The Many Roles of Ubiquitin in NF-κB Signaling


Abstract
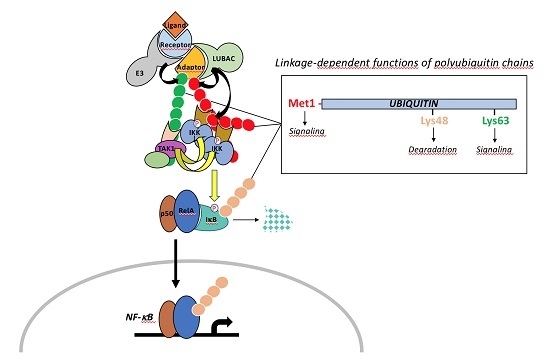
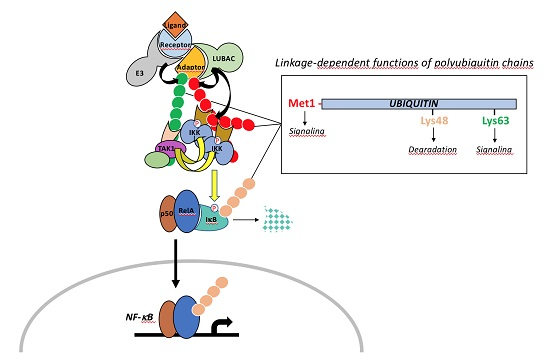{kind=link}
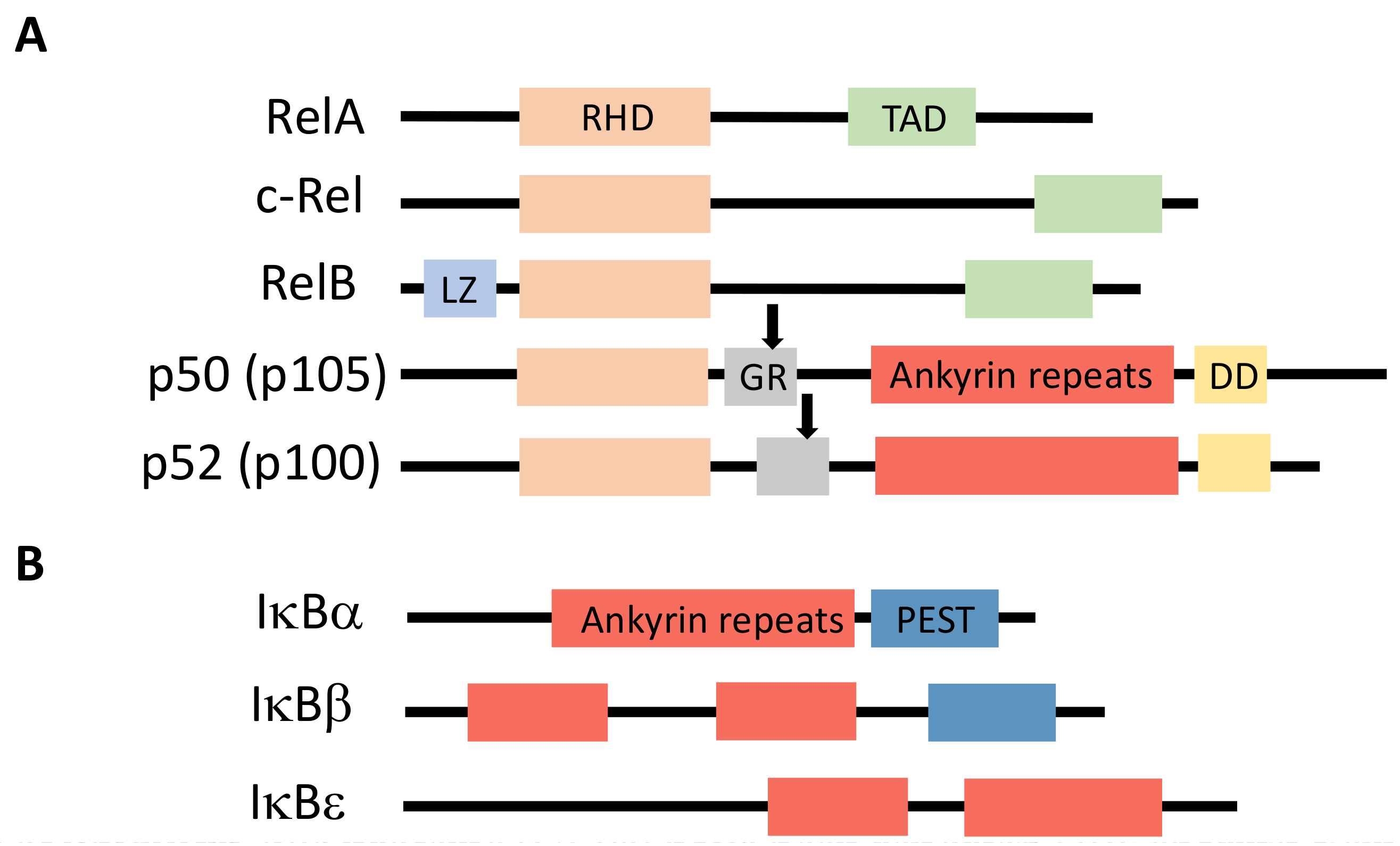{kind=link}
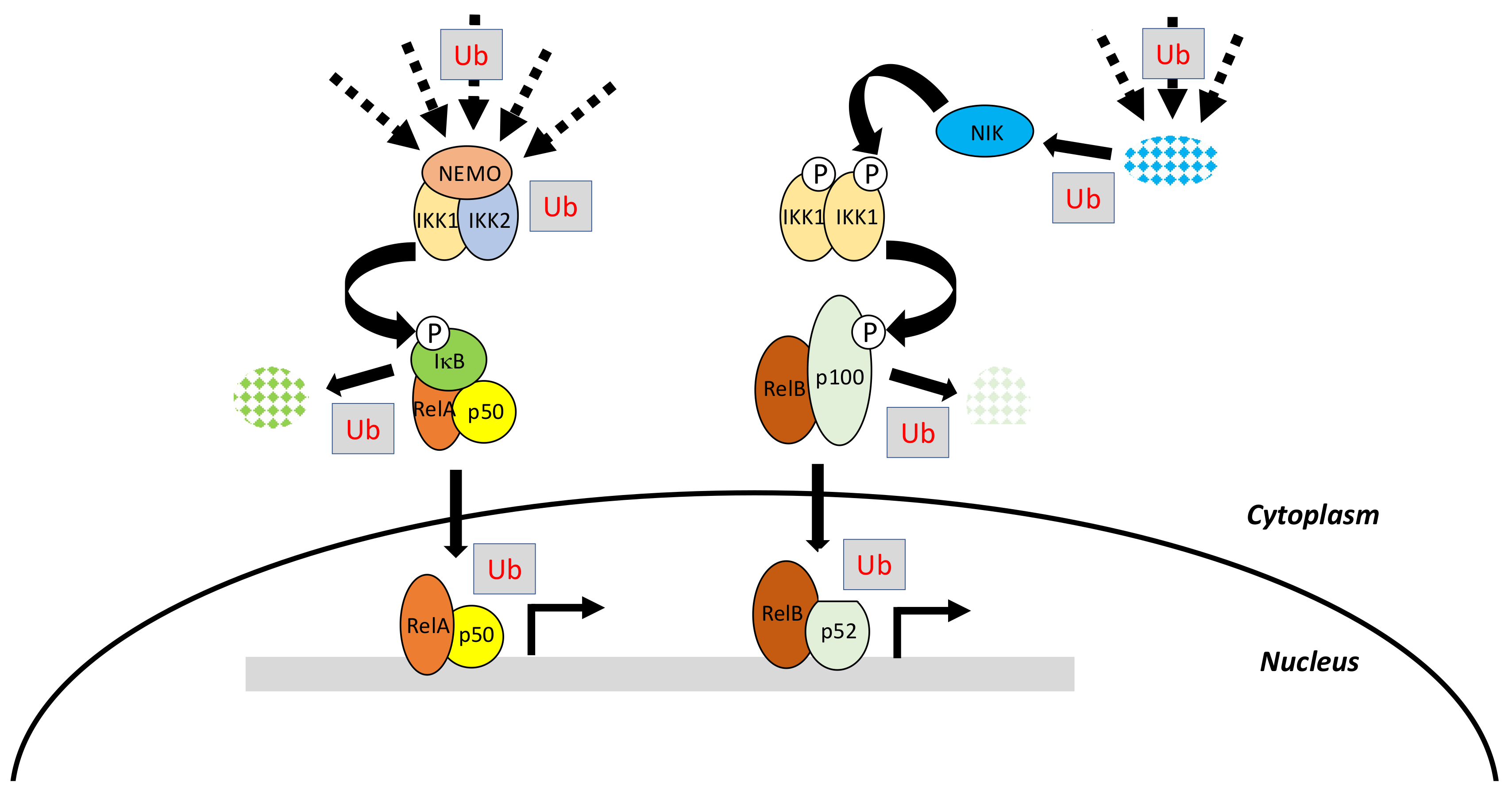{kind=link}
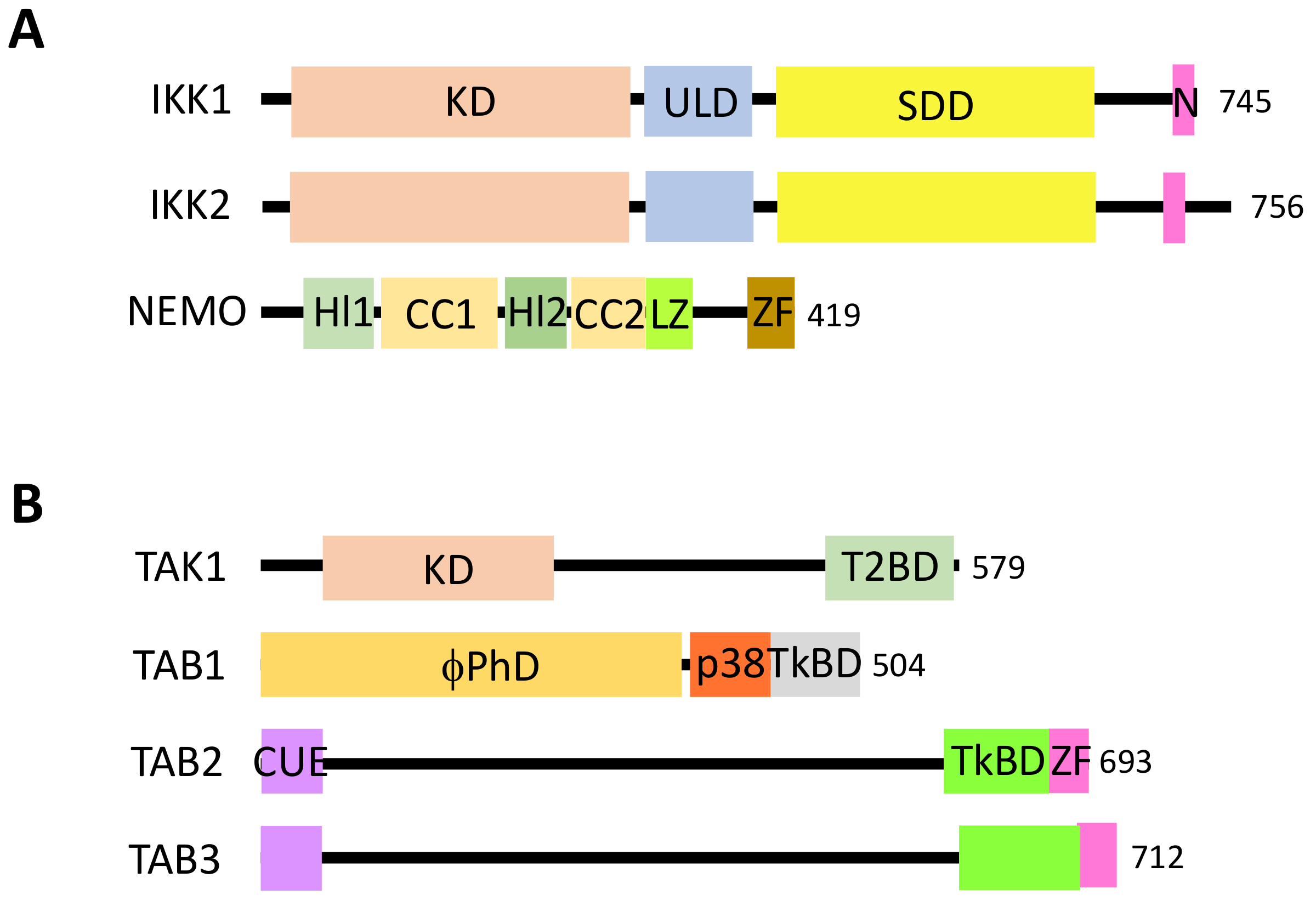{kind=link}
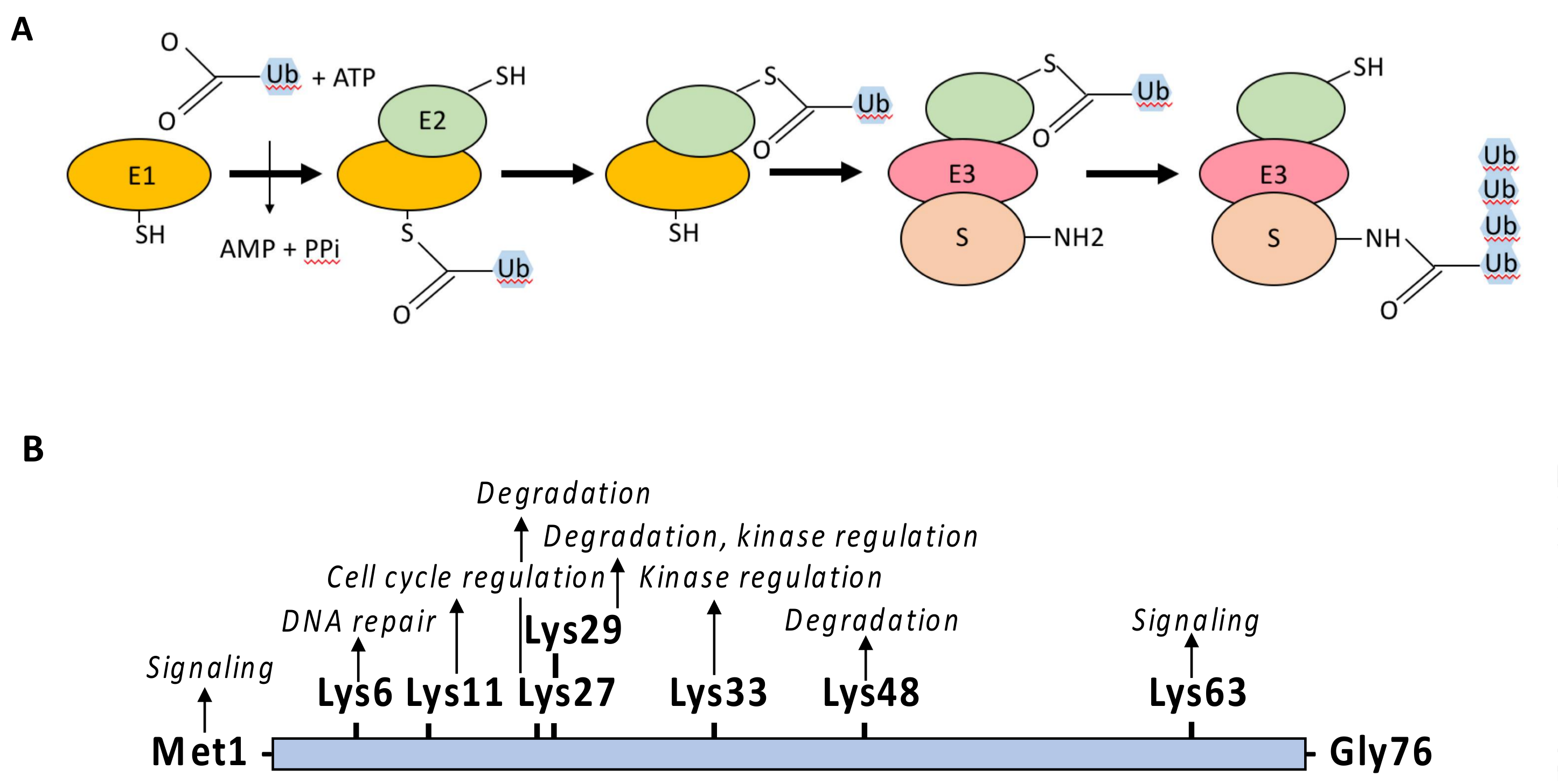{kind=link}
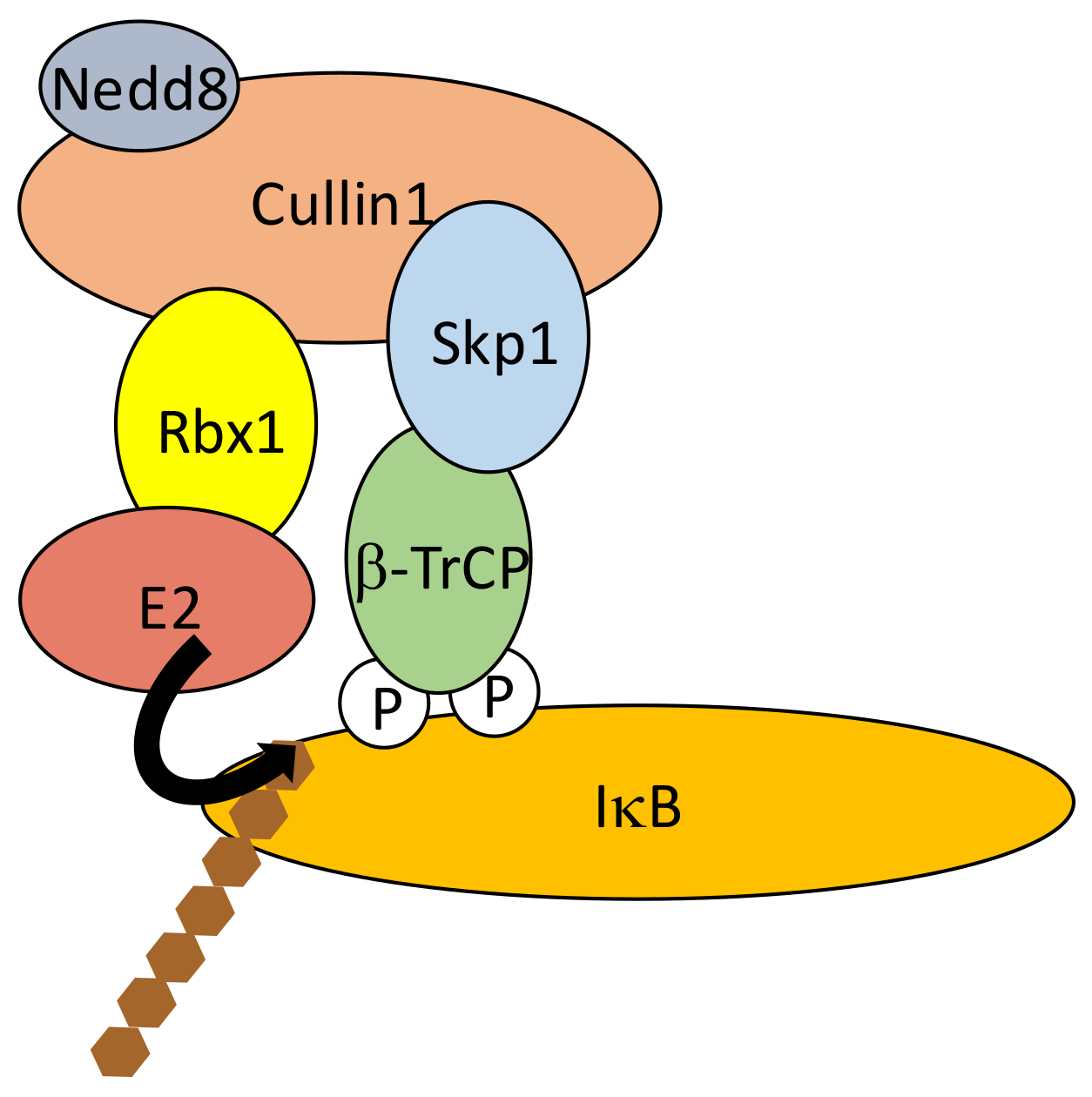{kind=link}
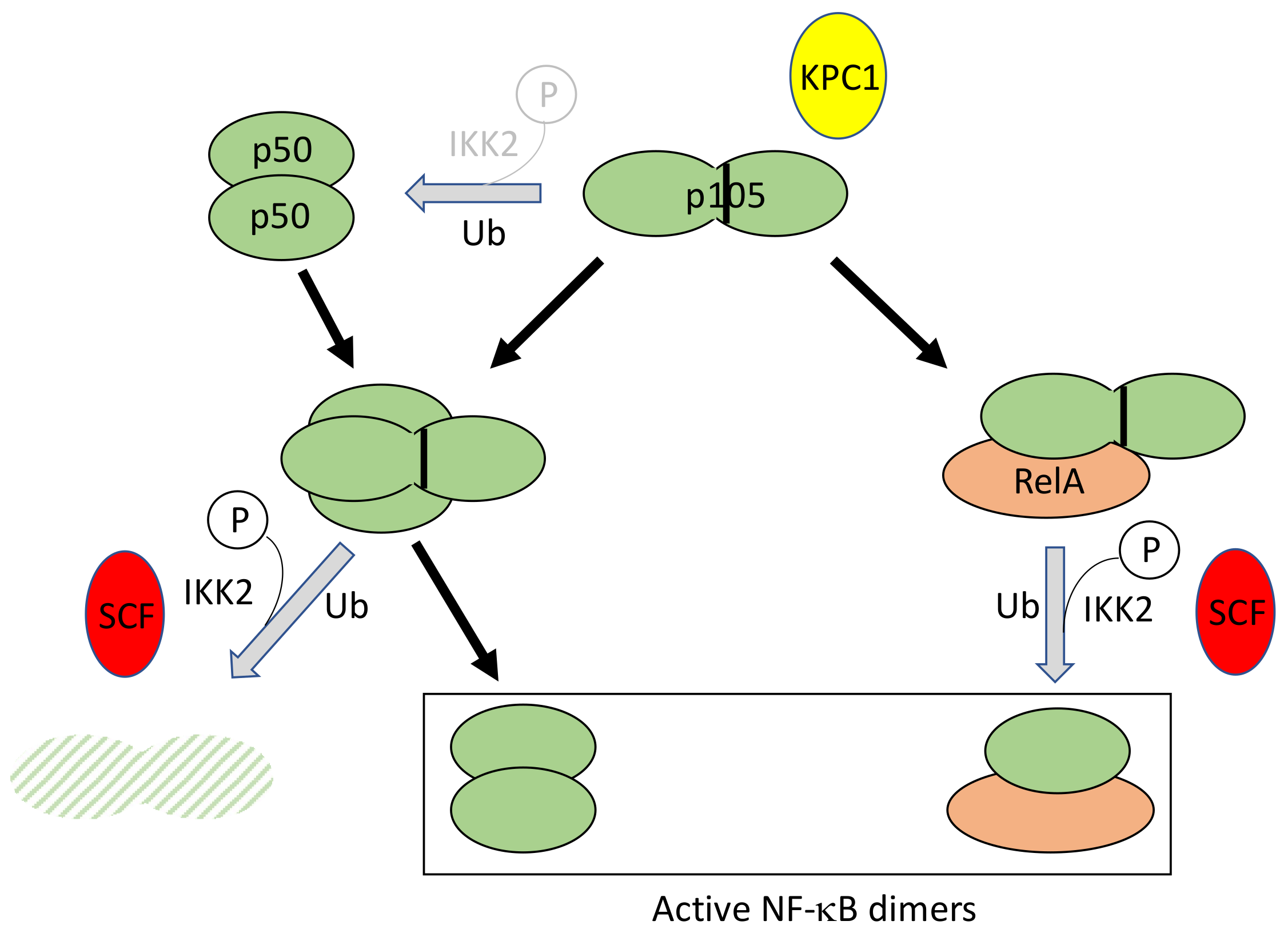{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. The NF-κB Signaling Pathway
2.1. The Canonical Pathway of NF-κB Activation
2.2. The Non-Canonical Pathway of NF-κB Activation
3. Ubiquitination: Players and Mechanisms
3.1. The Ubiquitination Process
3.2. E3 Ligase Families in NF-κB Signaling
3.2.1. TRAFs
3.2.2. TRIMs
3.2.3. LUBAC
3.3. Ubiquitin Binding Domains in NF-κB Signaling
4. Regulated Ubiquitination of IκBs and NF-κB Precursors
4.1. Regulated Ubiquitination of IκBs
4.2. Regulated Ubiquitination of p105
4.3. Regulated Ubiquitination of p100
5. Regulated Ubiquitination during Intracellular Signal Transduction
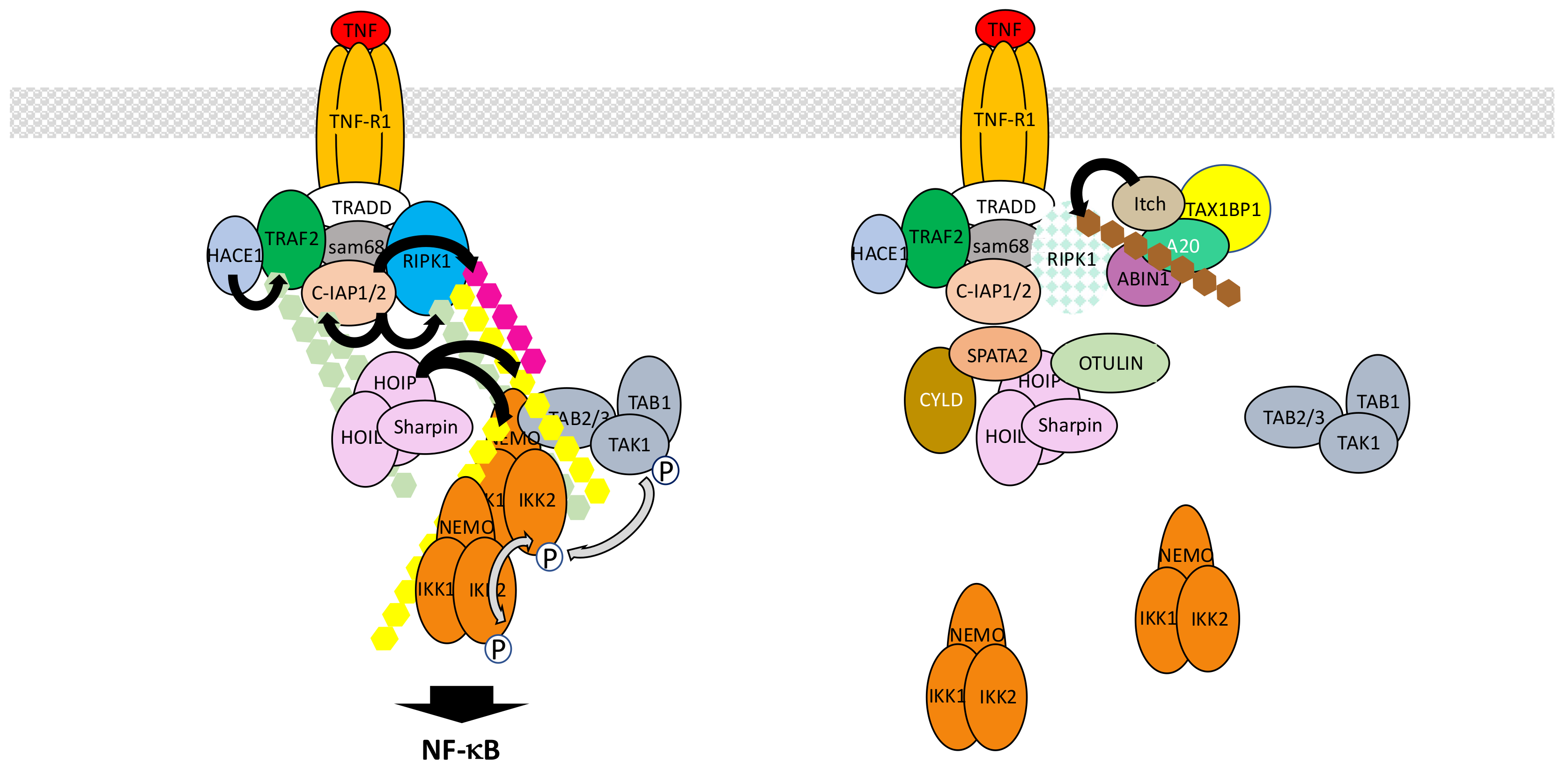
5.1. The TNF-R1 Signaling Pathway
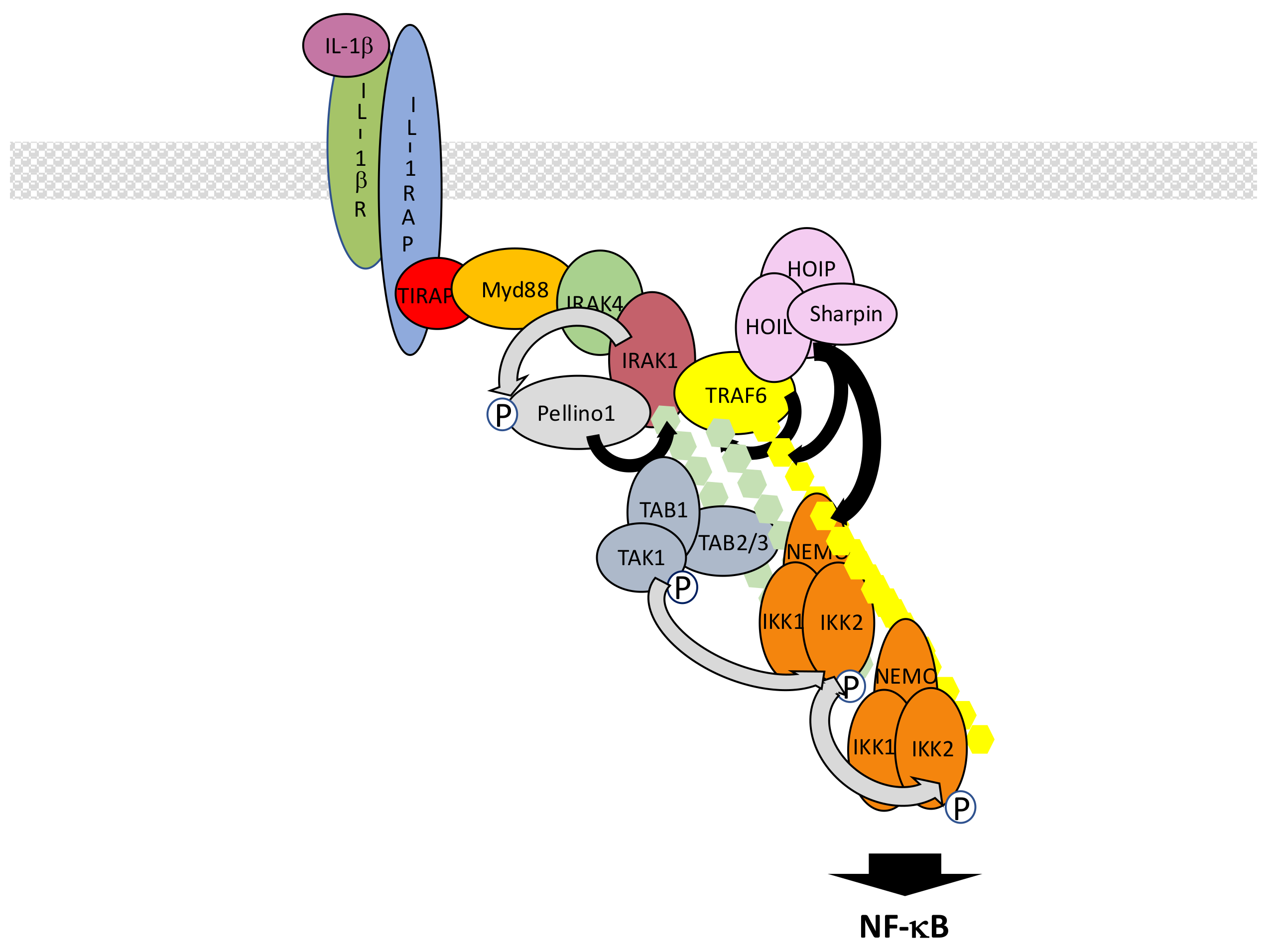
5.2. The IL-1β R/TLR Signaling Pathways
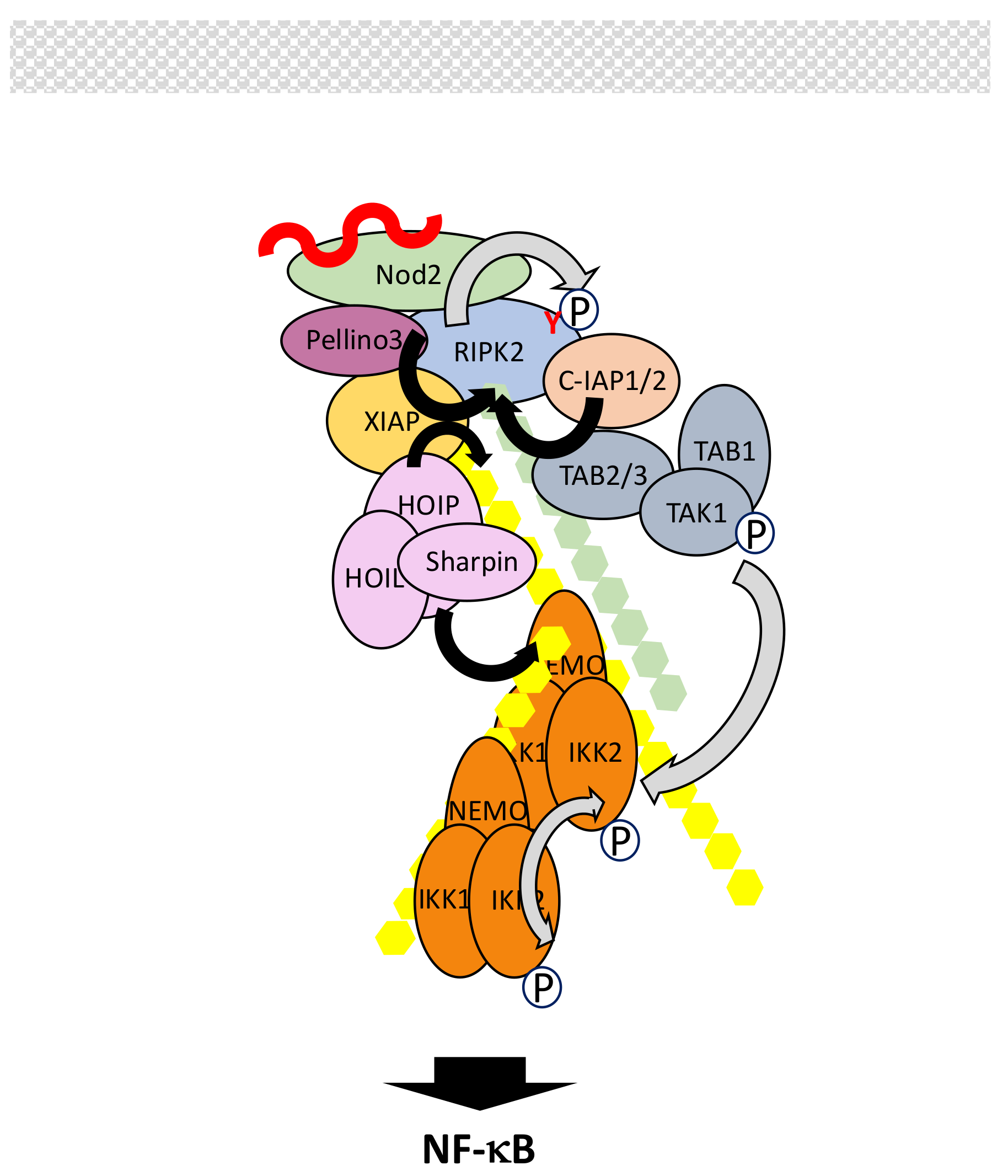
5.3. The Nod1/Nod2 Signaling Pathway
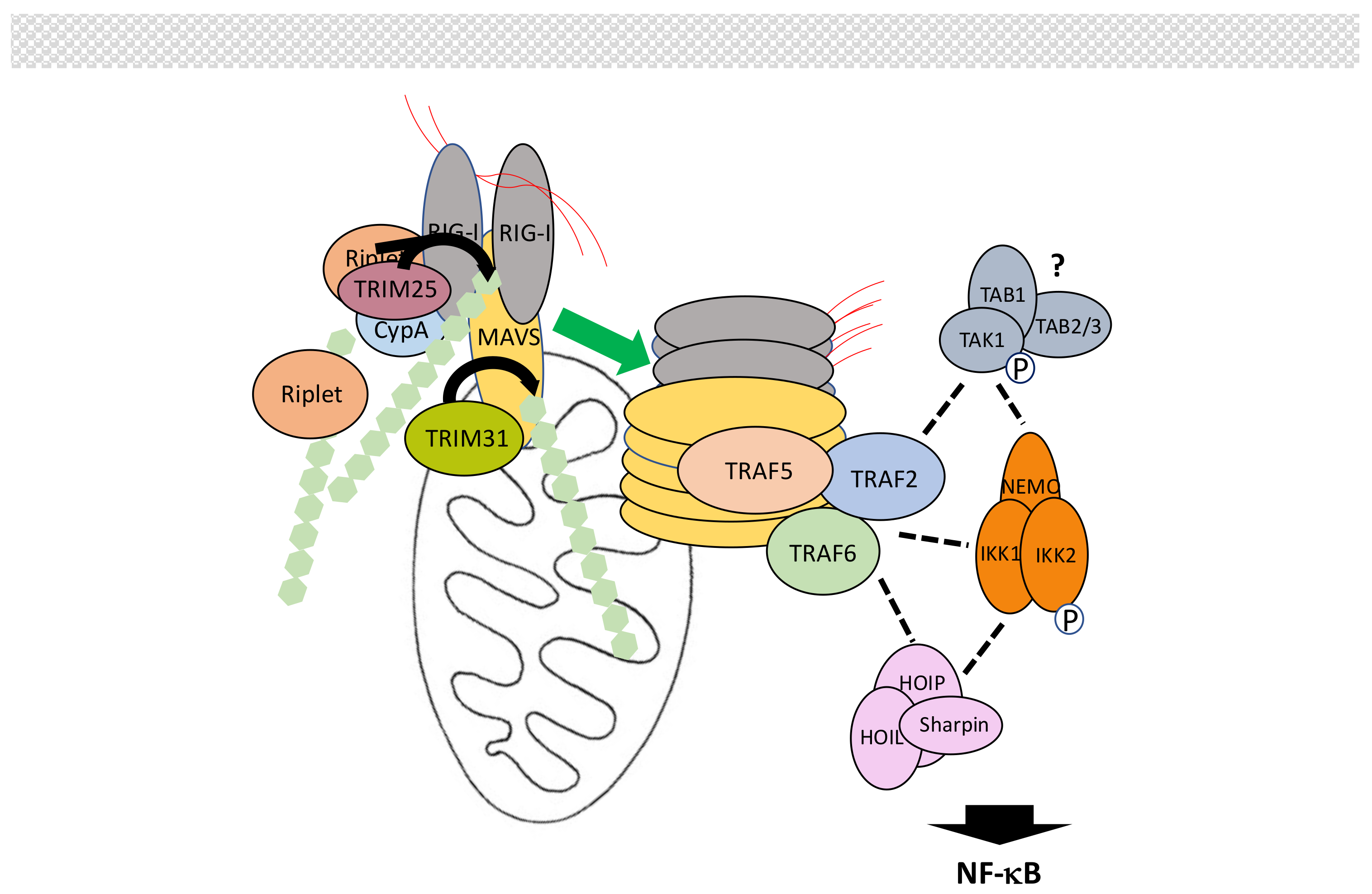
5.4. The MAVS Pathway
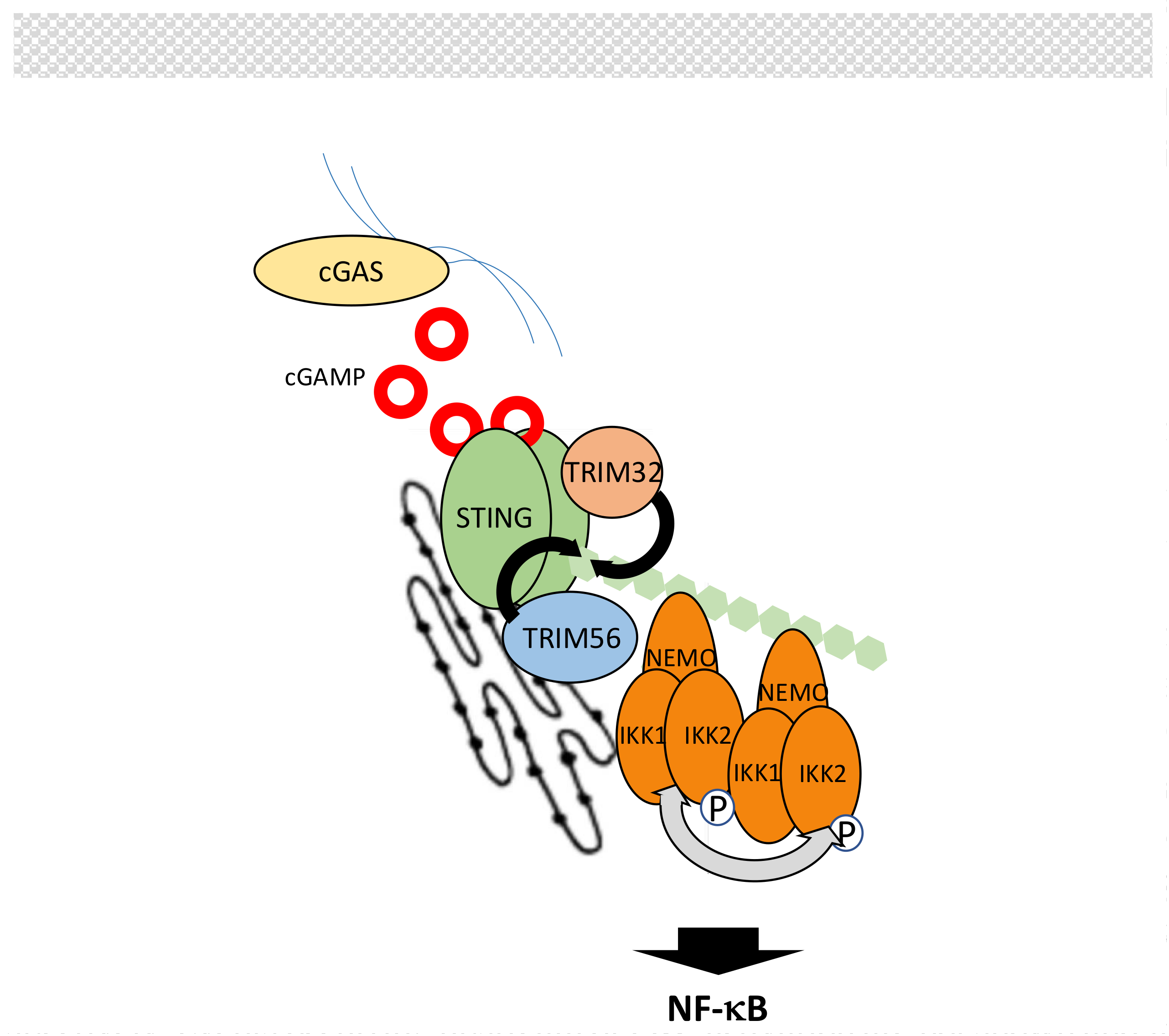
5.5. The cGAS/STING Pathway
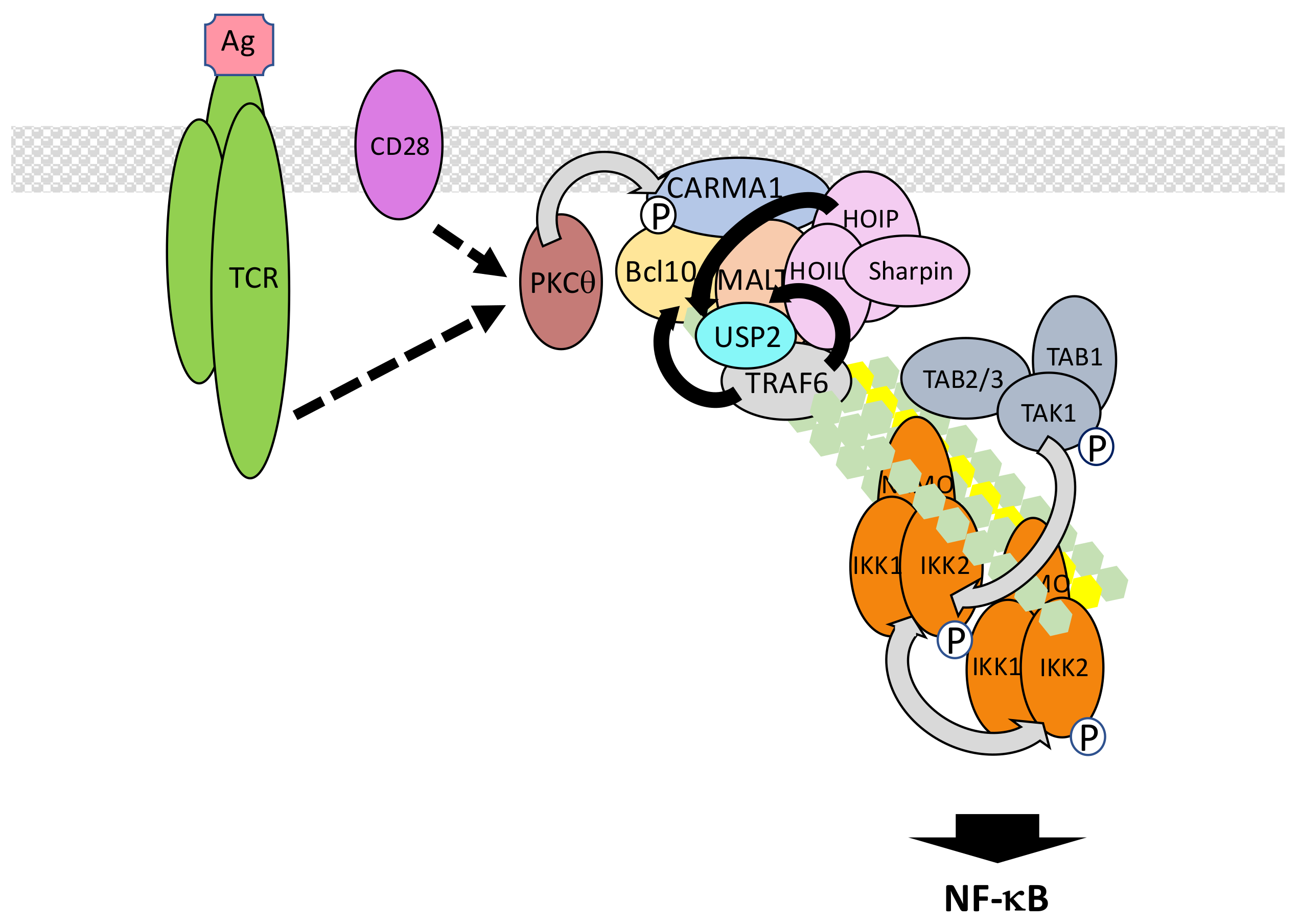
5.6. The TCR/BCR Pathway
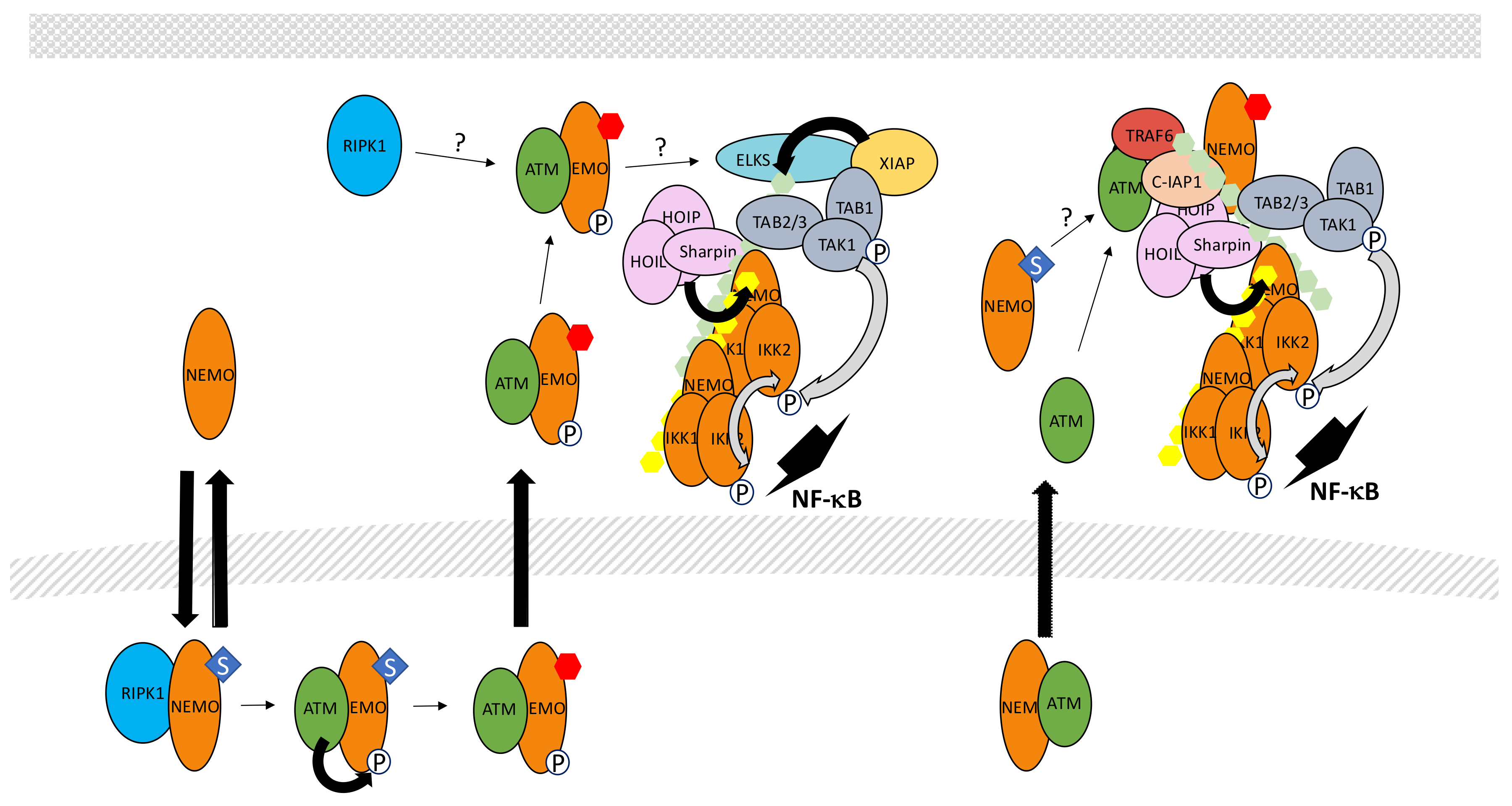
5.7. The Genotoxic Stress Pathway
6. Regulated Ubiquitination of TAK1 and IKK Complexes
6.1. Regulated Ubiquitination of TAK1 Complex Components
6.2. Regulated Ubiquitination of IKK Complex Components
7. Regulated Ubiquitination in the Non-Canonical Pathway of NF-κB Activation
8. Regulated Ubiquitination of NF-κB Proteins
9. In Vivo Relevance of Ubiquitin-Dependent NF-κB Processes
9.1. NEMO Mutations
9.2. LUBAC Mutations
9.3. OTULIN Mutations
10. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| ABIN | A20-Binding Inhibitor of NF-κB activation |
| AIP4 | Atrophin-1-Interacting Protein 4 |
| ATM | Ataxia Telangiectasia Mutated |
| BAFF-R | B-cell Activating Factor Receptor |
| Bcl | B cell lymphoma |
| BCR | B Cell Receptor |
| β-TrCP | β-Transducing repeat-Containing Protein |
| CARD | Caspase Recruitment Domain |
| CARMA1 | CARD-containing a Membrane-Associated Guanylate Kinase (MAGUK) protein 1 |
| CBM | CARMA1/Bcl10/MALT1 |
| CD | Cluster of Differentiation |
| CDC | Cell Division Cycle |
| 2′-5′-cGAMP | 2′-5′-cyclic Guanosine Adenosine MonoPhosphate |
| cGAS | cyclic Guanosine MonoPhosphate (GMP)-Adenosine MonoPhosphate (AMP) Synthase |
| CHIP | Carboxy terminus of Hsc70 Interacting Protein |
| c-IAP | Cellular Inhibitor of Apoptosis Protein |
| COMMD1 | COpper Metabolism MURR1 Domain-containing 1 |
| COP | COnstitutive Photomorphogenesis |
| CUE | Coupling of Ubiquitin conjugation to Endoplasmic reticulum-associated degradation |
| CYLD | CYLinDromatosis |
| DD | Death Domain |
| DEAD | DEAth effector Domain |
| DUB | DeUBiquitinase |
| EDA-ID | anhidrotic ectodermal dysplasia with immunodeficiency |
| EGLN3 | EGL Nine homolog 3 |
| ER | Endoplasmic Reticulum |
| ERK | Extracellular signal-Regulated Kinase |
| FADD | Fas-Associated protein with Death Domain |
| Fbw | F-box/WD repeat-containing protein |
| FHA | ForkHead Associated |
| GSK3 | Glycogen Synthase Kinase-3 |
| HACE1 | HECT domain and Ankyrin repeat Containing E3 ubiquitin protein ligase 1 |
| HECT | Homologous to the E6-AP Carboxyl Terminus |
| HOIL-1L | Haem-Oxydized IRP2 ubiquitin Ligase 1L |
| HOIP | HOIL-Interacting Protein |
| iE-DAP | γ-d-glutamyl-mesoDiAminoPimelic acid |
| IFN | Interferon |
| IKK | IκB Kinase |
| IκB | Inhibitory κB |
| IL-1βR | Interleukin-1β Receptor |
| IL1RAP | IL1 Receptor Accessory Protein |
| Imd | immune deficiency |
| IP | incontinentia pigmenti |
| IpaH | Invasion-plasmid antigen-H protein |
| IRAK | Interleukin-1 Receptor Associated Kinase |
| iRhom2 | inactive Rhomboid 2 |
| JAMM | JAB1/MPN/Mv34 metalloenzyme |
| KPC1 | Kipl ubiquitylation-Promoting Complex 1 |
| LDD | Linear ubiquitin chain Determining Domain |
| LGP2 | Laboratory of Genetics and Physiology 2 |
| LUBAC | Linear UBiquitin chain Assembly Complex |
| LZ | Leucine Zipper |
| MAGUK | Membrane-Associated Guanylate Kinase |
| MALT | Mucosa-Associated Lymphoid Tissue |
| MALT1 | MALT lymphoma associated translocation protein 1 |
| MARCH | Membrane Associated RING-CH |
| MAVS | Mitochondrial AntiViral-Signaling Protein |
| MCPIP1 | Monocyte Chemotactic Protein-Induced Protein-1 |
| MDA5 | Melanoma Differentiation Associated gene 5 |
| Mdm2 | Mouse double minute 2 homolog |
| MDP | Muramyl DiPeptide |
| MD2 | Myeloid Differentiation factor 2 |
| MEKK | Mitogen-activated protein/ERK Kinase Kinase |
| MEX3C | MEX-3 homolog C (C. elegans) |
| MJD | Machado-Joseph Diseases protease |
| MIB2 | MIndBomb 2 |
| Mindy | MIU-containing Novel DUB familY |
| MLKL | Mixed Lineage Kinase domain Like pseudokinase |
| Myd88 | Myeloid differentiation primary response 88 |
| Ndfip1 | Nedd4 family interacting protein 1 |
| NEDD8 | Neural precursor cell Expressed, Developmentally Down-Regulated 8 |
| NEMO | NF-κB Essential Modulator |
| NF-κB | Nuclear Factor κB |
| NIK | NF-κB Inducing Kinase |
| NLRC | NOD-LRR (Leucine-Rich Repeat) family with CARD |
| NLRP | Nod-Like Receptor Protein |
| Nod | Nucleotide-binding oligomerization domain |
| NSP | Non Structural Protein |
| NZF | Npl4 Zinc Finger |
| ORAS | OTULIN-Related Autoinflammatory Syndrome |
| OUT | Ovarian TUmor proteases |
| OTULIN | OTU deubiquitinase with LINear linkage specificity |
| PAMPs | Pathogen Associated Molecular Patterns |
| PARP-1 | Poly[ADP-Ribose (PAR)] Polymerase 1 |
| PPCBP2 | Poly(RC) Binding Protein 2 |
| PDLIM2 | PDZ and LIM domain 2 |
| PIASy. | Protein Inhibitor of Activated STAT (Signal Transducer and Activator of Transcription) y |
| PIDD | P53-Induced Protein with a Death Domain |
| PKC | Protein Kinase C |
| PPAR | Peroxisome Proliferator-Activated Receptor |
| PTM | Post-Translational Modifications |
| PUB | PNGase/UBA or UBX-containing proteins |
| RBCC | Ring, B-box, Coiled-Coil |
| Rbx1 | RING-box protein 1 |
| Rel | avian Reticuloendotheliosis |
| RBR | Ring Between Ring fingers |
| RHIM | RIP Homotypic Interaction Motif |
| RING | Really Interesting New Gene |
| RIPK1 | Receptor-Interacting serine/threonine-Protein Kinase 1 |
| RLRs | Retinoic-Inducible Gene-I (RIG-I) Like Receptors |
| RNF | RiNg Finger protein |
| Sam68 | Src-Associated in Mitosis 68 kDa |
| SCF | Skp, Cullin, F-box |
| SENP2 | SUMO-specific Protease 2 |
| SHARPIN | SHANK-associated RH domain interacting ProteIN |
| SHIP | SH2-containing Inositol phosphatase |
| Skp1 | S-phase kinase-associated protein 1 |
| Smurf1 | Smad ubiquitin regulatory factor 1 |
| SOCS1 | Suppressor Of Cytokine Signaling 1 |
| SPATA2 | SPermATogenesis-Associated protein 2 |
| Sphk1 | Sphingosine kinase 1 |
| STAT | Signal Transducer and Activator of Transcription |
| STING | STimulator of INterferon Genes |
| SUMO | Small Ubiquitin-like Modifier |
| TAB | TAK1 Binding protein |
| TAD | transcriptional activator domain |
| TAK1 | Tumour growth factor-Activated Kinase 1 |
| TANK | TRAF family member-Associated NF-κB activator |
| TCR | T cell receptor |
| TIR | Toll-Interleukin Receptor |
| TIRAP | Toll-Interleukin Receptor Adaptor Protein |
| TLR | Toll-Like Receptor |
| TNFAIP3 | TNF Alpha Induced Protein 3 |
| TRADD | Tumor necrosis factor Receptor type 1-Associated Death Domain protein |
| TRAF | TNF Receptor Associated Factor |
| TRAM | TRIF-Related Adaptor Molecule |
| TRIF | TIR domain-containing adaptor-inducing Interferon-β |
| TRIM | TRIpartite Motif protein |
| UBA | UBiquitin-Associated |
| UBASH3A | UBiquitin Associated and SH3 domain containing 3A |
| Ubc13 | Ubiquitin-conjugating 13 |
| UBD | Ubiquitin-Binding Domain |
| UBE2L3 | UBiquitin-conjugating Enzyme E2 L3 |
| UBL | Ubiquitin-Like |
| UCH | Ubiquitin C-terminal Hydrolases |
| Uev1A | Ubiquitin-conjugating enzyme variant 1A |
| ULP | Ubiquitin-Like Proteins |
| USP | Ubiquitin Specific Protease |
| WD40 | TrpAsp 40 amino acids |
| WWP | WW domain-containing Protein ligase |
| XIAP | X-linked Inhibitor of Apoptosis Protein |
| ZF | Zinc Finger |
| ZNRF | Zinc aNd Ring Finger |
References
- Karin, M.; Ben-Neriah, Y. Phosphorylation meets ubiquitination: The control of NF-κB activity. Annu. Rev. Immunol. 2000, 18, 621–663. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B.; Takada, Y.; Shishodia, S.; Gutierrez, A.M.; Oommen, O.V.; Ichikawa, H.; Baba, Y.; Kumar, A. Nuclear transcription factor NF-κB: Role in biology and medicine. Indian J. Exp. Biol. 2004, 42, 341–353. [Google Scholar] [PubMed]
- Hinz, M.; Scheidereit, C. The IκB kinase complex in NF-κB regulation and beyond. EMBO Rep. 2014, 15, 46–61. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Aye Thu, C.; Liu, X.Y.; Xi, J.; Cheung, P.C. TAK1, more than just innate immunity. IUBMB Life 2012, 64, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C. The noncanonical NF-κB pathway. Immunol. Rev. 2012, 246, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Pickart, C.M.; Eddins, M.J. Ubiquitin: Structures, functions, mechanisms. Biochim. Biophys. Acta 2004, 1695, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Komander, D. The emerging complexity of protein ubiquitination. Biochem. Soc. Trans. 2009, 37, 937–953. [Google Scholar] [CrossRef] [PubMed]
- Clague, M.J.; Heride, C.; Urbé, S. The demographics of the ubiquitin system. Trends Cell Biol. 2015, 25, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.D.; Ritterhoff, T.; Klevit, R.E.; Brzovic, P.S. E2 enzymes: More than just middle men. Cell Res. 2016, 26, 423–440. [Google Scholar] [CrossRef] [PubMed]
- Metzger, M.B.; Hristova, V.A.; Weissman, A.M. HECT and RING finger families of E3 ubiquitin ligases at a glance. J. Cell Sci. 2012, 125, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Zheng, N.; Shabek, N. Ubiquitin Ligases: Structure, Function, and Regulation. Annu. Rev. Biochem. 2017, 86, 129–157. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, T. Multiubiquitinylation by E4 enzymes: “one size” doesn’t fit all. Trends Biochem. Sci. 2005, 30, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, F.; Tsuchiya, H. The emerging complexity of ubiquitin architecture. J. Biochem. 2017, 161, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.T.; Ciechanover, A. The Ubiquitin Code in the Ubiquitin-Proteasome System and Autophagy. Trends Biochem. Sci. 2017, 42, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Mevissen, T.E.T.; Komander, D. Mechanisms of Deubiquitinase Specificity and Regulation. Annu. Rev. Biochem. 2017, 86, 159–192. [Google Scholar] [CrossRef] [PubMed]
