Subunit-Specific Role of NF-κB in Cancer


 ,
,
Abstract
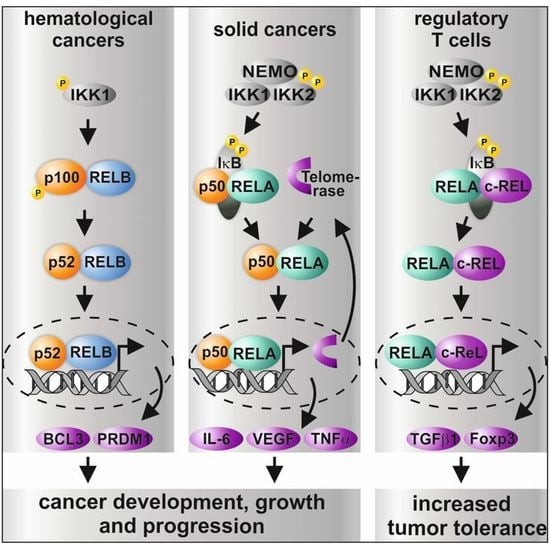
1. The NF-κB Family—An Introduction
2. NF-κB in Inflammation and Cancer
3. Autoregulation of NF-κB—A Potential Driver on the Road to Cancer Development?
4. Activity of Distinct NF-κB Upstream Kinases in Cancer
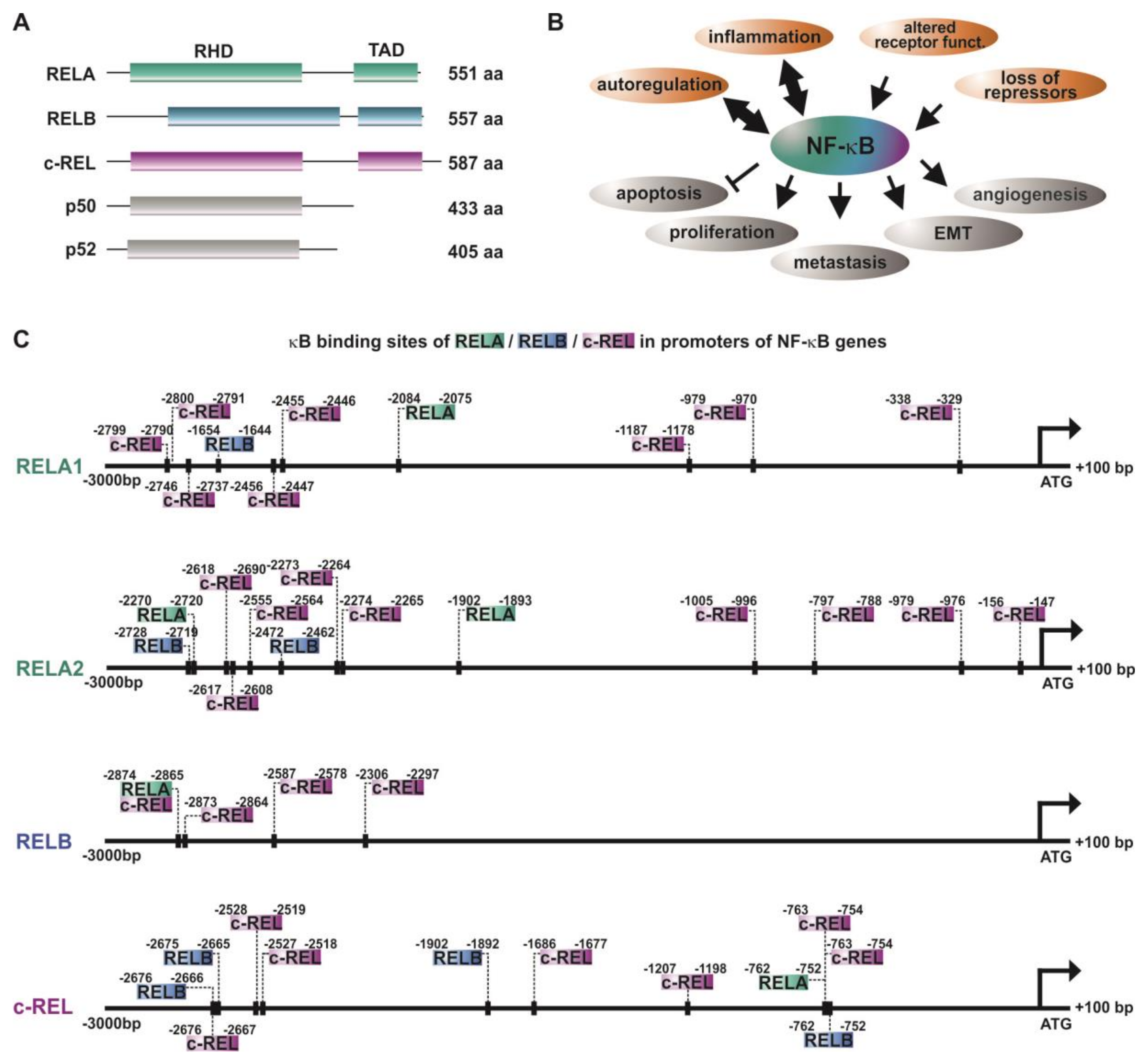
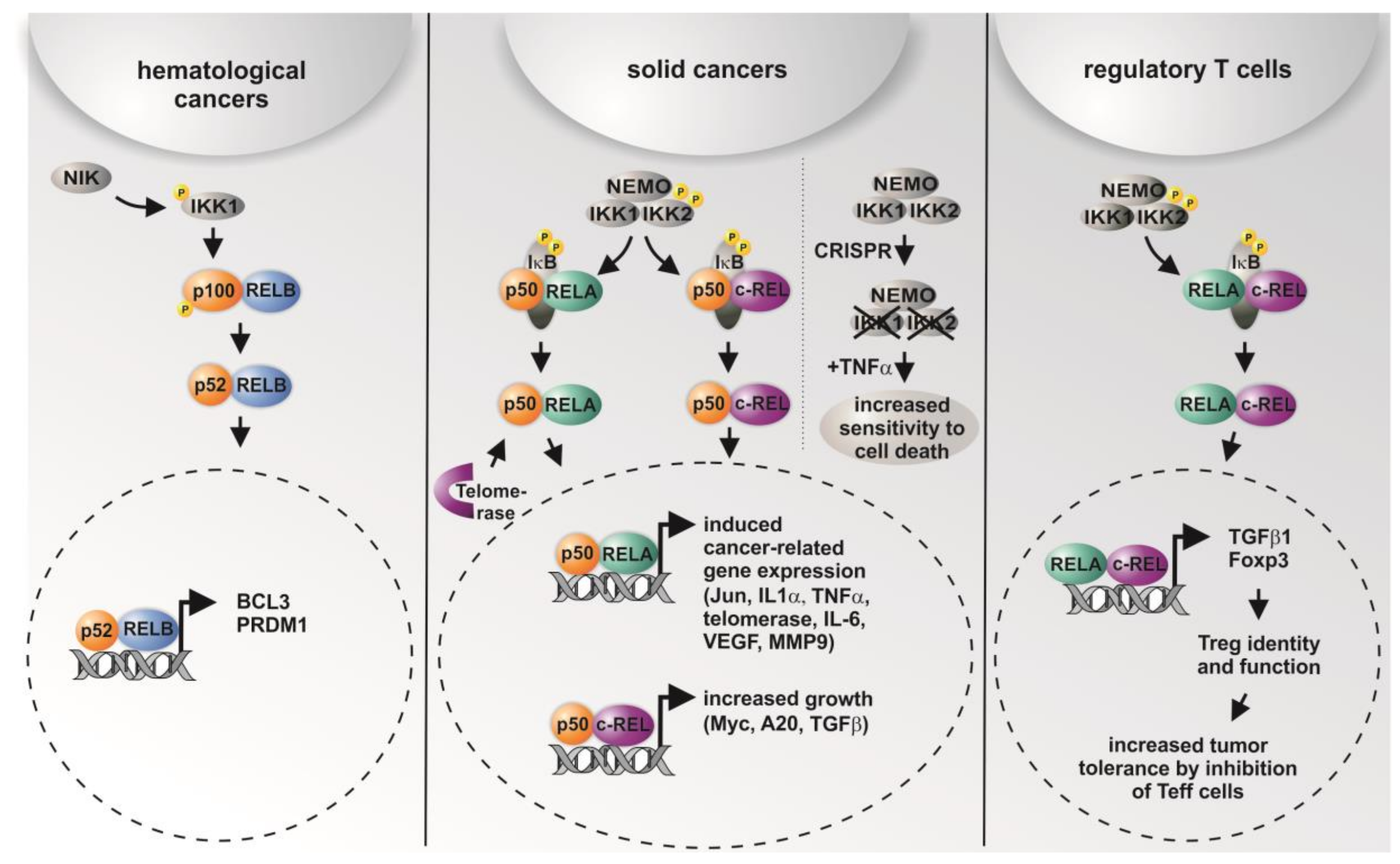
5. Differential Roles of NF-κB Subunits in Cancer
6. Targeting NF-κB Subunits via Genome Editing or Drugs—Therapeutic Implications
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Sen, R.; Baltimore, D. Inducibility of kappa immunoglobulin enhancer-binding protein NF-kappaB by a posttranslational mechanism. Cell 1986, 47, 921–928. [Google Scholar] [CrossRef]
- Sen, R.; Baltimore, D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell 1986, 46, 705–716. [Google Scholar] [CrossRef]
- Perkins, N.D. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat. Rev. Mol. Cell Biol. 2007, 8, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Kaltschmidt, B.; Kaltschmidt, C. NF-kappaB in the nervous system. Cold Spring Harb. Perspect. Biol. 2009, 1, a001271. [Google Scholar] [CrossRef] [PubMed]
- Kaltschmidt, B.; Kaltschmidt, C. NF-kappaB in long-term memory and structural plasticity in the adult mammalian brain. Front. Mol. Neurosci. 2015, 8, 69. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. NF-kappaB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef] [PubMed]
- Ben-Neriah, Y.; Karin, M. Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat. Immunol. 2011, 12, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Shen, S.; Verma, I.M. NF-kappaB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.E.; Huang, D.B.; Chen, Y.Q.; Ghosh, G. Crystal structure of p50/p65 heterodimer of transcription factor NF-kappaB bound to DNA. Nature 1998, 391, 410–413. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. The diverse and complex roles of NF-kappaB subunits in cancer. Nat. Rev. Cancer 2012, 12, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Slotta, C.; Schluter, T.; Ruiz-Perera, L.M.; Kadhim, H.M.; Tertel, T.; Henkel, E.; Hubner, W.; Greiner, J.F.W.; Huser, T.; Kaltschmidt, B.; et al. Crispr/cas9-mediated knockout of c-rel in hela cells results in profound defects of the cell cycle. PLoS ONE 2017, 12, e0182373. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Fornes, O.; Stigliani, A.; Gheorghe, M.; Castro-Mondragon, J.A.; van der Lee, R.; Bessy, A.; Cheneby, J.; Kulkarni, S.R.; Tan, G.; et al. Jaspar 2018: Update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 2018, 46, D260–D266. [Google Scholar] [CrossRef] [PubMed]
