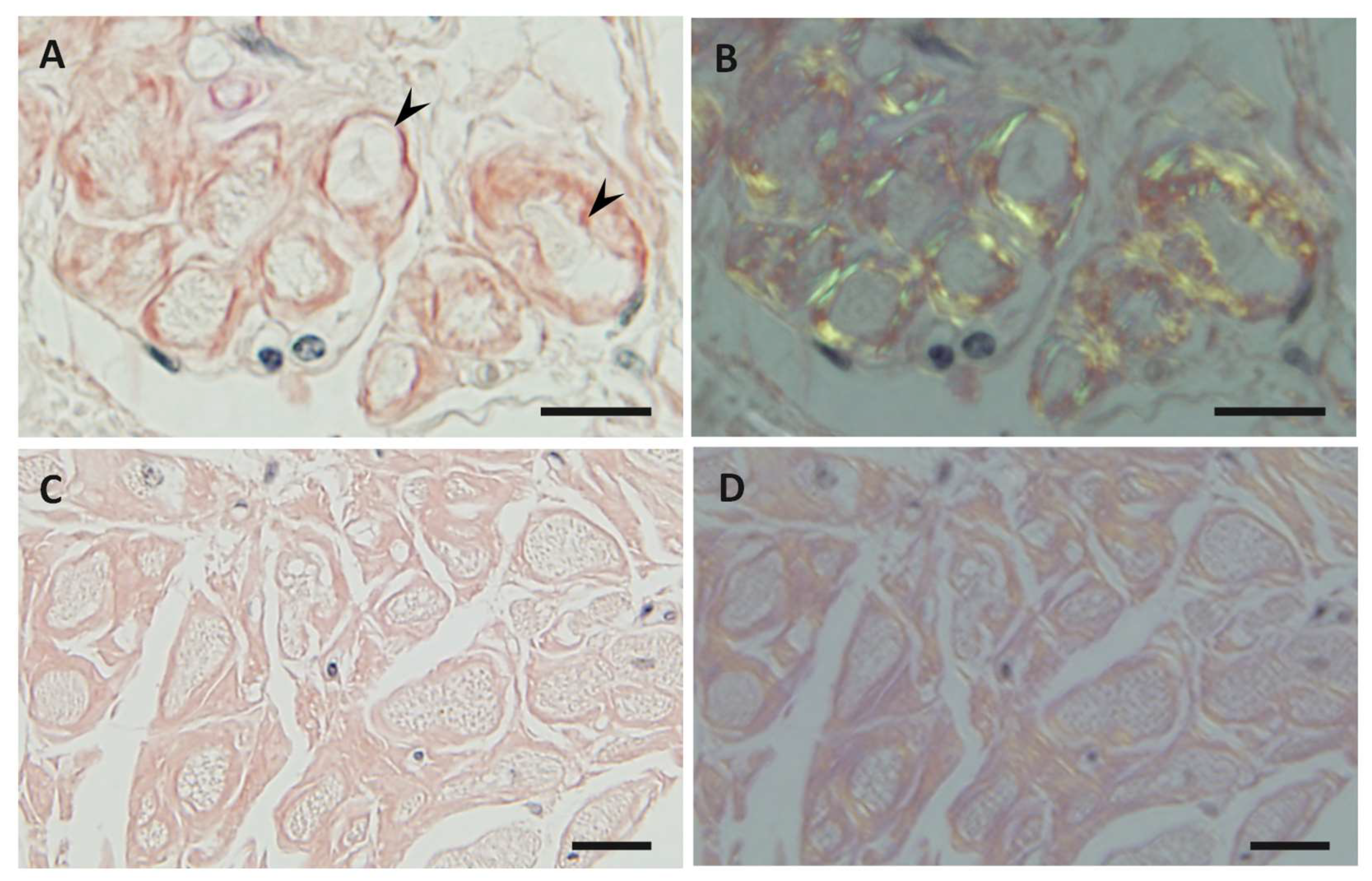
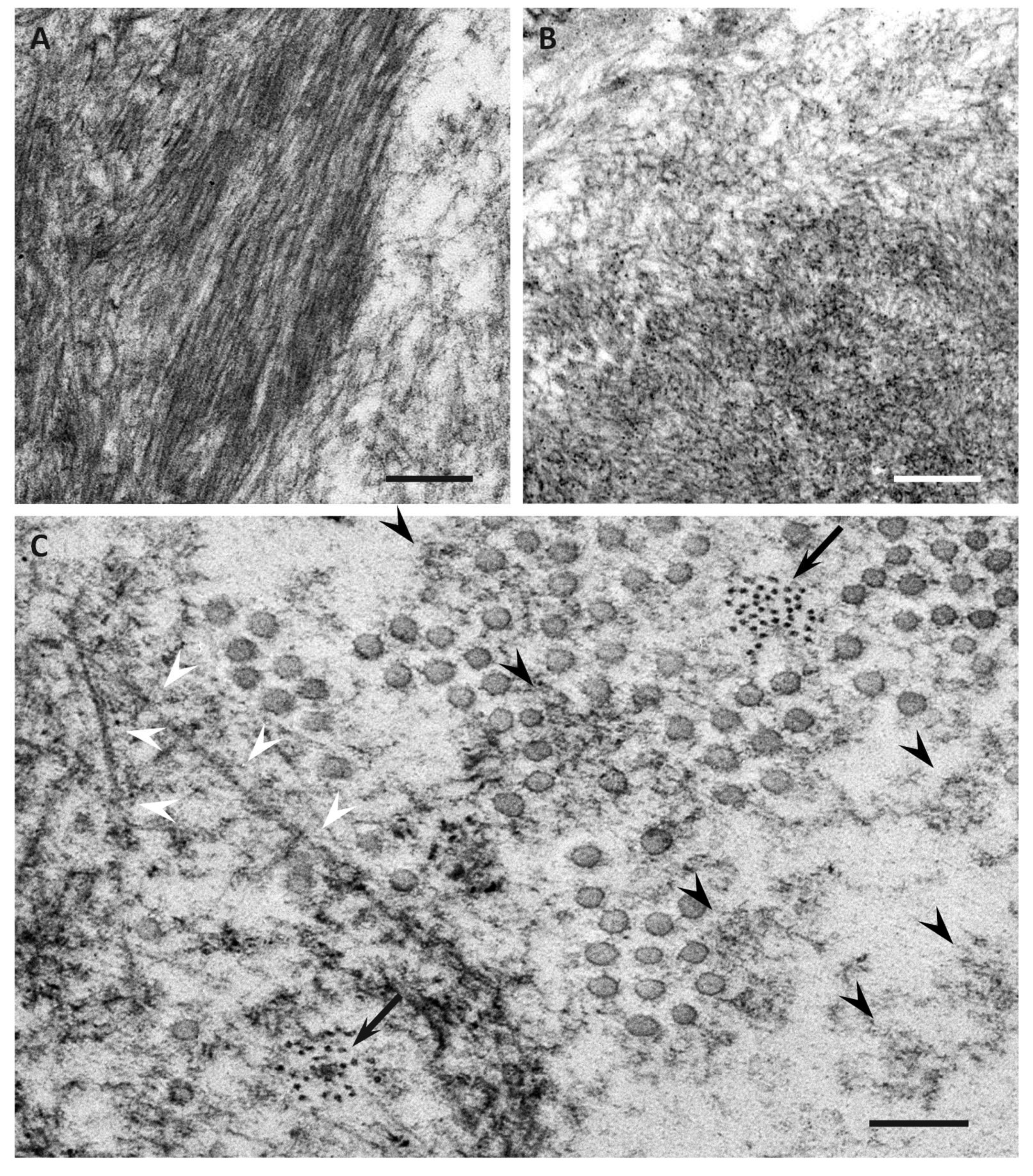
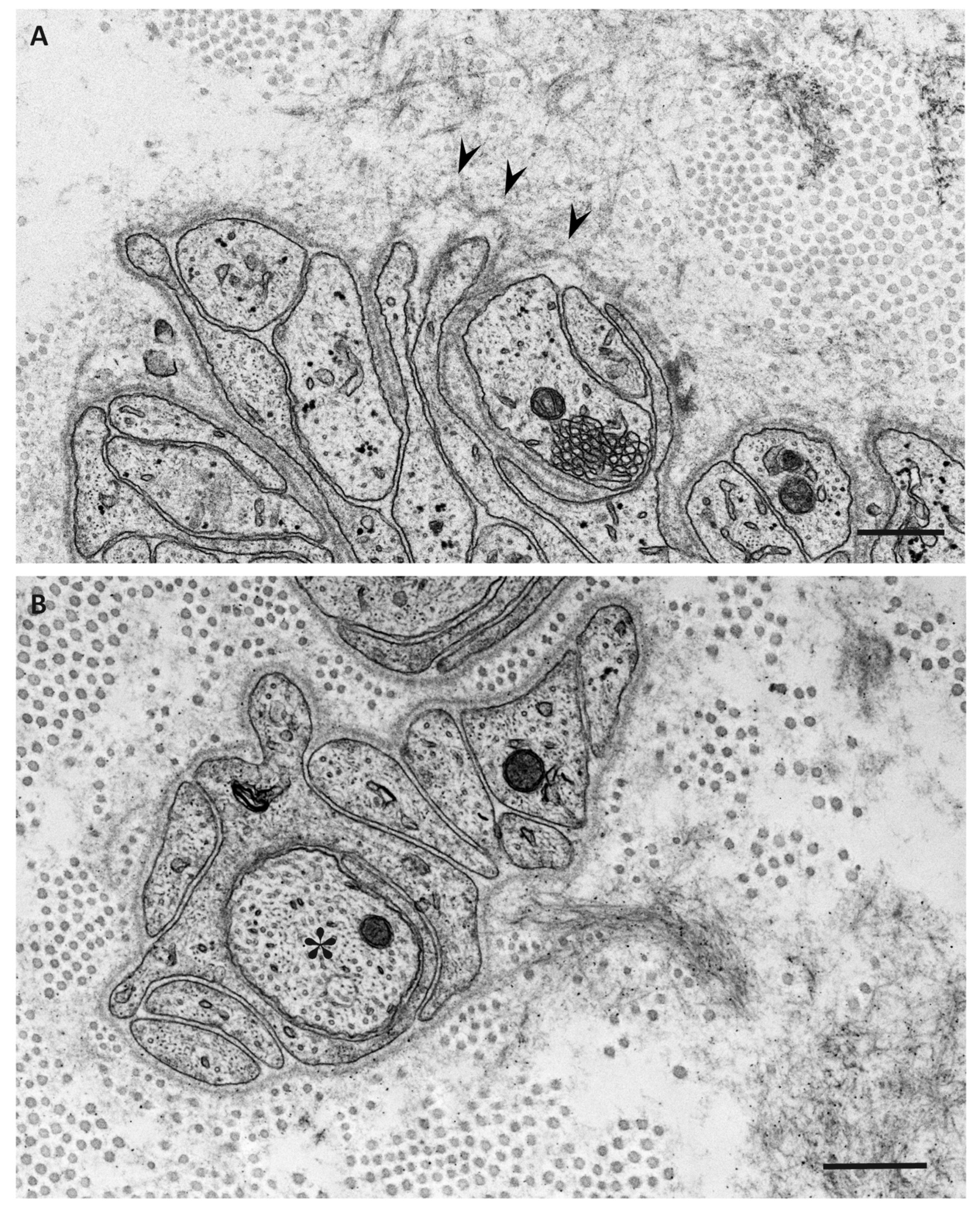

{kind=link}
{kind=link}
{kind=link}
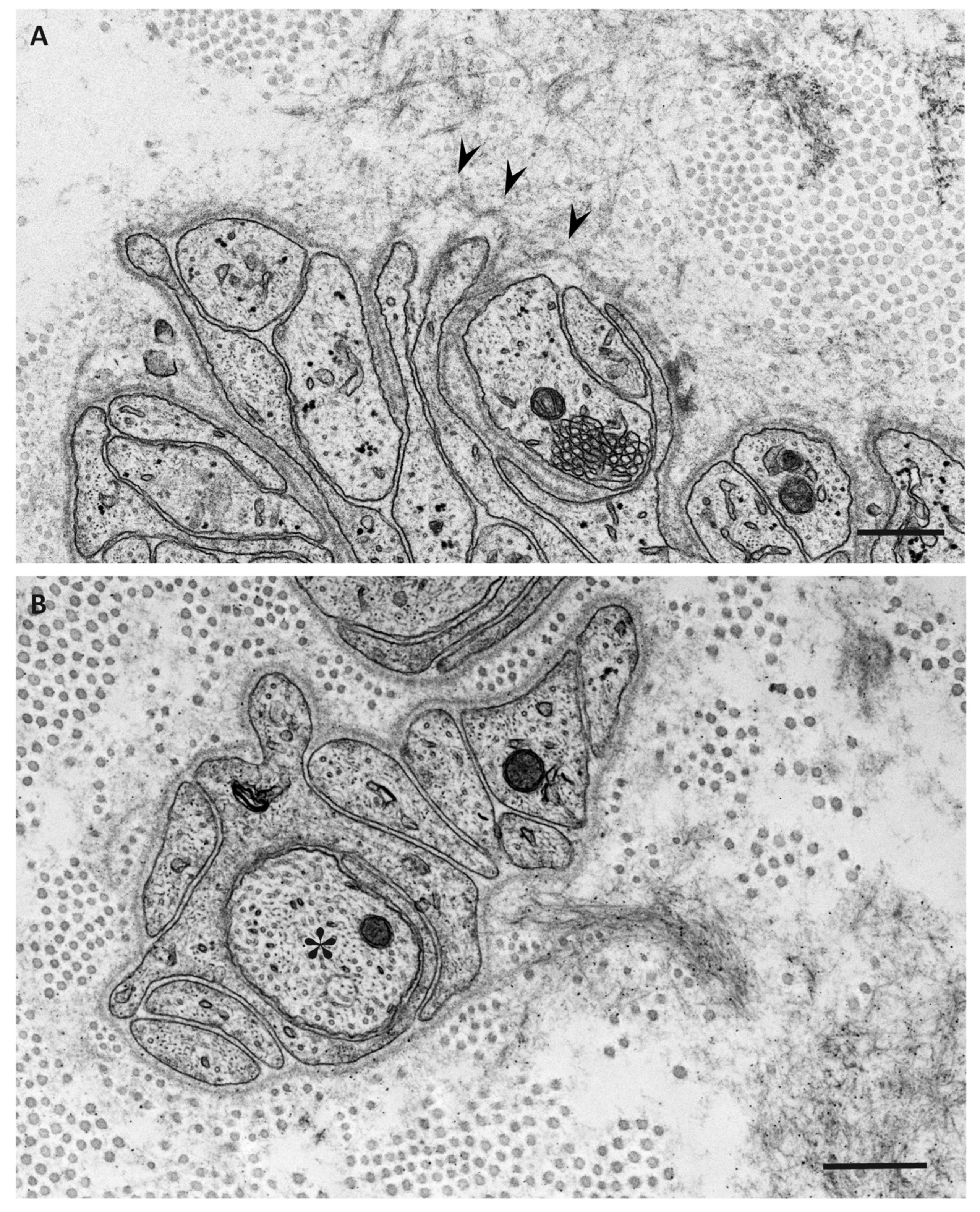{kind=link}
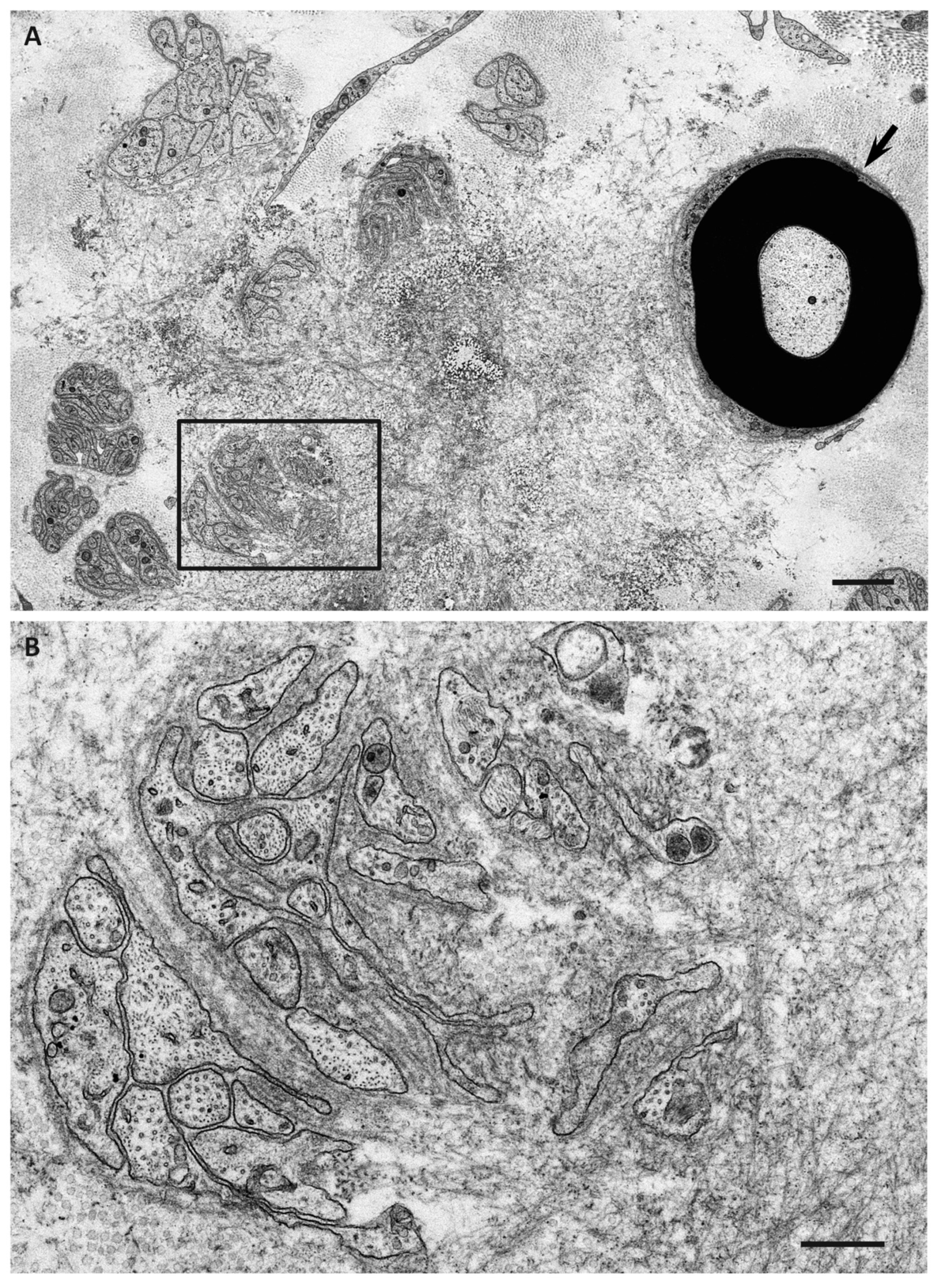{kind=link}
{kind=link}
| Features | Early-Onset Patients from Endemic Foci | Late-Onset Patients from Nonendemic Areas |