- Nijman, S.M.; Luna-Vargas, M.P.; Velds, A.; Brummelkamp, T.R.; Dirac, A.M.; Sixma, T.K.; Bernards, R. A genomic and functional inventory of deubiquitinating enzymes. Cell 2005, 123, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Leznicki, P.; Kulathu, Y. Mechanisms of regulation and diversification of deubiquitylating enzyme function. J. Cell Sci. 2017, 130, 1997–2006. [Google Scholar] [CrossRef] [PubMed]
- Sowa, M.E.; Bennett, E.J.; Gygi, S.P.; Wade Harper, J. Defining the human deubiquitinating enzyme interaction landscape. Cell 2009, 138, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Cappadocia, L.; Lima, C.D. Ubiquitin-like Protein Conjugation: Structures, Chemistry, and Mechanism. Chem. Rev. 2018, 118, 889–918. [Google Scholar] [CrossRef] [PubMed]
- Dohmen, R.J. SUMO protein modification. Biochim. Biophys. Acta 2004, 1695, 113–131. [Google Scholar] [CrossRef] [PubMed]
- Boase, N.A.; Kumar, S. NEDD4: The founding member of a family of ubiquitin-protein ligases. Gene 2015, 557, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Schwartz, A.L. The ubiquitin system: Pathogenesis of human diseases and drug targeting. Biochim. Biophys. Acta 2004, 1695, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Young, M.A.; Donato, N.J. Emerging potential of therapeutic targeting of ubiquitin-specific proteases in the treatment of cancer. Cancer Res. 2014, 74, 4955–4966. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Shaik, S.; Dai, X.; Wu, Q.; Zhou, X.; Wang, Z.; Wei, W. Targeting the ubiquitin pathway for cancer treatment. Biochim. Biophys. Acta 2015, 1855, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Xie, P. TRAF molecules in cell signaling and in human diseases. J. Mol. Signal. 2013, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Tomar, D.; Singh, R. TRIM family proteins: Emerging class of RING E3 ligases as regulator of NF-κB pathway. Biol. Cell 2015, 107, 22–40. [Google Scholar] [CrossRef] [PubMed]
- Van Tol, S.; Hage, A.; Giraldo, M.I.; Bharaj, P.; Rajsbaum, R. The TRIMendous Role of TRIMs in Virus-Host Interactions. Vaccines 2017, 5, 23. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, F.; Iwai, K. LUBAC, a novel ubiquitin ligase for linear ubiquitination, is crucial for inflammation and immune responses. Microbes Infect. 2012, 14, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Rittinger, K.; Ikeda, F. Linear ubiquitin chains: Enzymes, mechanisms and biology. Open Biol. 2017, 7, 170026. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Wang, C.; Spencer, E.; Yang, L.; Braun, A.; You, J.; Slaughter, C.; Pickart, C.; Chen, Z.J. Activation of the IκB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 2000, 103, 351–361. [Google Scholar] [CrossRef]
- Fukushima, T.; Matsuzawa, S.; Kress, C.L.; Bruey, J.M.; Krajewska, M.; Lefebvre, S.; Zapata, J.M.; Ronai, Z.; Reed, J.C. Ubiquitin-conjugating enzyme Ubc13 is a critical component of TNF receptor-associated factor (TRAF)-mediated inflammatory responses. Proc. Natl. Acad. Sci. USA 2007, 104, 6371–6376. [Google Scholar] [CrossRef] [PubMed]
- Fu, B.; Li, S.; Wang, L.; Berman, M.A.; Dorf, M.E. The ubiquitin conjugating enzyme UBE2L3 regulates TNFα-induced linear ubiquitination. Cell Res. 2014, 24, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Lewis, M.J.; Vyse, S.; Shields, A.M.; Boeltz, S.; Gordon, P.A.; Spector, T.D.; Lehner, P.J.; Walczak, H.; Vyse, T.J. UBE2L3 polymorphism amplifies NF-κB activation and promotes plasma cell development, linking linear ubiquitination to multiple autoimmune diseases. Am. J. Hum. Genet. 2015, 96, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.; Oldham, N.J.; Strachan, J.; Searle, M.S.; Layfield, R. Ubiquitin-binding domains: Mechanisms of ubiquitin recognition and use as tools to investigate ubiquitin-modified proteomes. Proteomics 2015, 15, 844–861. [Google Scholar] [CrossRef] [PubMed]
- Kulathu, Y.; Akutsu, M.; Bremm, A.; Hofmann, K.; Komander, D. Two-sided ubiquitin binding explains specificity of the TAB2 NZF domain. Nat. Struct. Mol. Biol. 2009, 16, 1328–1330. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Yoshikawa, A.; Yamashita, M.; Yamagata, A.; Fukai, S. Structural basis for specific recognition of Lys 63-linked polyubiquitin chains by NZF domains of TAB2 and TAB3. EMBO J. 2009, 28, 3903–3909. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.C.; Lin, S.C.; Rospigliosi, C.C.; Conze, D.B.; Wu, C.J.; Ashwell, J.D.; Eliezer, D.; Wu, H. Structural basis for recognition of diubiquitins by NEMO. Mol. Cell 2009, 33, 602–615. [Google Scholar] [CrossRef] [PubMed]
- Rahighi, S.; Ikeda, F.; Kawasaki, M.; Akutsu, M.; Suzuki, N.; Kato, R.; Kensche, T.; Uejima, T.; Bloor, S.; Komander, D.; et al. Specific recognition of linear ubiquitin chains by NEMO is important for NF-κB activation. Cell 2009, 136, 1098–1109. [Google Scholar] [CrossRef] [PubMed]
- Ngadjeua, F.; Chiaravalli, J.; Traincard, F.; Raynal, B.; Fontan, E.; Agou, F. Two-sided ubiquitin binding of NF-κB essential modulator (NEMO) zinc finger unveiled by a mutation associated with anhidrotic ectodermal dysplasia with immunodeficiency syndrome. J. Biol. Chem. 2013, 288, 33722–33737. [Google Scholar] [CrossRef] [PubMed]
- Laplantine, E.; Fontan, E.; Chiaravalli, J.; Lopez, T.; Lakisic, G.; Véron, M.; Agou, F.; Israël, A. NEMO specifically recognizes K63-linked poly-ubiquitin chains through a new bipartite ubiquitin-binding domain. EMBO J. 2009, 28, 2885–2895. [Google Scholar] [CrossRef] [PubMed]
- Kensche, T.; Tokunaga, F.; Ikeda, F.; Goto, E.; Iwai, K.; Dikic, I. Analysis of nuclear factor-κB (NF-κB) essential modulator (NEMO) binding to linear and lysine-linked ubiquitin chains and its role in the activation of NF-κB. J. Biol. Chem. 2012, 287, 23626–23634. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Fujita, H.; Sasaki, Y.; Tsuruyama, T.; Fukuda, K.; Iwai, K. Differential Involvement of the Npl4 Zinc Finger Domains of SHARPIN and HOIL-1L in Linear Ubiquitin Chain Assembly Complex-Mediated Cell Death Protection. Mol. Cell. Biol. 2016, 36, 1569–1583. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Fujita, H.; Yoshikawa, A.; Yamashita, M.; Yamagata, A.; Kaiser, S.E.; Iwai, K.; Fukai, S. Specific recognition of linear ubiquitin chains by the Npl4 zinc finger (NZF) domain of the HOIL-1L subunit of the linear ubiquitin chain assembly complex. Proc. Natl. Acad. Sci. USA 2011, 108, 20520–20525. [Google Scholar] [CrossRef] [PubMed]
- Kanarek, N.; London, N.; Schueler-Furman, O.; Ben-Neriah, Y. Ubiquitination and degradation of the inhibitors of NF-κB. Cold Spring Harb. Perspect. Biol. 2010, 2, a000166. [Google Scholar] [CrossRef] [PubMed]
- Spencer, E.; Jiang, J.; Chen, Z.J. Signal-induced ubiquitination of IκBα by the F-box protein Slimb/β-TrCP. Genes Dev. 1999, 13, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Winston, J.T.; Strack, P.; Beer-Romero, P.; Chu, C.Y.; Elledge, S.J.; Harper, J.W. The SCFβ-TRCP-ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IκBα and β-catenin and stimulates IκBα ubiquitination in vitro. Genes Dev. 1999, 13, 270–283. [Google Scholar] [CrossRef] [PubMed]
- Gonen, H.; Bercovich, B.; Orian, A.; Carrano, A.; Takizawa, C.; Yamanaka, K.; Pagano, M.; Iwai, K.; Ciechanover, A. Identification of the ubiquitin carrier proteins, E2s, involved in signal-induced conjugation and subsequent degradation of IκBα. J. Biol. Chem. 1999, 274, 14823–14830. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Ghosh, S. Differential phosphorylation of the signal-responsive domain of IκBα and IκBβ by IκB kinases. J. Biol. Chem. 2003, 278, 31980–31987. [Google Scholar] [CrossRef] [PubMed]
- Read, M.A.; Brownell, J.E.; Gladysheva, T.B.; Hottelet, M.; Parent, L.A.; Coggins, M.B.; Pierce, J.W.; Podust, V.N.; Luo, R.S.; Chau, V.; et al. Nedd8 modification of cul-1 activates SCF(β(TrCP))-dependent ubiquitination of IκBα. Mol. Cell. Biol. 2000, 20, 2326–2333. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, T.; Chiba, T.; Suzuki, T.; Iwai, K.; Yamanaka, K.; Minato, N.; Suzuki, H.; Shimbara, N.; Hidaka, Y.; Osaka, F.; et al. NEDD8 recruits E2-ubiquitin to SCF E3 ligase. EMBO J. 2001, 20, 4003–4012. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Cho, H.; Inn, K.S.; Yang, A.; Zhao, Z.; Liang, Q.; Versteeg, G.A.; Amini-Bavil-Olyaee, S.; Wong, L.Y.; et al. Negative regulation of NF-κB activity by brain-specific TRIpartite Motif protein 9. Nat. Commun. 2014, 5, 4820. [Google Scholar] [CrossRef] [PubMed]
- Di Fiore, I.J.; Pane, J.A.; Holloway, G.; Coulson, B.S. NSP1 of human rotaviruses commonly inhibits NF-κB signalling by inducing β-TrCP degradation. J. Gen. Virol. 2015, 96, 1768–1776. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Tan, X.; Shi, Y.; Xu, G.; Mao, R.; Gu, X.; Fan, Y.; Yu, Y.; Burlingame, S.; Zhang, H.; et al. USP11 negatively regulates TNFα-induced NF-κB activation by targeting on IκBα. Cell Signal. 2010, 22, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, K.; Bozko, P.M.; Dubiel, W.; Naumann, M. CSN controls NF-kB by deubiquitinylation of IκBα. EMBO J. 2007, 26, 1532–1541. [Google Scholar] [CrossRef] [PubMed]
- Da Silva-Ferrada, E.; Torres-Ramos, M.; Aillet, F.; Campagna, M.; Matute, C.; Rivas, C.; Rodríguez, M.S.; Lang, V. Role of monoubiquitylation on the control of IκBα degradation and NF-κB activity. PLoS ONE 2011, 6, e25397. [Google Scholar] [CrossRef] [PubMed]
- Desterro, J.M.; Rodriguez, M.S.; Hay, R.T. SUMO-1 modification of IκBα inhibits NF-κB activation. Mol. Cell 1998, 2, 233–239. [Google Scholar] [CrossRef]
- Orian, A.; Schwartz, A.L.; Israël, A.; Whiteside, S.; Kahana, C.; Ciechanover, A. Structural motifs involved in ubiquitin-mediated processing of the NF-κB precursor p105: Roles of the glycine-rich region and a downstream ubiquitination domain. Mol. Cell. Biol. 1999, 19, 3664–3673. [Google Scholar] [CrossRef] [PubMed]
- Kravtsova-Ivantsiv, Y.; Shomer, I.; Cohen-Kaplan, V.; Snijder, B.; Superti-Furga, G.; Gonen, H.; Sommer, T.; Ziv, T.; Admon, A.; Naroditsky, I.; et al. KPC1-mediated ubiquitination and proteasomal processing of NF-κB1 p105 to p50 restricts tumor growth. Cell 2015, 161, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Heissmeyer, V.; Krappmann, D.; Hatada, E.N.; Scheidereit, C. Shared pathways of Iκkinase-induced SCF(βTrCP)-mediated ubiquitination and degradation for the NF-κB precursor p105 and IκBα. Mol. Cell. Biol. 2001, 21, 1024–1035. [Google Scholar] [CrossRef] [PubMed]
- Orian, A.; Gonen, H.; Bercovich, B.; Fajerman, I.; Eytan, E.; Israël, A.; Mercurio, F.; Iwai, K.; Schwartz, A.L.; Ciechanover, A. SCF(β-TrCP) ubiquitin ligase-mediated processing of NF-κB p105 requires phosphorylation of its C-terminus by IκB kinase. EMBO J. 2000, 19, 2580–2591. [Google Scholar] [CrossRef] [PubMed]