- Senftleben, U.; Cao, Y.; Xiao, G.; Greten, F.R.; Krahn, G.; Bonizzi, G.; Chen, Y.; Hu, Y.; Fong, A.; Sun, S.C.; et al. Activation by ikkalpha of a second, evolutionary conserved, nf-kappa b signaling pathway. Science 2001, 293, 1495–1499. [Google Scholar] [CrossRef] [PubMed]
- Hoesel, B.; Schmid, J.A. The complexity of NF-kappaB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Alberti, C.; Pinciroli, P.; Valeri, B.; Ferri, R.; Ditto, A.; Umezawa, K.; Sensi, M.; Canevari, S.; Tomassetti, A. Ligand-dependent egfr activation induces the co-expression of il-6 and pai-1 via the nfkb pathway in advanced-stage epithelial ovarian cancer. Oncogene 2012, 31, 4139–4149. [Google Scholar] [CrossRef] [PubMed]
- Greiner, J.F.; Muller, J.; Zeuner, M.T.; Hauser, S.; Seidel, T.; Klenke, C.; Grunwald, L.M.; Schomann, T.; Widera, D.; Sudhoff, H.; et al. 1,8-cineol inhibits nuclear translocation of NF-kappaB p65 and NF-kappaB-dependent transcriptional activity. Biochim. Biophys. Acta 2013, 1833, 2866–2878. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Greiner, J.F.; Zeuner, M.; Brotzmann, V.; Schafermann, J.; Wieters, F.; Widera, D.; Sudhoff, H.; Kaltschmidt, B.; Kaltschmidt, C. 1,8-cineole potentiates irf3-mediated antiviral response in human stem cells and in an ex vivo model of rhinosinusitis. Clin. Sci. 2016, 130, 1339–1352. [Google Scholar] [CrossRef] [PubMed]
- Shakhov, A.N.; Collart, M.A.; Vassalli, P.; Nedospasov, S.A.; Jongeneel, C.V. Kappa b-type enhancers are involved in lipopolysaccharide-mediated transcriptional activation of the tumor necrosis factor alpha gene in primary macrophages. J. Exp. Med. 1990, 171, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Collart, M.A.; Baeuerle, P.; Vassalli, P. Regulation of tumor necrosis factor alpha transcription in macrophages: Involvement of four kappa B-like motifs and of constitutive and inducible forms of NF-kappaB. Mol. Cell. Biol. 1990, 10, 1498–1506. [Google Scholar] [CrossRef] [PubMed]
- Mori, N.; Prager, D. Transactivation of the interleukin-1alpha promoter by human t-cell leukemia virus type i and type ii tax proteins. Blood 1996, 87, 3410–3417. [Google Scholar] [PubMed]
- Serfling, E.; Barthelmas, R.; Pfeuffer, I.; Schenk, B.; Zarius, S.; Swoboda, R.; Mercurio, F.; Karin, M. Ubiquitous and lymphocyte-specific factors are involved in the induction of the mouse interleukin 2 gene in t lymphocytes. EMBO J. 1989, 8, 465–473. [Google Scholar] [PubMed]