|---|---|---|
| Age of onset | Late 20s to early 40s | ≥50 years |
| Sex | Male = female | Male > female |
| Family history | Common | Frequently absent |
| Penetrance rate | High | Low |
| Cardiac involvement | Conduction defects | Heart failure |
| Sensory dissociation | Common | Rare |
| Autonomic dysfunction | Severe | Mild |
| in early disease stage | ||
| Modality of nerve fiber loss | Small > large | Small = large |
| Amount of amyloid deposits | Large | Small |
| in the peripheral nervous system | ||
| Length of amyloid fibrils | Long | Short |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koike, H.; Katsuno, M. Ultrastructure in Transthyretin Amyloidosis: From Pathophysiology to Therapeutic Insights. Biomedicines 2019, 7, 11. https://doi.org/10.3390/biomedicines7010011
Koike H, Katsuno M. Ultrastructure in Transthyretin Amyloidosis: From Pathophysiology to Therapeutic Insights. Biomedicines. 2019; 7(1):11. https://doi.org/10.3390/biomedicines7010011
Chicago/Turabian StyleKoike, Haruki, and Masahisa Katsuno. 2019. "Ultrastructure in Transthyretin Amyloidosis: From Pathophysiology to Therapeutic Insights" Biomedicines 7, no. 1: 11. https://doi.org/10.3390/biomedicines7010011
APA StyleKoike, H., & Katsuno, M. (2019). Ultrastructure in Transthyretin Amyloidosis: From Pathophysiology to Therapeutic Insights. Biomedicines, 7(1), 11. https://doi.org/10.3390/biomedicines7010011