- Amir, R.E.; Iwai, K.; Ciechanover, A. The NEDD8 pathway is essential for SCF(β-TrCP)-mediated ubiquitination and processing of the NF-κB precursor p105. J. Biol. Chem. 2002, 277, 23253–23259. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Holmgren, R.A.; Matouschek, A. A conserved processing mechanism regulates the activity of transcription factors Cubitus interruptus and NF-κB. Nat. Struct. Mol. Biol. 2005, 12, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Lapid, D.; Lahav-Baratz, S.; Cohen, S. A20 inhibits both the degradation and limited processing of the NF-κB p105 precursor: A novel additional layer to its regulator role. Biochem. Biophys. Res. Commun. 2017, 493, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Heusch, M.; Lin, L.; Geleziunas, R.; Greene, W.C. The generation of nfκb2 p52: Mechanism and efficiency. Oncogene 1999, 18, 6201–6208. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Harhaj, E.W.; Sun, S.C. NF-κB-inducing kinase regulates the processing of NF-κB2 p100. Mol. Cell 2001, 7, 401–409. [Google Scholar] [CrossRef]
- Xiao, G.; Fong, A.; Sun, S.C. Induction of p100 processing by NF-κB-inducing kinase involves docking IκB kinase α (IKKα) to p100 and IKKα-mediated phosphorylation. J. Biol. Chem. 2004, 279, 30099–30105. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Zhang, M.; Sun, S.C. β-TrCP binding and processing of NF-κB2/p100 involve its phosphorylation at serines 866 and 870. Cell Signal. 2006, 18, 1309–1317. [Google Scholar] [CrossRef] [PubMed]
- Amir, R.E.; Haecker, H.; Karin, M.; Ciechanover, A. Mechanism of processing of the NF-κB2 p100 precursor: Identification of the specific polyubiquitin chain-anchoring lysine residue and analysis of the role of NEDD8-modification on the SCF(β-TrCP) ubiquitin ligase. Oncogene 2004, 23, 2540–2547. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, H.; Matsumoto, A.; Inuzuka, H.; Zhai, B.; Lau, A.W.; Wan, L.; Gao, D.; Shaik, S.; Yuan, M.; Gygi, S.P.; et al. SCF(Fbw7) modulates the NFκB signaling pathway by targeting NFκB2 for ubiquitination and destruction. Cell Rep. 2012, 1, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Busino, L.; Millman, S.E.; Scotto, L.; Kyratsous, C.A.; Basrur, V.; O’Connor, O.; Hoffmann, A.; Elenitoba-Johnson, K.S.; Pagano, M. Fbxw7α- and GSK3-mediated degradation of p100 is a pro-survival mechanism in multiple myeloma. Nat. Cell Biol. 2012, 14, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Silke, J. The regulation of TNF signalling: What a tangled web we weave. Curr. Opin. Immunol. 2011, 23, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Kupka, S.; Reichert, M.; Draber, P.; Walczak, H. Formation and removal of poly-ubiquitin chains in the regulation of tumor necrosis factor-induced gene activation and cell death. FEBS. J. 2016, 283, 2626–2639. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, P.; Baltimore, D. Sam68 is required for both NF-κB activation and apoptosis signaling by the TNF receptor. Mol. Cell 2011, 43, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Witt, A.; Vucic, D. Diverse ubiquitin linkages regulate RIP kinases-mediated inflammatory and cell death signaling. Cell Death Differ. 2017, 24, 1160–1171. [Google Scholar] [CrossRef] [PubMed]
- Vince, J.E.; Pantaki, D.; Feltham, R.; Mace, P.D.; Cordier, S.M.; Schmukle, A.C.; Davidson, A.J.; Callus, B.A.; Wong, W.W.; et al. TRAF2 must bind to cellular inhibitors of apoptosis for tumor necrosis factor (TNF) to efficiently activate NF-κB and to prevent TNF-induced apoptosis. J. Biol. Chem. 2009, 284, 35906–35915. [Google Scholar] [CrossRef] [PubMed]
- Haas, T.L.; Emmerich, C.H.; Gerlach, B.; Schmukle, A.C.; Cordier, S.M.; Rieser, E.; Feltham, R.; Vince, J.; Warnken, U.; Wenger, T.; et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol. Cell 2009, 36, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Emmerich, C.H.; Bakshi, S.; Kelsall, I.R.; Ortiz-Guerrero, J.; Shpiro, N.; Cohen, P. Lys63/Met1-hybrid ubiquitin chains are commonly formed during the activation of innate immune signaling. Biochem. Biophys. Res. Commun. 2016, 474, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Clark, K.; Lawrence, T.; Peggie, M.W.; Cohen, P. An unexpected twist to the activation of IKKβ: TAK1 primes IKKβ for activation by autophosphorylation. Biochem. J. 2014, 461, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Peltzer, N.; Darding, M.; Walczak, H. Holding RIPK1 on the Ubiquitin Leash in TNFR1 Signaling. Trends Cell Biol. 2016, 26, 445–461. [Google Scholar] [CrossRef] [PubMed]
- Dondelinger, Y.; Jouan-Lanhouet, S.; Divert, T.; Theatre, E.; Bertin, J.; Gough, P.J.; Giansanti, P.; Heck, A.J.; Dejardin, E.; Vandenabeele, P.; et al. NF-κB-Independent Role of IKKα/IKKβ in Preventing RIPK1 Kinase-Dependent Apoptotic and Necroptotic Cell Death during TNF Signaling. Mol. Cell 2015, 60, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Grootjans, S.; Vanden Berghe, T.; Vandenabeele, P. Initiation and execution mechanisms of necroptosis: An overview. Cell Death Differ. 2017, 24, 1184–1195. [Google Scholar] [CrossRef] [PubMed]
- Lork, M.; Verhelst, K.; Beyaert, R. CYLD, A20 and OTULIN deubiquitinases in NF-κB signaling and cell death: So similar, yet so different. Cell Death Differ. 2017, 24, 1172–1183. [Google Scholar] [CrossRef] [PubMed]
- Wertz, I.E.; O’Rourke, K.M.; Zhou, H.; Eby, M.; Aravind, L.; Seshagiri, S.; Wu, P.; Wiesmann, C.; Baker, R.; Boone, D.L.; et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-κB signalling. Nature 2004, 430, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Shembade, N.; Parvatiyar, K.; Harhaj, N.S.; Harhaj, E.W. The ubiquitin-editing enzyme A20 requires RNF11 to downregulate NF-κB signalling. EMBO J. 2009, 28, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Wertz, I.E.; Newton, K.; Seshasayee, D.; Kusam, S.; Lam, C.; Zhang, J.; Popovych, N.; Helgason, E.; Schoeffler, A.; Jeet, S.; et al. Phosphorylation and linear ubiquitin direct A20 inhibition of inflammation. Nature 2015, 528, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Dziedzic, S.A.; Su, Z.; Jean Barrett, V.; Najafov, A.; Mookhtiar, A.K.; Amin, P.; Pan, H.; Sun, L.; Zhu, H.; Ma, A.; et al. ABIN-1 regulates RIPK1 activation by linking Met1 ubiquitylation with Lys63 deubiquitylation in TNF-RSC. Nat. Cell Biol. 2018, 20, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C. CYLD: A tumor suppressor deubiquitinase regulating NF-κB activation and diverse biological processes. Cell Death Differ. 2010, 17, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Moquin, D.M.; McQuade, T.; Chan, F.K. CYLD deubiquitinates RIP1 in the TNFα-induced necrosome to facilitate kinase activation and programmed necrosis. PLoS ONE 2013, 8, e76841. [Google Scholar] [CrossRef] [PubMed]
- Kovalenko, A.; Chable-Bessia, C.; Cantarella, G.; Israël, A.; Wallach, D.; Courtois, G. The tumour suppressor CYLD negatively regulates NF-κB signalling by deubiquitination. Nature 2003, 424, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Schlicher, L.; Maurer, U. SPATA2: New insights into the assembly of the TNFR signaling complex. Cell Cycle 2017, 16, 11–12. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Massoumi, R. CYLD: A deubiquitination enzyme with multiple roles in cancer. Future Oncol. 2011, 7, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Keusekotten, K.; Elliott, P.R.; Glockner, L.; Fiil, B.K.; Damgaard, R.B.; Kulathu, Y.; Wauer, T.; Hospenthal, M.K.; Gyrd-Hansen, M.; Krappmann, D.; et al. OTULIN antagonizes LUBAC signaling by specifically hydrolyzing Met1-linked polyubiquitin. Cell 2013, 153, 1312–1326. [Google Scholar] [CrossRef] [PubMed]
- Fiil, B.K.; Damgaard, R.B.; Wagner, S.A.; Keusekotten, K.; Fritsch, M.; Bekker-Jensen, S.; Mailand, N.; Choudhary, C.; Komander, D.; Gyrd-Hansen, M. OTULIN restricts Met1-linked ubiquitination to control innate immune signaling. Mol. Cell 2013, 50, 818–830. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, V.; Akutsu, M.; Olma, M.H.; Gomes, L.C.; Kawasaki, M.; Dikic, I. Binding of OTULIN to the PUB domain of HOIP controls NF-κB signaling. Mol. Cell 2014, 54, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Elliott, P.R.; Nielsen, S.V.; Marco-Casanova, P.; Fiil, B.K.; Keusekotten, K.; Mailand, N.; Freund, S.M.; Gyrd-Hansen, M.; Komander, D. Molecular basis and regulation of OTULIN-LUBAC interaction. Mol. Cell 2014, 54, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Mahul-Mellier, A.L.; Pazarentzos, E.; Datler, C.; Iwasawa, R.; AbuAli, G.; Lin, B.; Grimm, S. De-ubiquitinating protease USP2a targets RIP1 and TRAF2 to mediate cell death by TNF. Cell Death Differ. 2012, 19, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Metzig, M.; Nickles, D.; Falschlehner, C.; Lehmann-Koch, J.; Straub, B.K.; Roth, W.; Boutros, M. An RNAi screen identifies USP2 as a factor required for TNF-α-induced NF-κB signaling. Int. J. Cancer 2011, 129, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Wang, L.; Zhang, L.; Pan, X.; Zhao, W. Ubiquitin-specific protease 4 promotes TNF-α-induced apoptosis by deubiquitination of RIP1 in head and neck squamous cell carcinoma. FEBS Lett. 2013, 587, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Xiao, N.; Li, H.; Luo, J.; Wang, R.; Chen, H.; Chen, J.; Wang, P. Ubiquitin-specific protease 4 (USP4) targets TRAF2 and TRAF6 for deubiquitination and inhibits TNFα-induced cancer cell migration. Biochem. J. 2012, 441, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Tan, X.; Wang, H.; Sun, W.; Shi, Y.; Burlingame, S.; Gu, X.; Cao, G.; Zhang, T.; Qin, J.; et al. Ubiquitin-specific peptidase 21 inhibits tumor necrosis factor α-induced nuclear factor κB activation via binding to and deubiquitinating receptor-interacting protein 1. J. Biol. Chem. 2010, 285, 969–978. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, L.; Dorf, M.E. PKC phosphorylation of TRAF2 mediates IKKα/β recruitment and K63-linked polyubiquitination. Mol. Cell 2009, 33, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Tortola, L.; Nitsch, R.; Bertrand, M.J.M.; Kogler, M.; Redouane, Y.; Kozieradzki, I.; Uribesalgo, I.; Fennell, L.M.; Daugaard, M.; Klug, H.; et al. The Tumor Suppressor Hace1 Is a Critical Regulator of TNFR1-Mediated Cell Fate. Cell Rep. 2016, 15, 1481–1492. [Google Scholar] [CrossRef] [PubMed]
- Jang, K.W.; Lee, K.H.; Kim, S.H.; Jin, T.; Choi, E.Y.; Jeon, H.J.; Kim, E.; Han, Y.S.; Chung, J.H. Ubiquitin ligase CHIP induces TRAF2 proteasomal degradation and NF-κB inactivation to regulate breast cancer cell invasion. J. Cell Biochem. 2011, 112, 3612–3620. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Smac Mimetics to Therapeutically Target IAP Proteins in Cancer. Int. Rev. Cell. Mol. Biol. 2017, 330, 157–169. [Google Scholar] [PubMed]