- Kunsch, C.; Rosen, C.A. NF-kappaB subunit-specific regulation of the interleukin-8 promoter. Mol. Cell. Biol. 1993, 13, 6137–6146. [Google Scholar] [CrossRef] [PubMed]
- Disis, M.L. Immune regulation of cancer. J. Clin. Oncol. 2010, 28, 4531–4538. [Google Scholar] [CrossRef] [PubMed]
- Nakshatri, H.; Bhat-Nakshatri, P.; Martin, D.A.; Goulet, R.J., Jr.; Sledge, G.W., Jr. Constitutive activation of NF-kappaB during progression of breast cancer to hormone-independent growth. Mol. Cell. Biol. 1997, 17, 3629–3639. [Google Scholar] [CrossRef] [PubMed]
- Baud, V.; Karin, M. Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 2009, 8, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Terzic, J.; Grivennikov, S.; Karin, E.; Karin, M. Inflammation and colon cancer. Gastroenterology 2010, 138, 2101–2114. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Pettaway, C.A.; Uehara, H.; Bucana, C.D.; Fidler, I.J. Blockade of NF-kappaB activity in human prostate cancer cells is associated with suppression of angiogenesis, invasion, and metastasis. Oncogene 2001, 20, 4188–4197. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Karin, M. NF-kappaB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018. [Google Scholar] [CrossRef]
- Hannink, M.; Temin, H.M. Structure and autoregulation of the c-rel promoter. Oncogene 1990, 5, 1843–1850. [Google Scholar] [PubMed]
- Bren, G.D.; Solan, N.J.; Miyoshi, H.; Pennington, K.N.; Pobst, L.J.; Paya, C.V. Transcription of the relb gene is regulated by NF-kappaB. Oncogene 2001, 20, 7722–7733. [Google Scholar] [CrossRef] [PubMed]
- Linker, R.A.; Baeuerle, P.A.; Kaltschmidt, C. Cloning of the murine rela (p65 nf-kappa b) gene and comparison to the human gene reveals a distinct first intron. Gene 1996, 176, 119–124. [Google Scholar] [CrossRef]
- Capobianco, A.J.; Gilmore, T.D. Repression of the chicken c-rel promoter by vrel in chicken embryo fibroblasts is not mediated through a consensus NF-kappaB binding site. Oncogene 1991, 6, 2203–2210. [Google Scholar] [PubMed]
- Lombardi, L.; Ciana, P.; Cappellini, C.; Trecca, D.; Guerrini, L.; Migliazza, A.; Maiolo, A.T.; Neri, A. Structural and functional characterization of the promoter regions of the NFKB2 gene. Nucleic Acids Res. 1995, 23, 2328–2336. [Google Scholar] [CrossRef] [PubMed]
- Ten, R.M.; Paya, C.V.; Israel, N.; Le Bail, O.; Mattei, M.G.; Virelizier, J.L.; Kourilsky, P.; Israel, A. The characterization of the promoter of the gene encoding the p50 subunit of NF-kappaB indicates that it participates in its own regulation. EMBO J. 1992, 11, 195–203. [Google Scholar] [PubMed]