- Goncharov, T.; Niessen, K.; de Almagro, M.C.; Izrael-Tomasevic, A.; Fedorova, A.V.; Varfolomeev, E.; Arnott, D.; Deshayes, K.; Kirkpatrick, D.S.; Vucic, D. OTUB1 modulates c-IAP1 stability to regulate signalling pathways. EMBO J. 2013, 32, 1103–1114. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.; Hahn, A.A.; Hu, S.; Yang, X. The USP19 deubiquitinase regulates the stability of c-IAP1 and c-IAP2. J. Biol. Chem. 2011, 286, 35380–35387. [Google Scholar] [CrossRef] [PubMed]
- De Jong, M.F.; Liu, Z.; Chen, D.; Alto, N.M. Shigella flexneri suppresses NF-κB activation by inhibiting linear ubiquitin chain ligation. Nat. Microbiol. 2016, 1, 16084. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.W.; Gentle, I.E.; Nachbur, U.; Anderton, H.; Vaux, D.L.; Silke, J. RIPK1 is not essential for TNFR1-induced activation of NF-κB. Cell Death Differ. 2010, 17, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Skaug, B.; Zeng, W.; Chen, Z.J. A ubiquitin replacement strategy in human cells reveals distinct mechanisms of IKK activation by TNFα and IL-1β. Mol. Cell 2009, 36, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, K.; Zhang, L.; Workman, L.M.; Ting, A.T.; Iwai, K.; Habelhah, H. Two coordinated mechanisms underlie tumor necrosis factor a-induced immediate and delayed IκB kinase activation. Mol. Cell. Biol. 2013, 33, 1901–1915. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, S.E.; Harikumar, K.B.; Hait, N.C.; Allegood, J.; Strub, G.M.; Kim, E.Y.; Maceyka, M.; Jiang, H.; Luo, C.; Kordula, T.; et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 2010, 465, 1084–1088. [Google Scholar] [CrossRef] [PubMed]
- Etemadi, N.; Chopin, M.; Anderton, H.; Tanzer, M.C.; Rickard, J.A.; Abeysekera, W.; Hall, C.; Spall, S.K.; Wang, B.; Xiong, Y.; et al. TRAF2 regulates TNF and NF-κB signalling to suppress apoptosis and skin inflammation independently of Sphingosine kinase 1. Elife 2015, 4, E10592. [Google Scholar] [CrossRef] [PubMed]
- Tada, K.; Okazaki, T.; Sakon, S.; Kobarai, T.; Kurosawa, K.; Yamaoka, S.; Hashimoto, H.; Mak, T.W.; Yagita, H.; Okumura, K.; et al. Critical roles of TRAF2 and TRAF5 in tumor necrosis factor-induced NF-κB activation and protection from cell death. J. Biol. Chem. 2001, 276, 36530–36534. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Blackwell, K.; Thomas, G.S.; Sun, S.; Yeh, W.C.; Habelhah, H. TRAF2 suppresses basal IKK activity in resting cells and TNFα can activate IKK in TRAF2 and TRAF5 double knockout cells. J. Mol. Biol. 2009, 389, 495–510. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.U.; Wesche, H. Summary and comparison of the signaling mechanisms of the Toll/interleukin-1 receptor family. Biochim. Biophys. Acta 2002, 1592, 265–280. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.; Bowie, A.G. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 2007, 7, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Motshwene, P.G.; Moncrieffe, M.C.; Grossmann, J.G.; Kao, C.; Ayaluru, M.; Sandercock, A.M.; Robinson, C.V.; Latz, E.; Gay, N.J. An oligomeric signaling platform formed by the Toll-like receptor signal transducers MyD88 and IRAK-4. J. Biol. Chem. 2009, 284, 25404–25411. [Google Scholar] [CrossRef] [PubMed]
- Ferrao, R.; Zhou, H.; Shan, Y.; Liu, Q.; Li, Q.; Shaw, D.E.; Li, X.; Wu, H. IRAK4 dimerization and trans-autophosphorylation are induced by Myddosome assembly. Mol. Cell 2014, 55, 891–903. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Arron, J.R.; Lamothe, B.; Cirilli, M.; Kobayashi, T.; Shevde, N.K.; Segal, D.; Dzivenu, O.K.; Vologodskaia, M.; Yim, M.; et al. Distinct molecular mechanism for initiating TRAF6 signalling. Nature 2002, 418, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Moynagh, P.N. The roles of Pellino E3 ubiquitin ligases in immunity. Nat. Rev. Immunol. 2014, 14, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Medvedev, A.E.; Murphy, M.; Zhou, H.; Li, X. E3 ubiquitin ligases Pellinos as regulators of pattern recognition receptor signaling and immune response. Immunol. Rev. 2015, 266, 109–122. [Google Scholar] [PubMed]
- Lin, C.C.; Huoh, Y.S.; Schmitz, K.R.; Jensen, L.E.; Ferguson, K.M. Pellino proteins contain a cryptic FHA domain that mediates interaction with phosphorylated IRAK1. Structure 2008, 16, 1806–1816. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Strickson, S. The role of hybrid ubiquitin chains in the Myd88 and other innate immune signaling pathways. Cell Death Differ. 2017, 24, 1153–1159. [Google Scholar] [PubMed]
- Xia, Z.P.; Sun, L.; Chen, X.; Pineda, G.; Jiang, X.; Adhikari, A.; Zeng, W.; Chen, Z.J. Direct activation of protein kinases by unanchored polyubiquitin chains. Nature 2009, 461, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Xiao, N.; Xiao, H.; Zhou, H.; Yu, M.; Gu, J.; Li, X. β-TrCP-mediated IRAK1 degradation releases TAK1-TRAF6 from the membrane to the cytosol for TAK1-dependent NF-κB activation. Mol. Cell. Biol. 2012, 32, 3990–4000. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa, A.; Tseng, P.H.; Vallabhapurapu, S.; Luo, J.L.; Zhang, W.; Wang, H.; Vignali, D.A.; Gallagher, E.; Karin, M. Essential cytoplasmic translocation of a cytokine receptor-assembled signaling complex. Science 2008, 321, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Kagan, J.C.; Su, T.; Horng, T.; Chow, A.; Akira, S.; Medzhitov, R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-β. Nat. Immunol. 2008, 9, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Sasai, M.; Tatematsu, M.; Oshiumi, H.; Funami, K.; Matsumoto, M.; Hatakeyama, S.; Seya, T. Direct binding of TRAF2 and TRAF6 to TICAM-1/TRIF adaptor participates in activation of the Toll-like receptor 3/4 pathway. Mol. Immunol. 2010, 47, 1283–1291. [Google Scholar] [CrossRef] [PubMed]
- Cusson-Hermance, N.; Khurana, S.; Lee, T.H.; Fitzgerald, K.A.; Kelliher, M.A. Rip1 mediates the Trif-dependent toll-like receptor 3- and 4-induced NF-κB activation but does not contribute to interferon regulatory factor 3 activation. J. Biol. Chem. 2005, 280, 36560–36566. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, W.J.; Offermann, M.K. Apoptosis induced by the toll-like receptor adaptor TRIF is dependent on its receptor interacting protein homotypic interaction motif. J. Immunol. 2005, 174, 4942–4952. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.; Jin, W.; Sun, S.C. Peli1 facilitates TRIF-dependent Toll-like receptor signaling and proinflammatory cytokine production. Nat. Immunol. 2009, 10, 1089–1095. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Li, M.; Zhang, Y.; Zhou, Q.; Shu, H.B. The E3 ubiquitin ligase MARCH8 negatively regulates IL-1β-induced NF-κB activation by targeting the IL1RAP coreceptor for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2012, 109, 14128–14133. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Mudge, M.C.; Soll, J.M.; Rodrigues, R.B.; Byrum, A.K.; Schwarzkopf, E.A.; Bradstreet, T.R.; Gygi, S.P.; Edelson, B.T.; Mosammaparast, N. OTUD4 Is a Phospho-Activated K63 Deubiquitinase That Regulates MyD88-Dependent Signaling. Mol. Cell 2018, 69, 505–516. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Li, Y.; Li, C.; Liu, L.J.; Zhang, X.D.; Liu, Y.; Shu, H.B. USP2a negatively regulates IL-1β- and virus-induced NF-κB activation by deubiquitinating TRAF6. J. Mol. Cell. Biol. 2013, 5, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Zhang, X.; van Dam, H.; Ten Dijke, P.; Huang, H.; Zhang, L. Ubiquitin-specific protease 4 mitigates Toll-like/interleukin-1 receptor signaling and regulates innate immune activation. J. Biol. Chem. 2012, 287, 11002–11010. [Google Scholar] [CrossRef] [PubMed]
- Yasunaga, J.; Lin, F.C.; Lu, X.; Jeang, K.T. Ubiquitin-specific peptidase 20 targets TRAF6 and human T cell leukemia virus type 1 tax to negatively regulate NF-κB signaling. J. Virol. 2011, 85, 6212–6219. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Jono, H.; Kai, H.; Li, J.D. The tumor suppressor cylindromatosis (CYLD) acts as a negative regulator for toll-like receptor 2 signaling via negative cross-talk with TRAF6 and TRAF7. J. Biol. Chem. 2005, 280, 41111–41121. [Google Scholar] [CrossRef] [PubMed]
- Heyninck, K.; Beyaert, R. The cytokine-inducible zinc finger protein A20 inhibits IL-1-induced NF-κB activation at the level of TRAF6. FEBS Lett. 1999, 442, 147–150. [Google Scholar] [CrossRef]
- Yuk, J.M.; Shin, D.M.; Lee, H.M.; Kim, J.J.; Kim, S.W.; Jin, H.S.; Yang, C.S.; Park, K.A.; Chanda, D.; Kim, D.K.; et al. The orphan nuclear receptor SHP acts as a negative regulator in inflammatory signaling triggered by Toll-like receptors. Nat. Immunol. 2011, 12, 742–751. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Huang, X.; Xin, H.B.; Fu, M.; Xue, A.; Wu, Z.H. TRAF Family Member-associated NF-κB Activator (TANK) Inhibits Genotoxic Nuclear Factor κB Activation by Facilitating Deubiquitinase USP10-dependent Deubiquitination of TRAF6 Ligase. J. Biol. Chem. 2015, 290, 13372–13385. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Su, Z.; Lin, M.; Ou, J.; Zhao, W.; Cui, J.; Wang, R.F. NLRP11 attenuates Toll-like receptor signalling by targeting TRAF6 for degradation via the ubiquitin ligase RNF19A. Nat. Commun. 2017, 8, 1977. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.W.; Xu, W.C.; Luo, J.G.; Guo, X.J.; Sun, T.; Zhao, X.L.; Fu, Z.J. WW domain containing E3 ubiquitin protein ligase 1 (WWP1) negatively regulates TLR4-mediated TNF-α and IL-6 production by proteasomal degradation of TNF receptor associated factor 6 (TRAF6). PLoS ONE 2013, 8, e67633. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Wang, L.; Zhang, M.; Yuan, C.; Gao, C. E3 ubiquitin ligase tripartite motif 38 negatively regulates TLR-mediated immune responses by proteasomal degradation of TNF receptor-associated factor 6 in macrophages. J. Immunol. 2012, 188, 2567–2574. [Google Scholar] [CrossRef] [PubMed]
- Zhong, B.; Liu, X.; Wang, X.; Liu, X.; Li, H.; Darnay, B.G.; Lin, X.; Sun, S.C.; Dong, C. Ubiquitin-specific protease 25 regulates TLR4-dependent innate immune responses through deubiquitination of the adaptor protein TRAF3. Sci. Signal. 2013, 6, ra35. [Google Scholar] [CrossRef] [PubMed]
- Caruso, R.; Warner, N.; Inohara, N.; Núñez, G. NOD1 and NOD2: Signaling, host defense, and inflammatory disease. Immunity 2014, 41, 898–908. [Google Scholar] [CrossRef] [PubMed]
- Boyle, J.P.; Parkhouse, R.; Monie, T.P. Insights into the molecular basis of the NOD2 signalling pathway. Open Biol. 2014, 4, 140178. [Google Scholar] [CrossRef] [PubMed]
- Tigno-Aranjuez, J.T.; Abbott, D.W. Ubiquitination and phosphorylation in the regulation of NOD2 signaling and NOD2-mediated disease. Biochim. Biophys. Acta 2012, 1823, 2022–2028. [Google Scholar] [CrossRef] [PubMed]
- Tigno-Aranjuez, J.T.; Asara, J.M.; Abbott, D.W. Inhibition of RIP2′s tyrosine kinase activity limits NOD2-driven cytokine responses. Genes Dev. 2010, 24, 2666–2677. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yin, C.; Pandey, A.; Abbott, D.; Sassetti, C.; Kelliher, M.A. NOD2 pathway activation by MDP or Mycobacterium tuberculosis infection involves the stable polyubiquitination of Rip2. J. Biol. Chem. 2007, 282, 36223–36229. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Fujimoto, Y.; Lucas, P.C.; Nakano, H.; Fukase, K.; Núñez, G.; Inohara, N. A critical role of RICK/RIP2 polyubiquitination in Nod-induced NF-κB activation. EMBO J. 2008, 27, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, M.J.; Doiron, K.; Labbé, K.; Korneluk, R.G.; Barker, P.A.; Saleh, M. Cellular inhibitors of apoptosis cIAP1 and cIAP2 are required for innate immunity signaling by the pattern recognition receptors NOD1 and NOD2. Immunity 2009, 30, 789–801. [Google Scholar] [PubMed]
- Krieg, A.; Correa, R.G.; Garrison, J.B.; Le Negrate, G.; Welsh, K.; Huang, Z.; Knoefel, W.T.; Reed, J.C. XIAP mediates NOD signaling via interaction with RIP2. Proc. Natl. Acad. Sci. USA 2009, 106, 14524–14529. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wang, B.; Humphries, F.; Jackson, R.; Healy, M.E.; Bergin, R.; Aviello, G.; Hall, B.; McNamara, D.; Darby, T.; et al. Pellino3 ubiquitinates RIP2 and mediates Nod2-induced signaling and protective effects in colitis. Nat. Immunol. 2013, 14, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Damgaard, R.B.; Nachbur, U.; Yabal, M.; Wong, W.W.; Fiil, B.K.; Kastirr, M.; Rieser, E.; Rickard, J.A.; Bankovacki, A.; Peschel, C.; et al. The ubiquitin ligase XIAP recruits LUBAC for NOD2 signaling in inflammation and innate immunity. Mol. Cell 2012, 46, 746–758. [Google Scholar] [CrossRef] [PubMed]
- Damgaard, R.B.; Fiil, B.K.; Speckmann, C.; Yabal, M.; zur Stadt, U.; Bekker-Jensen, S.; Jost, P.J.; Ehl, S.; Mailand, N.; Gyrd-Hansen, M. Disease-causing mutations in the XIAP BIR2 domain impair NOD2-dependent immune signalling. EMBO Mol. Med. 2013, 5, 1278–1295. [Google Scholar] [CrossRef] [PubMed]
- Abbott, D.W.; Yang, Y.; Hutti, J.E.; Madhavarapu, S.; Kelliher, M.A.; Cantley, L.C. Coordinated regulation of Toll-like receptor and NOD2 signaling by K63-linked polyubiquitin chains. Mol. Cell. Biol. 2007, 27, 6012–6025. [Google Scholar] [CrossRef] [PubMed]
- Clark, N.M.; Marinis, J.M.; Cobb, B.A.; Abbott, D.W. MEKK4 sequesters RIP2 to dictate NOD2 signal specificity. Curr. Biol. 2008, 18, 1402–1408. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Draber, P.; Kupka, S.; Reichert, M.; Draberova, H.; Lafont, E.; de Miguel, D.; Spilgies, L.; Surinova, S.; Taraborrelli, L.; Hartwig, T.; et al. LUBAC-Recruited CYLD and A20 Regulate Gene Activation and Cell Death by Exerting Opposing Effects on Linear Ubiquitin in Signaling Complexes. Cell Rep. 2015, 13, 2258–2272. [Google Scholar] [CrossRef] [PubMed]
- Hrdinka, M.; Fiil, B.K.; Zucca, M.; Leske, D.; Bagola, K.; Yabal, M.; Elliott, P.R.; Damgaard, R.B.; Komander, D.; Jost, P.J.; et al. CYLD Limits Lys63- and Met1-Linked Ubiquitin at Receptor Complexes to Regulate Innate Immune Signaling. Cell Rep. 2016, 14, 2846–2858. [Google Scholar] [PubMed]
- Tao, M.; Scacheri, P.C.; Marinis, J.M.; Harhaj, E.W.; Matesic, L.E.; Abbott, D.W. ITCH K63-ubiquitinates the NOD2 binding protein, RIP2, to influence inflammatory signaling pathways. Curr. Biol. 2009, 19, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Hitotsumatsu, O.; Ahmad, R.C.; Tavares, R.; Wang, M.; Philpott, D.; Turer, E.E.; Lee, B.L.; Shiffin, N.; Advincula, R.; Malynn, B.A.; et al. The ubiquitin-editing enzyme A20 restricts nucleotide-binding oligomerization domain containing 2-triggered signals. Immunity 2008, 28, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Bist, P.; Cheong, W.S.; Ng, A.; Dikshit, N.; Kim, B.H.; Pulloor, N.K.; Khameneh, H.J.; Hedl, M.; Shenoy, A.R.; Balamuralidhar, V.; et al. E3 Ubiquitin ligase ZNRF4 negatively regulates NOD2 signalling and induces tolerance to MDP. Nat. Commun. 2017, 8, 15865. [Google Scholar] [CrossRef] [PubMed]
- Zurek, B.; Schoultz, I.; Neerincx, A.; Napolitano, L.M.; Birkner, K.; Bennek, E.; Sellge, G.; Lerm, M.; Meroni, G.; Söderholm, J.D. TRIM27 negatively regulates NOD2 by ubiquitination and proteasomal degradation. PLoS ONE 2012, 7, e41255. [Google Scholar] [CrossRef] [PubMed]
- Condé, C.; Rambout, X.; Lebrun, M.; Lecat, A.; Di Valentin, E.; Dequiedt, F.; Piette, J.; Gloire, G.; Legrand, S. The inositol phosphatase SHIP-1 inhibits NOD2-induced NF-κB activation by disturbing the interaction of XIAP with RIP2. PLoS ONE 2012, 7, e41005. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Fujita, T. RNA recognition and signal transduction by RIG-I-like receptors. Immunol. Rev. 2009, 227, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, C.; Gale, M., Jr. Recognition of viruses by cytoplasmic sensors. Curr. Opin. Immunol. 2010, 22, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Goubau, D.; Deddouche, S.; Reis e Sousa, C. Cytosolic sensing of viruses. Immunity 2013, 38, 855–869. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Kato, H.; Kumagai, Y.; Yoneyama, M.; Sato, S.; Matsushita, K.; Tsujimura, T.; Fujita, T.; Akira, S.; Takeuchi, O. LGP2 is a positive regulator of RIG-I- and MDA5-mediated antiviral responses. Proc. Natl. Acad. Sci. USA 2010, 107, 1512–1517. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.; Gack, M.U. Post-translational Control of Intracellular Pathogen Sensing Pathways. Trends Immunol. 2017, 38, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Gao, C. Regulation of MAVS activation through post-translational modifications. Curr. Opin. Immunol. 2017, 50, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Gack, M.U.; Shin, Y.C.; Joo, C.H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 2007, 446, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Li, J.; Zheng, W.; Shang, Y.; Zhao, Z.; Wang, S.; Bi, Y.; Zhang, S.; Xu, C.; Duan, Z. Cyclophilin A-regulated ubiquitination is critical for RIG-I-mediated antiviral immune responses. Elife 2017, 6, E24425. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Kinch, L.N.; Brautigam, C.A.; Chen, X.; Du, F.; Grishin, N.V.; Chen, Z.J. Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity 2012, 36, 959–973. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Sun, L.; Jiang, X.; Chen, X.; Hou, F.; Adhikari, A.; Xu, M.; Chen, Z.J. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell 2010, 141, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Oshiumi, H.; Miyashita, M.; Matsumoto, M.; Seya, T. A distinct role of Riplet-mediated K63-Linked polyubiquitination of the RIG-I repressor domain in human antiviral innate immune responses. PLoS Pathog. 2013, 9, e1003533. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Li, Q.; Mao, A.P.; Hu, M.M.; Shu, H.B. TRIM4 modulates type I interferon induction and cellular antiviral response by targeting RIG-I for K63-linked ubiquitination. J. Mol. Cell. Biol. 2014, 6, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Kuniyoshi, K.; Takeuchi, O.; Pandey, S.; Satoh, T.; Iwasaki, H.; Akira, S.; Kawai, T. Pivotal role of RNA-binding E3 ubiquitin ligase MEX3C in RIG-I-mediated antiviral innate immunity. Proc. Natl. Acad. Sci. USA 2014, 111, 5646–5651. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Xian, H.; Tian, S.; Sun, T.; Qin, Y.; Zhang, S.; Cui, J. A Hierarchical Mechanism of RIG-I Ubiquitination Provides Sensitivity, Robustness and Synergy in Antiviral Immune Responses. Sci. Rep. 2016, 6, 29263. [Google Scholar] [CrossRef] [PubMed]
- Lang, X.; Tang, T.; Jin, T.; Ding, C.; Zhou, R.; Jiang, W. TRIM65-catalized ubiquitination is essential for MDA5-mediated antiviral innate immunity. J. Exp. Med. 2017, 214, 459–473. [Google Scholar] [CrossRef] [PubMed]
- Hou, F.; Sun, L.; Zheng, H.; Skaug, B.; Jiang, Q.X.; Chen, Z.J. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 2011, 146, 448–461. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Chen, J.; Cai, X.; Wu, J.; Chen, X.; Wu, Y.T.; Sun, L.; Chen, Z.J. MAVS recruits multiple ubiquitin E3 ligases to activate antiviral signaling cascades. Elife 2013, 2, e00785. [Google Scholar] [CrossRef] [PubMed]
- Fang, R.; Jiang, Q.; Zhou, X.; Wang, C.; Guan, Y.; Tao, J.; Xi, J.; Feng, J.M.; Jiang, Z. MAVS activates TBK1 and IKKε through TRAFs in NEMO dependent and independent manner. PLoS Pathog. 2017, 13, e1006720. [Google Scholar] [CrossRef] [PubMed]
- Tang, E.D.; Wang, C.Y. TRAF5 is a downstream target of MAVS in antiviral innate immune signaling. PLoS ONE 2010, 5, e9172. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, S.S.; Jensen, S.B.; Chiliveru, S.; Melchjorsen, J.; Julkunen, I.; Gaestel, M.; Arthur, J.S.; Flavell, R.A.; Ghosh, S.; Paludan, S.R. RIG-I-mediated activation of p38 MAPK is essential for viral induction of interferon and activation of dendritic cells: Dependence on TRAF2 and TAK1. J. Biol. Chem. 2009, 284, 10774–10782. [Google Scholar] [PubMed]
- Pauli, E.K.; Chan, Y.K.; Davis, M.E.; Gableske, S.; Wang, M.K.; Feister, K.F.; Gack, M.U. The ubiquitin-specific protease USP15 promotes RIG-I-mediated antiviral signaling by deubiquitylating TRIM25. Sci. Signal. 2014, 7, ra3. [Google Scholar] [CrossRef] [PubMed]
- Inn, K.S.; Gack, M.U.; Tokunaga, F.; Shi, M.; Wong, L.Y.; Iwai, K.; Jung, J.U. Linear ubiquitin assembly complex negatively regulates RIG-I- and TRIM25-mediated type I interferon induction. Mol. Cell 2011, 41, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Song, Y.; Li, Y.; Zhu, Q.; Tan, P.; Qin, Y.; Wang, H.Y.; Wang, R.F. USP3 inhibits type I interferon signaling by deubiquitinating RIG-I-like receptors. Cell Res. 2014, 24, 400–416. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Mao, R.; Yu, Y.; Liu, S.; Shi, Z.; Cheng, J.; Zhang, H.; An, L.; Zhao, Y.; Xu, X.; et al. USP21 negatively regulates antiviral response by acting as a RIG-I deubiquitinase. J. Exp. Med. 2014, 211, 313–328. [Google Scholar] [CrossRef] [PubMed]
- Friedman, C.S.; O’Donnell, M.A.; Legarda-Addison, D.; Ng, A.; Cárdenas, W.B.; Yount, J.S.; Moran, T.M.; Basler, C.F.; Komuro, A.; Horvath, C.M.; et al. The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep. 2008, 9, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Zhang, J.; Lin, H.; Li, Z.; Sun, X.; Xin, D.; Yang, M.; Sun, L.; Li, L.; Wang, H.; et al. Syndecan-4 negatively regulates antiviral signalling by mediating RIG-I deubiquitination via CYLD. Nat. Commun. 2016, 7, 11848. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Jiang, M.; Liu, S.; Zhang, S.; Liu, W.; Ma, Y.; Zhang, L.; Zhang, J.; Cao, X. RNF122 suppresses antiviral type I interferon production by targeting RIG-I CARDs to mediate RIG-I degradation. Proc. Natl. Acad. Sci. USA 2016, 113, 9581–9586. [Google Scholar] [CrossRef] [PubMed]
- Arimoto, K.; Takahashi, H.; Hishiki, T.; Konishi, H.; Fujita, T.; Shimotohno, K. Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc. Natl. Acad. Sci. USA 2007, 104, 7500–7505. [Google Scholar] [CrossRef] [PubMed]