- Forbes, S.A.; Beare, D.; Gunasekaran, P.; Leung, K.; Bindal, N.; Boutselakis, H.; Ding, M.; Bamford, S.; Cole, C.; Ward, S.; et al. Cosmic: Exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015, 43, D805–D811.38. [Google Scholar] [CrossRef] [PubMed]
- Slotta, C.; Storm, J.; Pfisterer, N.; Henkel, E.; Kleinwachter, S.; Pieper, M.; Ruiz-Perera, L.M.; Greiner, J.F.W.; Kaltschmidt, B.; Kaltschmidt, C. Ikk1/2 protect human cells from tnf-mediated ripk1-dependent apoptosis in an nf-kappab-independent manner. Biochim. Biophys. Acta 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Yeddula, N.; Leblanc, M.; Ke, E.; Zhang, Y.; Oldfield, E.; Shaw, R.J.; Verma, I.M. Reduced cell proliferation by ikk2 depletion in a mouse lung-cancer model. Nat. Cell Biol. 2012, 14, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Van Antwerp, D.; Mercurio, F.; Lee, K.F.; Verma, I.M. Severe liver degeneration in mice lacking the ikappab kinase 2 gene. Science 1999, 284, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.W.; Chu, W.; Hu, Y.; Delhase, M.; Deerinck, T.; Ellisman, M.; Johnson, R.; Karin, M. The ikkbeta subunit of ikappab kinase (ikk) is essential for nuclear factor kappab activation and prevention of apoptosis. J. Exp. Med. 1999, 189, 1839–1845. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. Ikkbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004, 118, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Vlantis, K.; Wullaert, A.; Sasaki, Y.; Schmidt-Supprian, M.; Rajewsky, K.; Roskams, T.; Pasparakis, M. Constitutive ikk2 activation in intestinal epithelial cells induces intestinal tumors in mice. J. Clin. Investig. 2011, 121, 2781–2793. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Ogata, H.; Nishigaki, R.; Broide, D.H.; Karin, M. Tobacco smoke promotes lung tumorigenesis by triggering ikkbeta- and jnk1-dependent inflammation. Cancer Cell 2010, 17, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.A.; Azoitei, N.; Baumann, B.; Grunert, S.; Sommer, A.; Pehamberger, H.; Kraut, N.; Beug, H.; Wirth, T. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J. Clin. Investig. 2004, 114, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Sterling, J.A.; Edwards, J.R.; DeGraff, D.J.; Lee, C.; Park, S.I.; Matusik, R.J. Activation of nf-kappa b signaling promotes growth of prostate cancer cells in bone. PLoS ONE 2013, 8, e60983. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.L.; Tan, W.; Ricono, J.M.; Korchynskyi, O.; Zhang, M.; Gonias, S.L.; Cheresh, D.A.; Karin, M. Nuclear cytokine-activated ikkalpha controls prostate cancer metastasis by repressing maspin. Nature 2007, 446, 690–694. [Google Scholar] [CrossRef] [PubMed]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Brandl, C.; Florian, C.; Driemel, O.; Weber, B.H.; Morsczeck, C. Identification of neural crest-derived stem cell-like cells from the corneal limbus of juvenile mice. Exp. Eye Res. 2009, 89, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Kaltschmidt, B.; Kaltschmidt, C.; Hehner, S.P.; Droge, W.; Schmitz, M.L. Repression of NF-kappaB impairs hela cell proliferation by functional interference with cell cycle checkpoint regulators. Oncogene 1999, 18, 3213–3225. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, K.A.; Kaergel, E.; Heinig, M.; Fontaine, J.F.; Patone, G.; Muro, E.M.; Mathas, S.; Hummel, M.; Andrade-Navarro, M.A.; Hubner, N.; et al. A roadmap of constitutive NF-kappaB activity in hodgkin lymphoma: Dominant roles of p50 and p52 revealed by genome-wide analyses. Genome Med. 2016, 8, 28. [Google Scholar] [CrossRef] [PubMed]
- Keats, J.J.; Fonseca, R.; Chesi, M.; Schop, R.; Baker, A.; Chng, W.J.; Van Wier, S.; Tiedemann, R.; Shi, C.X.; Sebag, M.; et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell 2007, 12, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Toma, J.G.; Akhavan, M.; Fernandes, K.J.; Barnabe-Heider, F.; Sadikot, A.; Kaplan, D.R.; Miller, F.D. Isolation of multipotent adult stem cells from the dermis of mammalian skin. Nat. Cell Biol. 2001, 3, 778–784. [Google Scholar] [CrossRef] [PubMed]
- Techawattanawisal, W.; Nakahama, K.; Komaki, M.; Abe, M.; Takagi, Y.; Morita, I. Isolation of multipotent stem cells from adult rat periodontal ligament by neurosphere-forming culture system. Biochem. Biophys. Res. Commun. 2007, 357, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Q.; Kameswaran, V.; Tone, Y.; Li, L.; Liou, H.C.; Greene, M.I.; Tone, M.; Chen, Y.H. Development of foxp3(+) regulatory T cells is driven by the c-Rel enhanceosome. Immunity 2009, 31, 932–940. [Google Scholar] [CrossRef] [PubMed]
- Libermann, T.A.; Baltimore, D. Activation of interleukin-6 gene expression through the NF-kappaB transcription factor. Mol. Cell. Biol. 1990, 10, 2327–2334. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; MacLennan, G.T.; Fu, P.; Patel, J.; Marengo, S.R.; Resnick, M.I.; Gupta, S. Nuclear factor-kappaB/p65 (Rel A) is constitutively activated in human prostate adenocarcinoma and correlates with disease progression. Neoplasia 2004, 6, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Saginc, G.; Leow, S.C.; Khattar, E.; Shin, E.M.; Yan, T.D.; Wong, M.; Zhang, Z.; Li, G.; Sung, W.K.; et al. Telomerase directly regulates NF-kappaB-dependent transcription. Nat. Cell Biol. 2012, 14, 1270–1281. [Google Scholar] [CrossRef] [PubMed]