- Hao, Q.; Jiao, S.; Shi, Z.; Li, C.; Meng, X.; Zhang, Z.; Wang, Y.; Song, X.; Wang, W.; Zhang, R.; et al. A non-canonical role of the p97 complex in RIG-I antiviral signaling. EMBO J. 2015, 34, 2903–2920. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhao, W.; Zhang, M.; Wang, P.; Zhao, K.; Zhao, X.; Yang, S.; Gao, C. USP4 positively regulates RIG-I-mediated antiviral response through deubiquitination and stabilization of RIG-I. J. Virol. 2013, 87, 4507–4515. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tong, X.; Ye, X. Ndfip1 negatively regulates RIG-I-dependent immune signaling by enhancing E3 ligase Smurf1-mediated MAVS degradation. J. Immunol. 2012, 189, 5304–5313. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Li, R.; Meng, J.L.; Mao, H.T.; Zhang, Y.; Zhang, J. Smurf2 negatively modulates RIG-I-dependent antiviral response by targeting VISA/MAVS for ubiquitination and degradation. J. Immunol. 2014, 192, 4758–4764. [Google Scholar] [CrossRef] [PubMed]
- Yoo, Y.S.; Park, Y.Y.; Kim, J.H.; Cho, H.; Kim, S.H.; Lee, H.S.; Kim, T.H.; Sun Kim, Y.; Lee, Y.; Kim, C.J.; et al. The mitochondrial ubiquitin ligase MARCH5 resolves MAVS aggregates during antiviral signalling. Nat. Commun. 2015, 6, 7910. [Google Scholar] [CrossRef] [PubMed]
- Zhong, B.; Zhang, Y.; Tan, B.; Liu, T.T.; Wang, Y.Y.; Shu, H.B. The E3 ubiquitin ligase RNF5 targets virus-induced signaling adaptor for ubiquitination and degradation. J. Immunol. 2010, 184, 6249–6255. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.W.; Li, S.; Li, C.; Zheng, Z.Q.; Cao, P.; Tong, Z.; Lian, H.; Wang, S.Y.; Shu, H.B.; Wang, Y.Y. iRhom2 is essential for innate immunity to RNA virus by antagonizing ER- and mitochondria-associated degradation of VISA. PLoS Pathog. 2017, 13, e1006693. [Google Scholar] [CrossRef] [PubMed]
- You, F.; Sun, H.; Zhou, X.; Sun, W.; Liang, S.; Zhai, Z.; Jiang, Z. PCBP2 mediates degradation of the adaptor MAVS via the HECT ubiquitin ligase AIP4. Nat. Immunol. 2009, 10, 1300–1308. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar]
- Xia, P.; Wang, S.; Gao, P.; Gao, G.; Fan, Z. DNA sensor cGAS-mediated immune recognition. Protein Cell 2016, 7, 777–791. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Damania, B. The cGAS-STING Defense Pathway and Its Counteraction by Viruses. Cell Host Microbe 2016, 19, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Barber, G.N. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-κB activation through TBK1. J. Virol. 2014, 88, 5328–5341. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Hu, M.M.; Wang, Y.Y.; Shu, H.B. TRIM32 protein modulates type I interferon induction and cellular antiviral response by targeting MITA/STING protein for K63-linked ubiquitination. J. Biol. Chem. 2012, 287, 28646–28655. [Google Scholar] [CrossRef] [PubMed]
- Fang, R.; Wang, C.; Jiang, Q.; Lv, M.; Gao, P.; Yu, X.; Mu, P.; Zhang, R.; Bi, S.; Feng, J.M.; et al. NEMO-IKKβ Are Essential for IRF3 and NF-κB Activation in the cGAS-STING Pathway. J. Immunol. 2017, 199, 3222–3233. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zhang, Q.; Jing, Y.Y.; Zhang, M.; Wang, H.Y.; Cai, Z.; Liuyu, T.; Zhang, Z.D.; Xiong, T.C.; Wu, Y. USP13 negatively regulates antiviral responses by deubiquitinating STING. Nat. Commun. 2017, 8, 15534. [Google Scholar] [CrossRef] [PubMed]
- Zhong, B.; Zhang, L.; Lei, C.; Li, Y.; Mao, A.P.; Yang, Y.; Wang, Y.Y.; Zhang, X.L.; Shu, H.B. The ubiquitin ligase RNF5 regulates antiviral responses by mediating degradation of the adaptor protein MITA. Immunity 2009, 30, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lian, Q.; Yang, B.; Yan, S.; Zhou, H.; He, L.; Lin, G.; Lian, Z.; Jiang, Z.; Sun, B. TRIM30α Is a Negative-Feedback Regulator of the Intracellular DNA and DNA Virus-Triggered Response by Targeting STING. PLoS Pathog. 2015, 11, e1005012. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, M.X.; Zhang, Q.; Zhu, G.F.; Yuan, L.; Zhang, D.E.; Zhu, Q.; Yao, J.; Shu, H.B.; Zhong, B. USP18 recruits USP20 to promote innate antiviral response through deubiquitinating STING/MITA. Cell Res. 2016, 26, 1302–1319. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.M.; Yang, Q.; Xie, X.Q.; Liao, C.Y.; Lin, H.; Liu, T.T.; Yin, L.; Shu, H.B. Sumoylation Promotes the Stability of the DNA Sensor cGAS and the Adaptor STING to Regulate the Kinetics of Response to DNA Virus. Immunity 2016, 45, 555–569. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Huang, L.; Hong, Z.; Lv, Z.; Mao, Z.; Tang, Y.; Kong, X.; Li, S.; Cui, Y.; Liu, H.; et al. The E3 ubiquitin ligase RNF185 facilitates the cGAS-mediated innate immune response. PLoS Pathog. 2017, 13, e1006264. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Meng, Q.; Qin, Y.; Liang, P.; Tan, P.; He, L.; Zhou, Y.; Chen, Y.; Huang, J.; Wang, R.F.; et al. TRIM14 Inhibits cGAS Degradation Mediated by Selective Autophagy Receptor p62 to Promote Innate Immune Responses. Mol. Cell 2016, 64, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Courtney, A.H.; Lo, W.L.; Weiss, A. TCR Signaling: Mechanisms of Initiation and Propagation. Trends Biochem. Sci. 2018, 43, 108–123. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Schaefer, B.C. A new look at T cell receptor signaling to nuclear factor-κB. Trends Immunol. 2013, 34, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Meininger, I.; Krappmann, D. Lymphocyte signaling and activation by the CARMA1-BCL10-MALT1 signalosome. Biol. Chem. 2016, 397, 1315–1333. [Google Scholar] [CrossRef] [PubMed]
- Sommer, K.; Guo, B.; Pomerantz, J.L.; Bandaranayake, A.D.; Moreno-García, M.E.; Ovechkina, Y.L.; Rawlings, D.J. Phosphorylation of the CARMA1 linker controls NF-κB activation. Immunity 2005, 23, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Deng, L.; Ea, C.K.; Xia, Z.P.; Chen, Z.J. The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes. Mol. Cell 2004, 14, 289–301. [Google Scholar] [CrossRef]
- Li, Y.; He, X.; Wang, S.; Shu, H.B.; Liu, Y. USP2a positively regulates TCR-induced NF-κB activation by bridging MALT1-TRAF6. Protein Cell 2013, 4, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, G.; Le Guelte, A.; Demian, C.; Vazquez, A.; Gavard, J.; Bidère, N. Interplay between BCL10, MALT1 and IκBα during T-cell-receptor-mediated NFκB activation. J. Cell Sci. 2010, 123, 2375–2380. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Traver, M.K.; Kashyap, A.K.; Washington, M.A.; Latoche, J.R.; Schaefer, B.C. T cell receptor signals to NF-κB are transmitted by a cytosolic p62-Bcl10-Malt1-IKK signalosome. Sci. Signal. 2014, 7, ra45. [Google Scholar] [CrossRef] [PubMed]
- King, C.G.; Kobayashi, T.; Cejas, P.J.; Kim, T.; Yoon, K.; Kim, G.K.; Chiffoleau, E.; Hickman, S.P.; Walsh, P.T.; et al. TRAF6 is a T cell-intrinsic negative regulator required for the maintenance of immune homeostasis. Nat. Med. 2006, 12, 1088–1092. [Google Scholar]
- Stempin, C.C.; Chi, L.; Giraldo-Vela, J.P.; High, A.A.; Häcker, H.; Redecke, V. The E3 ubiquitin ligase mind bomb-2 (MIB2) protein controls B-cell CLL/lymphoma 10 (BCL10)-dependent NF-κB activation. J. Biol. Chem. 2011, 286, 37147–37157. [Google Scholar] [CrossRef] [PubMed]
- Dubois, S.M.; Alexia, C.; Wu, Y.; Leclair, H.M.; Leveau, C.; Schol, E.; Fest, T.; Tarte, K.; Chen, Z.J.; Gavard, J.; et al. A catalytic-independent role for the LUBAC in NF-κB activation upon antigen receptor engagement and in lymphoma cells. Blood 2014, 123, 2199–2203. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.K.; Yang, C.; Chan, W.; Wang, Z.; Deibel, K.E.; Pomerantz, J.L. Molecular Determinants of Scaffold-induced Linear Ubiquitinylation of B Cell Lymphoma/Leukemia 10 (Bcl10) during T Cell Receptor and Oncogenic Caspase Recruitment Domain-containing Protein 11 (CARD11) Signaling. J. Biol. Chem. 2016, 291, 25921–25936. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Kelly, P.; Shaffer, A.L., III; Schmitz, R.; Yoo, H.M.; Liu, X.; Huang, D.W.; Webster, D.; Young, R.M.; Nakagawa, M.; et al. Targeting Non-proteolytic Protein Ubiquitination for the Treatment of Diffuse B Cell Lymphoma. Cancer Cell 2016, 29, 494–507. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.Y.; Chi, H.; Xie, M.; Schneider, M.D.; Flavell, R.A. The kinase TAK1 integrates antigen and cytokine receptor signaling for T cell development, survival and function. Nat. Immunol. 2006, 7, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, H.; Yamasaki, S.; Maeda, S.; Saito, T.; Kurosaki, T. Regulation of NF-κB-dependent T cell activation and development by MEKK3. Int. Immunol. 2009, 21, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Düwel, M.; Welteke, V.; Oeckinghaus, A.; Baens, M.; Kloo, B.; Ferch, U.; Darnay, B.G.; Ruland, J.; Marynen, P.; Krappmann, D. A20 negatively regulates T cell receptor signaling to NF-κB by cleaving Malt1 ubiquitin chains. J. Immunol. 2009, 182, 7718–7728. [Google Scholar] [CrossRef] [PubMed]
- Coornaert, B.; Baens, M.; Heyninck, K.; Bekaert, T.; Haegman, M.; Staal, J.; Sun, L.; Chen, Z.J.; Marynen, P.; Beyaert, R. T cell antigen receptor stimulation induces MALT1 paracaspase-mediated cleavage of the NF-κB inhibitor A20. Nat. Immunol. 2008, 9, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Reiley, W.W.; Jin, W.; Lee, A.J.; Wright, A.; Wu, X.; Tewalt, E.F.; Leonard, T.O.; Norbury, C.C.; Fitzpatrick, L.; Zhang, M.; et al. Deubiquitinating enzyme CYLD negatively regulates the ubiquitin-dependent kinase Tak1 and prevents abnormal T cell responses. J. Exp. Med. 2007, 204, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Staal, J.; Driege, Y.; Bekaert, T.; Demeyer, A.; Muyllaert, D.; Van Damme, P.; Gevaert, K.; Beyaert, R. T-cell receptor-induced JNK activation requires proteolytic inactivation of CYLD by MALT1. EMBO J. 2011, 30, 1742–1752. [Google Scholar] [CrossRef] [PubMed]
- Klein, T.; Fung, S.Y.; Renner, F.; Blank, M.A.; Dufour, A.; Kang, S.; Bolger-Munro, M.; Scurll, J.M.; Priatel, J.J.; Schweigler, P.; et al. The paracaspase MALT1 cleaves HOIL1 reducing linear ubiquitination by LUBAC to dampen lymphocyte NF-κB signalling. Nat. Commun. 2015, 6, 8777. [Google Scholar] [CrossRef] [PubMed]
- Poalas, K.; Hatchi, E.M.; Cordeiro, N.; Dubois, S.M.; Leclair, H.M.; Leveau, C.; Alexia, C.; Gavard, J.; Vazquez, A.; Bidère, N. Negative regulation of NF-κB signaling in T lymphocytes by the ubiquitin-specific protease USP34. Cell. Commun. Signal. 2013, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Paisie, T.K.; Newman, J.R.B.; McIntyre, L.M.; Concannon, P. UBASH3A Mediates Risk for Type 1 Diabetes Through Inhibition of T-Cell Receptor-Induced NF-κB Signaling. Diabetes 2017, 66, 2033–2043. [Google Scholar] [CrossRef] [PubMed]
- Hadian, K.; Krappmann, D. Signals from the nucleus: Activation of NF-κB by cytosolic ATM in the DNA damage response. Sci. Signal. 2011, 4, pe2. [Google Scholar] [CrossRef] [PubMed]
- McCool, K.W.; Miyamoto, S. DNA damage-dependent NF-κB activation: NEMO turns nuclear signaling inside out. Immunol. Rev. 2012, 246, 311–326. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.T.; Wuerzberger-Davis, S.M.; Wu, Z.H.; Miyamoto, S. Sequential modification of NEMO/IKKγ by SUMO-1 and ubiquitin mediates NF-κB activation by genotoxic stress. Cell 2003, 115, 565–576. [Google Scholar] [CrossRef]
- Mabb, A.M.; Wuerzberger-Davis, S.M.; Miyamoto, S. PIASy mediates NEMO sumoylation and NF-κB activation in response to genotoxic stress. Nat. Cell Biol. 2006, 8, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Janssens, S.; Tinel, A.; Lippens, S.; Tschopp, J. PIDD mediates NF-κB activation in response to DNA damage. Cell 2005, 123, 1079–1092. [Google Scholar] [CrossRef] [PubMed]
- Bock, F.J.; Krumschnabel, G.; Manzl, C.; Peintner, L.; Tanzer, M.C.; Hermann-Kleiter, N.; Baier, G.; Llacuna, L.; Yelamos, J.; Villunger, A. Loss of PIDD limits NF-κB activation and cytokine production but not cell survival or transformation after DNA damage. Cell Death Differ. 2013, 20, 546–557. [Google Scholar] [CrossRef] [PubMed]
- Stilmann, M.; Hinz, M.; Arslan, S.C.; Zimmer, A.; Schreiber, V.; Scheidereit, C. A nuclear poly(ADP-ribose)-dependent signalosome confers DNA damage-induced IκB kinase activation. Mol. Cell 2009, 36, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Banin, S.; Ouyang, H.; Li, G.C.; Courtois, G.; Shiloh, Y.; Karin, M.; Rotman, G. ATM is required for IκB kinase (IKK) activation in response to DNA double strand breaks. J. Biol. Chem. 2001, 276, 8898–8903. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.H.; Shi, Y.; Tibbetts, R.S.; Miyamoto, S. Molecular linkage between the kinase ATM and NF-κB signaling in response to genotoxic stimuli. Science 2006, 311, 1141–1146. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.H.; Wong, E.T.; Shi, Y.; Niu, J.; Chen, Z.; Miyamoto, S.; Tergaonkar, V. ATM- and NEMO-dependent ELKS ubiquitination coordinates TAK1-mediated IKK activation in response to genotoxic stress. Mol. Cell 2010, 40, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xia, F.; Hermance, N.; Mabb, A.; Simonson, S.; Morrissey, S.; Gandhi, P.; Munson, M.; Miyamoto, S.; Kelliher, M.A. A cytosolic ATM/NEMO/RIP1 complex recruits TAK1 to mediate the NF-κB and p38 mitogen-activated protein kinase (MAPK)/MAPK-activated protein 2 responses to DNA damage. Mol. Cell. Biol. 2011, 31, 2774–2786. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Shi, Y.; Iwai, K.; Wu, Z.H. LUBAC regulates NF-κB activation upon genotoxic stress by promoting linear ubiquitination of NEMO. EMBO J. 2011, 30, 3741–3753. [Google Scholar] [CrossRef] [PubMed]
- Hinz, M.; Stilmann, M.; Arslan, S.Ç.; Khanna, K.K.; Dittmar, G.; Scheidereit, C. A cytoplasmic ATM-TRAF6-cIAP1 module links nuclear DNA damage signaling to ubiquitin-mediated NF-κB activation. Mol. Cell 2010, 40, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.S.; Lee, D.H.; Kim, D.H.; Chung, J.H.; Lee, S.J.; Lee, T.H. cIAP1, cIAP2, and XIAP act cooperatively via nonredundant pathways to regulate genotoxic stress-induced nuclear factor-κB activation. Cancer Res. 2009, 69, 1782–1791. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Mabb, A.M.; Gill, G.B.; Yeh, E.T.; Miyamoto, S. NF-κB induction of the SUMO protease SENP2: A negative feedback loop to attenuate cell survival response to genotoxic stress. Mol. Cell 2011, 43, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Shi, Y.; Xue, J.; Miao, R.; Huang, S.; Wang, T.; Wu, J.; Fu, M.; Wu, Z.H. USP10 inhibits genotoxic NF-κB activation by MCPIP1-facilitated deubiquitination of NEMO. EMBO J. 2013, 32, 3206–3219. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Takahashi, M.; Morishita, T.; Noguchi, T.; Matsuzawa, A. Post-Translational Modifications of the TAK1-TAB Complex. Int. J. Mol. Sci. 2017, 18, 205. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, A.; von Bulow, V.; Hamidi, R.; Winssinger, N.; Barluenga, S.; Heldin, C.H.; Landström, M. Polyubiquitination of transforming growth factor β (TGFβ)-associated kinase 1 mediates nuclear factor-κB activation in response to different inflammatory stimuli. J. Biol. Chem. 2012, 287, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Yu, Y.; Shi, Y.; Sun, W.; Xie, M.; Ge, N.; Mao, R.; Chang, A.; Xu, G.; Schneider, M.D.; et al. Lysine 63-linked polyubiquitination of TAK1 at lysine 158 is required for tumor necrosis factor α- and interleukin-1β-induced IKK/NF-κB and JNK/AP-1 activation. J. Biol. Chem. 2010, 285, 5347–5360. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Yan, J.; Mao, A.P.; Li, C.; Ran, Y.; Shu, H.B.; Wang, Y.Y. Tripartite motif 8 (TRIM8) modulates TNFα- and IL-1β-triggered NF-κB activation by targeting TAK1 for K63-linked polyubiquitination. Proc. Natl. Acad. Sci. USA 2011, 108, 19341–19346. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.H.; Yu, Y.; Mao, R.F.; Tan, X.J.; Xu, G.F.; Zhang, H.; Lu, X.B.; Fu, S.B.; Yang, J. USP4 targets TAK1 to downregulate TNFα-induced NF-κB activation. Cell Death Differ. 2011, 18, 1547–1560. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, H.; Zhong, B.; Blonska, M.; Gorjestani, S.; Yan, M.; Tian, Q.; Zhang, D.E.; Lin, X.; Dong, C. USP18 inhibits NF-κB and NFAT activation during Th17 differentiation by deubiquitinating the TAK1-TAB1 complex. J. Exp. Med. 2013, 210, 1575–1590. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Xian, H.; Hu, J.; Tian, S.; Qin, Y.; Wang, R.F.; Cui, J. USP18 negatively regulates NF-κB signaling by targeting TAK1 and NEMO for deubiquitination through distinct mechanisms. Sci. Rep. 2015, 5, 12738. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Qian, W.; Staschke, K.; Qian, Y.; Cui, G.; Deng, L.; Ehsani, M.; Wang, X.; Qian, Y.W.; Chen, Z.J.; et al. Pellino 3b negatively regulates interleukin-1-induced TAK1-dependent NF κB activation. J. Biol. Chem. 2008, 283, 14654–14664. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhou, C.; Wang, Y.; Liu, W.; Liu, C.; Wang, L.; Liu, Y.; Shang, Y.; Li, M.; Zhou, S.; et al. The E3 ubiquitin ligase RNF114 and TAB1 degradation are required for maternal-to-zygotic transition. EMBO Rep. 2017, 18, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Charlaftis, N.; Suddason, T.; Wu, X.; Anwar, S.; Karin, M.; Gallagher, E. The MEKK1 PHD ubiquitinates TAB1 to activate MAPKs in response to cytokines. EMBO J. 2014, 33, 2581–2596. [Google Scholar] [CrossRef] [PubMed]
- Theivanthiran, B.; Kathania, M.; Zeng, M.; Anguiano, E.; Basrur, V.; Vandergriff, T.; Pascual, V.; Wei, W.Z.; Massoumi, R.; et al. The E3 ubiquitin ligase Itch inhibits p38α signaling and skin inflammation through the ubiquitylation of Tab1. Sci. Signal. 2015, 8, ra22. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.M.; Yang, Q.; Zhang, J.; Liu, S.M.; Zhang, Y.; Lin, H.; Huang, Z.F.; Wang, Y.Y.; Zhang, X.D.; Zhong, B. TRIM38 inhibits TNFα- and IL-1β-triggered NF-κB activation by mediating lysosome-dependent degradation of TAB2/3. Proc. Natl. Acad. Sci. USA 2014, 111, 1509–1514. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Deng, W.; Bi, E.; Mao, K.; Ji, Y.; Lin, G.; Wu, X.; Tao, Z.; Li, Z.; Cai, X.; et al. TRIM30α negatively regulates TLR-mediated NF-κB activation by targeting TAB2 and TAB3 for degradation. Nat. Immunol. 2008, 9, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.; Mu, R.; Chang, Y.; Wang, Y.B.; Wu, M.; Tu, H.Q.; Zhang, Y.C.; Guo, S.S.; Qin, X.H.; Li, T.; et al. RNF4 negatively regulates NF-κB signaling by down-regulating TAB2. FEBS Lett. 2015, 589, 2850–2858. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Wertz, I.; O’Rourke, K.; Ultsch, M.; Seshagiri, S.; Eby, M.; Xiao, W.; Dixit, V.M. Bcl10 activates the NF-κB pathway through ubiquitination of NEMO. Nature 2004, 427, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Abbott, D.W.; Wilkins, A.; Asara, J.M.; Cantley, L.C. The Crohn’s disease protein, NOD2, requires RIP2 in order to induce ubiquitinylation of a novel site on NEMO. Curr. Biol. 2004, 14, 2217–2227. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, F.; Sakata, S.; Saeki, Y.; Satomi, Y.; Kirisako, T.; Kamei, K.; Nakagawa, T.; Kato, M.; Murata, S.; Yamaoka, S.; et al. Involvement of linear polyubiquitylation of NEMO in NF-κB activation. Nat. Cell. Biol. 2009, 11, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Ni, C.Y.; Wu, Z.H.; Florence, W.C.; Parekh, V.V.; Arrate, M.P.; Pierce, S.; Schweitzer, B.; Van Kaer, L.; Joyce, S.; Miyamoto, S.; et al. K63-linked polyubiquitination of NEMO modulates TLR signaling and inflammation in vivo. J. Immunol. 2008, 180, 7107–7111. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.C.; Kertesy, S.; Jones, M.B.; Marinis, J.M.; Cobb, B.A.; Tigno-Aranjuez, J.T.; Abbott, D.W. Innate immune-directed NF-κB signaling requires site-specific NEMO ubiquitination. Cell Rep. 2013, 4, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Supprian, M.; Bloch, W.; Courtois, G.; Addicks, K.; Israël, A.; Rajewsky, K.; Pasparakis, M. NEMO/IKKγ-deficient mice model incontinentia pigmenti. Mol. Cell 2000, 5, 981–992. [Google Scholar] [CrossRef]
- Arimoto, K.; Funami, K.; Saeki, Y.; Tanaka, K.; Okawa, K.; Takeuchi, O.; Akira, S.; Murakami, Y.; Shimotohno, K. Polyubiquitin conjugation to NEMO by triparite motif protein 23 (TRIM23) is critical in antiviral defense. Proc. Natl. Acad. Sci. USA 2010, 107, 15856–15861. [Google Scholar] [CrossRef] [PubMed]
- Zotti, T.; Uva, A.; Ferravante, A.; Vessichelli, M.; Scudiero, I.; Ceccarelli, M.; Vito, P.; Stilo, R. TRAF7 protein promotes Lys-29-linked polyubiquitination of IκB Kinase (IKKγ)/NF-κB Essential Modulator (NEMO) and p65/RelA protein and represses NF-κB activation. J. Biol. Chem. 2011, 286, 22924–22933. [Google Scholar] [CrossRef] [PubMed]
- Tsikitis, M.; Acosta-Alvear, D.; Blais, A.; Campos, E.I.; Lane, W.S.; Sánchez, I.; Dynlacht, B.D. Traf7, a MyoD1 transcriptional target regulates nuclear factor-κB activity during myogenesis. EMBO Rep. 2010, 11, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Taubman, M.B. EGLN3 inhibition of NF-κB is mediated by prolyl hydroxylase-independent inhibition of IκB kinase γ ubiquitination. Mol. Cell. Biol. 2013, 33, 3050–3061. [Google Scholar] [CrossRef] [PubMed]
- Tomar, D.; Singh, R. TRIM13 regulates ubiquitination and turnover of NEMO to suppress TNF induced NF-κB activation. Cell Signal. 2014, 26, 2606–2613. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Guan, J.; Li, S.; Zhang, X.; Zheng, X. HSCARG downregulates NF-κB signaling by interacting with USP7 and inhibiting NEMO ubiquitination. Cell Death Dis. 2014, 5, e1229. [Google Scholar] [CrossRef] [PubMed]
- Niida, M.; Tanaka, M.; Kamitani, T. Downregulation of active IKKβ by Ro52-mediated autophagy. Mol. Immunol. 2010, 47, 2378–2387. [Google Scholar] [CrossRef] [PubMed]