- Widera, D.; Zander, C.; Heidbreder, M.; Kasperek, Y.; Noll, T.; Seitz, O.; Saldamli, B.; Sudhoff, H.; Sader, R.; Kaltschmidt, C.; et al. Adult palatum as a novel source of neural crest-related stem cells. Stem Cells 2009, 27, 1899–1910. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rajasekhar, V.K.; Studer, L.; Gerald, W.; Socci, N.D.; Scher, H.I. Tumour-initiating stem-like cells in human prostate cancer exhibit increased NF-kappaB signalling. Nat. Commun. 2011, 2, 162. [Google Scholar] [CrossRef] [PubMed]
- Gannon, P.O.; Lessard, L.; Stevens, L.M.; Forest, V.; Begin, L.R.; Minner, S.; Tennstedt, P.; Schlomm, T.; Mes-Masson, A.M.; Saad, F. Large-scale independent validation of the nuclear factor-kappaB p65 prognostic biomarker in prostate cancer. Eur. J. Cancer 2013, 49, 2441–2448. [Google Scholar] [CrossRef] [PubMed]
- Basseres, D.S.; Ebbs, A.; Levantini, E.; Baldwin, A.S. Requirement of the NF-kappaB subunit p65/RelA for K-Ras-induced lung tumorigenesis. Cancer Res. 2010, 70, 3537–3546. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, T.; Roth, J.A.; Maxwell, S.A. Altered expression of the p50 subunit of the NF-kappaB transcription factor complex in non-small cell lung carcinoma. Oncogene 1995, 11, 999–1003. [Google Scholar] [PubMed]
- Ling, J.; Kang, Y.; Zhao, R.; Xia, Q.; Lee, D.F.; Chang, Z.; Li, J.; Peng, B.; Fleming, J.B.; Wang, H.; et al. Krasg12d-induced ikk2/beta/NF-kappaB activation by il-1alpha and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 105–120. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.; Venkatraman, M.; Maliekal, T.T.; Nair, B.; Karunagaran, D. NF-kappaB is constitutively activated in high-grade squamous intraepithelial lesions and squamous cell carcinomas of the human uterine cervix. Oncogene 2003, 22, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Cogswell, P.C.; Guttridge, D.C.; Funkhouser, W.K.; Baldwin, A.S., Jr. Selective activation of nf-kappa b subunits in human breast cancer: Potential roles for NF-kappaB2/p52 and for Bcl-3. Oncogene 2000, 19, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Shehata, M.; Shehata, F.; Pater, A. Apoptosis effects of xrel3 c-Rel/Nuclear factor-kappa B homolog in human cervical cancer cells. Cell Biol. Int. 2005, 29, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Grinberg-Bleyer, Y.; Oh, H.; Desrichard, A.; Bhatt, D.M.; Caron, R.; Chan, T.A.; Schmid, R.M.; Klein, U.; Hayden, M.S.; Ghosh, S. NF-kappaB c-Rel is crucial for the regulatory T cell immune checkpoint in cancer. Cell 2017, 170, 1096–1108. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, H.; Sakaguchi, S. Regulatory t cells in tumor immunity. Int. J. Cancer 2010, 127, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Jandus, C.; Bioley, G.; Speiser, D.E.; Romero, P. Selective accumulation of differentiated foxp3(+) cd4 (+) T cells in metastatic tumor lesions from melanoma patients compared to peripheral blood. Cancer Immunol. Immunother. 2008, 57, 1795–1805. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, J.M.; Gonzalez, R.; Lewis, K.D.; Robinson, W.A.; Richter, D.A.; Palmer, B.E.; Wilson, C.C.; McCarter, M.D. Increased survival from stage IV melanoma associated with fewer regulatory T cells. J. Surg. Res. 2009, 154, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.; Kauffman, M. Development of the proteasome inhibitor velcade (bortezomib). Cancer Investig. 2004, 22, 304–311. [Google Scholar] [CrossRef]
- Majumdar, S.; Lamothe, B.; Aggarwal, B.B. Thalidomide suppresses NF-kappa B activation induced by TNF and H2O2, but not that activated by ceramide, lipopolysaccharides, or phorbol ester. J. Immunol. 2002, 168, 2644–2651. [Google Scholar] [CrossRef] [PubMed]
- Singhal, S.; Mehta, J. Thalidomide in cancer. Biomed. Pharmacother. 2002, 56, 4–12. [Google Scholar] [CrossRef]
- Strasser, K.; Ludwig, H. Thalidomide treatment in multiple myeloma. Blood Rev. 2002, 16, 207–215. [Google Scholar] [CrossRef]
- Carcamo, J.M.; Pedraza, A.; Borquez-Ojeda, O.; Golde, D.W. Vitamin C suppresses TNF alpha-induced NF kappa B activation by inhibiting I kappa B alpha phosphorylation. Biochemistry 2002, 41, 12995–13002. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Cancer Tissue | RELA | RELB | c-REL | |||
|---|---|---|---|---|---|---|
| % Overexpressed | No. Tested | % Overexpressed | No. Tested | % Overexpressed | No. Tested | |
| Ovary | 11.65 | 266 | 3.38 | 266 | 7.52 | 266 |
| Lung | 2.36 | 1019 | 4.12 | 1019 | 7.26 | 1019 |
| Urinary tract | 2.45 | 408 | 4.41 | 408 | 7.11 | 408 |
| Endometrium | 1.99 | 602 | 8.8 | 602 | 6.81 | 602 |
| Pancreas | 2.79 | 179 | 6.7 | 179 | 6.7 | 179 |
| Haematopoietic and lymphoid | 4.07 | 221 | 1.36 | 221 | 6.33 | 221 |
| Soft tissue | 3.42 | 263 | 1.9 | 263 | 6.08 | 263 |
| Cervix | 1.3 | 307 | 7.17 | 307 | 5.86 | 307 |
| Upper aerodigestive tract | 2.49 | 522 | 4.02 | 522 | 5.75 | 522 |
| Kidney | 2.83 | 600 | 4.5 | 600 | 5.5 | 600 |
| Thyroid | 1.36 | 513 | 3.7 | 513 | 5.46 | 513 |
| Large intestine | 1.87 | 610 | 5.25 | 610 | 4.92 | 610 |
| Stomach | 7.02 | 285 | 7.37 | 285 | 4.91 | 285 |
| Liver | 3.75 | 373 | 6.97 | 373 | 4.83 | 373 |
| Central nervous system(CNS) | 4.45 | 697 | 3.73 | 697 | 4.73 | 697 |
| Prostate | 4.62 | 498 | 5.02 | 498 | 4.62 | 498 |
| Breast | 4.17 | 1104 | 4.26 | 1104 | 3.71 | 1104 |
| Skin | 6.34 | 473 | 4.23 | 473 | 3.59 | 473 |
| Oesophagus | 2.4 | 125 | 2.4 | 125 | 3.2 | 125 |
| Adrenal gland | 12.66 | 79 | 5.06 | 79 | 2.53 | 79 |
| Nervous system (NS) | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. |
| Bone | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. |
| Cancer Tissue | IKK1 | IKK2 | ||
|---|---|---|---|---|
| % Overexpressed | No. Tested | % Overexpressed | No. Tested | |
| Breast | 7.07 | 1104 | 9.6 | 1104 |
| Lung | 5.1 | 1019 | 7.16 | 1019 |
| Adrenal Gland | 5.06 | 79 | 1.27 | 79 |
| Endometrium | 4.98 | 602 | 13.12 | 602 |
| Oesophagus | 4.8 | 125 | 24.8 | 125 |
| Liver | 4.56 | 373 | 5.36 | 373 |
| Pancreas | 4.47 | 179 | 4.47 | 179 |
| Urinary tract | 4.41 | 408 | 4.9 | 408 |
| Stomach | 4.21 | 285 | 7.72 | 285 |
| Ovary | 4.14 | 266 | 7.52 | 266 |
| Thyroid | 4.09 | 513 | 2.34 | 513 |
| Prostate | 3.21 | 498 | 5.02 | 498 |
| Haematopoietic and lymphoid | 3.17 | 221 | 5.43 | 221 |
| Upper aerodigestive tract | 2.87 | 522 | 6.13 | 522 |
| Large intestine | 2.46 | 610 | 18.52 | 610 |
| Central nervous system(CNS) | 2.44 | 697 | 3.59 | 697 |
| Cervix | 1.95 | 307 | 5.54 | 307 |
| Soft tissue | 1.9 | 263 | 6.08 | 263 |
| Kidney | 1.83 | 600 | 3.33 | 600 |
| Skin | 1.48 | 473 | 8.25 | 473 |
| Biliary tract | n.a. | n.a. | n.a. | n.a. |
| Bone | n.a. | n.a. | n.a. | n.a. |
| Nervous system (NS) | n.a. | n.a. | n.a. | n.a. |
| Pituitary | n.a. | n.a. | n.a. | n.a. |
| Salivary gland | n.a. | n.a. | n.a. | n.a. |
| Testis | n.a. | n.a. | n.a. | n.a. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaltschmidt, B.; Greiner, J.F.W.; Kadhim, H.M.; Kaltschmidt, C. Subunit-Specific Role of NF-κB in Cancer. Biomedicines 2018, 6, 44. https://doi.org/10.3390/biomedicines6020044
Kaltschmidt B, Greiner JFW, Kadhim HM, Kaltschmidt C. Subunit-Specific Role of NF-κB in Cancer. Biomedicines. 2018; 6(2):44. https://doi.org/10.3390/biomedicines6020044
Chicago/Turabian StyleKaltschmidt, Barbara, Johannes F. W. Greiner, Hussamadin M. Kadhim, and Christian Kaltschmidt. 2018. "Subunit-Specific Role of NF-κB in Cancer" Biomedicines 6, no. 2: 44. https://doi.org/10.3390/biomedicines6020044
APA StyleKaltschmidt, B., Greiner, J. F. W., Kadhim, H. M., & Kaltschmidt, C. (2018). Subunit-Specific Role of NF-κB in Cancer. Biomedicines, 6(2), 44. https://doi.org/10.3390/biomedicines6020044