- Liao, G.; Zhang, M.; Harhaj, E.W.; Sun, S.C. Regulation of the NF-κB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. J. Biol. Chem. 2004, 279, 26243–26250. [Google Scholar] [CrossRef] [PubMed]
- Vallabhapurapu, S.; Matsuzawa, A.; Zhang, W.; Tseng, P.H.; Keats, J.J.; Wang, H.; Vignali, D.A.; Bergsagel, P.L.; Karin, M. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-κB signaling. Nat. Immunol. 2008, 9, 1364–1370. [Google Scholar] [CrossRef] [PubMed]
- Zarnegar, B.J.; Wang, Y.; Mahoney, D.J.; Dempsey, P.W.; Cheung, H.H.; He, J.; Shiba, T.; Yang, X.; Yeh, W.C.; Mak, T.W.; et al. Noncanonical NF-κB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat. Immunol. 2008, 9, 1371–1378. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Challa-Malladi, M.; Bratton, S.B.; Wright, C.W. Nuclear factor-κB-inducing kinase (NIK) contains an amino-terminal inhibitor of apoptosis (IAP)-binding motif (IBM) that potentiates NIK degradation by cellular IAP1 (c-IAP1). J. Biol. Chem. 2014, 289, 30680–30689. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.W.; Hostager, B.S.; Bishop, G.A. TRAF3, ubiquitination, and B-lymphocyte regulation. Immunol. Rev. 2015, 266, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.D.; Sun, S.C. Targeting signaling factors for degradation, an emerging mechanism for TRAF functions. Immunol. Rev. 2015, 266, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Fotin-Mleczek, M.; Henkler, F.; Hausser, A.; Glauner, H.; Samel, D.; Graness, A.; Scheurich, P.; Mauri, D.; Wajant, H. Tumor necrosis factor receptor-associated factor (TRAF) 1 regulates CD40-induced TRAF2-mediated NF-κB activation. J. Biol. Chem. 2004, 279, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Brittain, G.C.; Chang, J.H.; Puebla-Osorio, N.; Jin, J.; Zal, A.; Xiao, Y.; Cheng, X.; Chang, M.; Fu, Y.X.; et al. OTUD7B controls non-canonical NF-κB activation through deubiquitination of TRAF3. Nature 2013, 494, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.B.K.; Taunk, N.K.; et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 2018, 553, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Saccani, S.; Marazzi, I.; Beg, A.A.; Natoli, G. Degradation of promoter-bound p65/RelA is essential for the prompt termination of the nuclear factor κB response. J. Exp. Med. 2004, 200, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Maine, G.N.; Burstein, E. COMMD proteins and the control of the NFκB pathway. Cell Cycle 2007, 6, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Grusby, M.J.; Kaisho, T. PDLIM2-mediated termination of transcription factor NF-κB activation by intranuclear sequestration and degradation of the p65 subunit. Nat. Immunol. 2007, 8, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Ono, R.; Kaisho, T.; Tanaka, T. PDLIM1 inhibits NF-κB-mediated inflammatory signaling by sequestering the p65 subunit of NF-κB in the cytoplasm. Sci. Rep. 2015, 5, 18327. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Moreau, F.; Chadee, K. PPARγ is an E3 ligase that induces the degradation of NFκB/p65. Nat. Commun. 2012, 3, 1300. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wu, X.; Zhang, J.; Chen, Y.; Xu, J.; Xia, X.; He, S.; Qiang, F.; Li, A.; Shu, Y.; et al. CHIP functions as a novel suppressor of tumour angiogenesis with prognostic significance in human gastric cancer. Gut 2013, 62, 496–508. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, K.; Naumann, M. CSN-associated USP48 confers stability to nuclear NF-κB/RelA by trimming K48-linked Ub-chains. Biochim. Biophys. Acta 2015, 1853, 453–469. [Google Scholar] [CrossRef] [PubMed]
- Colleran, A.; Collins, P.E.; O’Carroll, C.; Ahmed, A.; Mao, X.; McManus, B.; Kiely, P.A.; Burstein, E.; Carmody, R.J. Deubiquitination of NF-κB by Ubiquitin-Specific Protease-7 promotes transcription. Proc. Natl. Acad. Sci. USA 2013, 110, 618–623. [Google Scholar] [CrossRef] [PubMed]
- Hochrainer, K.; Racchumi, G.; Zhang, S.; Iadecola, C.; Anrather, J. Monoubiquitination of nuclear RelA negatively regulates NF-κB activity independent of proteasomal degradation. Cell. Mol. Life Sci. 2012, 69, 2057–2073. [Google Scholar] [CrossRef] [PubMed]
- Carmody, R.J.; Ruan, Q.; Palmer, S.; Hilliard, B.; Chen, Y.H. Negative regulation of toll-like receptor signaling by NF-κB p50 ubiquitination blockade. Science 2007, 317, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.; Jin, W.; Chang, J.H.; Xiao, Y.; Brittain, G.C.; Yu, J.; Zhou, X.; Wang, Y.H.; Cheng, X.; Li, P.; et al. The ubiquitin ligase Peli1 negatively regulates T cell activation and prevents autoimmunity. Nat. Immunol. 2011, 12, 1002–1009. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.H.; Kang, S.G.; Huang, Z.; Wu, C.J.; Jin, H.Y.; Maine, C.J.; Liu, Y.; Shepherd, J.; Sabouri-Ghomi, M.; Gonzalez-Martin, A. A miR-155-Peli1-c-Rel pathway controls the generation and function of T follicular helper cells. J. Exp. Med. 2016, 213, 1901–1919. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Xiao, Y.; Hu, H.; Zou, Q.; Li, Y.; Gao, Y.; Ge, W.; Cheng, X.; Sun, S.C. Proinflammatory TLR signalling is regulated by a TRAF2-dependent proteolysis mechanism in macrophages. Nat. Commun. 2015, 6, 5930. [Google Scholar] [CrossRef] [PubMed]
- Leidner, J.; Palkowitsch, L.; Marienfeld, U.; Fischer, D.; Marienfeld, R. Identification of lysine residues critical for the transcriptional activity and polyubiquitination of the NF-κB family member RelB. Biochem. J. 2008, 416, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Marienfeld, R.; Berberich-Siebelt, F.; Berberich, I.; Denk, A.; Serfling, E.; Neumann, M. Signal-specific and phosphorylation-dependent RelB degradation: A potential mechanism of NF-κB control. Oncogene 2001, 20, 8142–8147. [Google Scholar] [CrossRef] [PubMed]
- Leidner, J.; Voogdt, C.; Niedenthal, R.; Möller, P.; Marienfeld, U.; Marienfeld, R.B. SUMOylation attenuates the transcriptional activity of the NF-κB subunit RelB. J. Cell. Biochem. 2014, 115, 1430–1440. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-κB: A Blossoming of Relevance to Human Pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef] [PubMed]
- Maubach, G.; Naumann, M. NEMO Links Nuclear Factor-κB to Human Diseases. Trends Mol. Med. 2017, 23, 1138–1155. [Google Scholar] [CrossRef] [PubMed]
- Sebban-Benin, H.; Pescatore, A.; Fusco, F.; Pascuale, V.; Gautheron, J.; Yamaoka, S.; Moncla, A.; Ursini, M.V.; Courtois, G. Identification of TRAF6-dependent NEMO polyubiquitination sites through analysis of a new NEMO mutation causing incontinentia pigmenti. Hum. Mol. Genet. 2007, 16, 2805–2815. [Google Scholar] [CrossRef] [PubMed]
- Bal, E.; Laplantine, E.; Hamel, Y.; Dubosclard, V.; Boisson, B.; Pescatore, A.; Picard, C.; Hadj-Rabia, S.; Royer, G.; Steffann, J.; et al. Lack of interaction between NEMO and SHARPIN impairs linear ubiquitination and NF-κB activation and leads to incontinentia pigmenti. J. Allergy Clin. Immunol. 2017, 140, 1671–1682. [Google Scholar] [CrossRef] [PubMed]
- Senegas, A.; Gautheron, J.; Gentil-Dit-Maurin, A.; Courtois, G. IKK-related genetic diseases: Probing NF-κB functions in humans and other matters. Cell. Mol. Life Sci. 2015, 72, 1275–1287. [Google Scholar] [CrossRef] [PubMed]
- Boisson, B.; Laplantine, E.; Prando, C.; Giliani, S.; Israelsson, E.; Xu, Z.; Abhyankar, A.; Israël, L.; Trevejo-Nunez, G.; Bogunovic, D.; et al. Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL-1 and LUBAC deficiency. Nat. Immunol. 2012, 13, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Boisson, B.; Laplantine, E.; Dobbs, K.; Cobat, A.; Tarantino, N.; Hazen, M.; Lidov, H.G.; Hopkins, G.; Du, L.; Belkadi, A.; et al. Human HOIP and LUBAC deficiency underlies autoinflammation, immunodeficiency, amylopectinosis, and lymphangiectasia. J. Exp. Med. 2015, 212, 939–951. [Google Scholar] [CrossRef] [PubMed]
- Damgaard, R.B.; Walker, J.A.; Marco-Casanova, P.; Morgan, N.V.; Titheradge, H.L.; Elliott, P.R.; McHale, D.; Maher, E.R.; McKenzie, A.N.J.; Komander, D. The Deubiquitinase OTULIN Is an Essential Negative Regulator of Inflammation and Autoimmunity. Cell. 2016, 166, 1215–1230. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Yu, X.; Demirkaya, E.; Deuitch, N.; Stone, D.; Tsai, W.L.; Kuehn, H.S.; Wang, H.; Yang, D.; Park, Y.H.; et al. Biallelic hypomorphic mutations in a linear deubiquitinase define otulipenia, an early-onset autoinflammatory disease. Proc. Natl. Acad. Sci. USA 2016, 113, 10127–10132. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lin, Y.; Guo, Z.; Cheng, J.; Huang, J.; Deng, L.; Liao, W.; Chen, Z.; Liu, Z.; Su, B. The essential role of MEKK3 in TNF-induced NF-κB activation. Nat. Immunol. 2001, 2, 620–624. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Yang, J.; Lin, Y.; Walker, C.; Cheng, J.; Liu, Z.G.; Su, B. Differential regulation of interleukin 1 receptor and Toll-like receptor signaling by MEKK3. Nat. Immunol. 2004, 5, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Kim, T.W.; Qin, J.; Jiang, Z.; Qian, Y.; Xiao, H.; Lu, Y.; Qian, W.; Gulen, M.F.; Sizemore, N. Interleukin-1 (IL-1)-induced TAK1-dependent versus MEKK3-dependent NFκB activation pathways bifurcate at IL-1 receptor-associated kinase modification. J. Biol. Chem. 2007, 282, 6075–6089. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, K.; Gohda, J.; Kanayama, A.; Miyamoto, Y.; Sakurai, H.; Yamamoto, M.; Akira, S.; Hayashi, H.; Su, B.; Inoue, J. Two mechanistically and temporally distinct NF-κB activation pathways in IL-1 signaling. Sci. Signal. 2009, 2, ra66. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Macartney, T.; Peggie, M.; Cohen, P. Interleukin-1 and TRAF6-dependent activation of TAK1 in the absence of TAB2 and TAB3. Biochem. J. 2017, 474, 2235–2248. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, F.; Saeki, Y.; Ishido, S.; Kanno, J.; Tanaka, K. The K48-K63 Branched Ubiquitin Chain Regulates NF-κB Signaling. Mol. Cell 2016, 64, 251–266. [Google Scholar] [CrossRef] [PubMed]
- Myllymäki, H.; Valanne, S.; Rämet, M. The Drosophila imd signaling pathway. J. Immunol. 2014, 192, 3455–3462. [Google Scholar] [CrossRef] [PubMed]













© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Courtois, G.; Fauvarque, M.-O. The Many Roles of Ubiquitin in NF-κB Signaling. Biomedicines 2018, 6, 43. https://doi.org/10.3390/biomedicines6020043
Courtois G, Fauvarque M-O. The Many Roles of Ubiquitin in NF-κB Signaling. Biomedicines. 2018; 6(2):43. https://doi.org/10.3390/biomedicines6020043
Chicago/Turabian StyleCourtois, Gilles, and Marie-Odile Fauvarque. 2018. "The Many Roles of Ubiquitin in NF-κB Signaling" Biomedicines 6, no. 2: 43. https://doi.org/10.3390/biomedicines6020043
APA StyleCourtois, G., & Fauvarque, M.-O. (2018). The Many Roles of Ubiquitin in NF-κB Signaling. Biomedicines, 6(2), 43. https://doi.org/10.3390/biomedicines6020043