Plant-Derived Anticancer Agents: Lessons from the Pharmacology of Geniposide and Its Aglycone, Genipin



Abstract
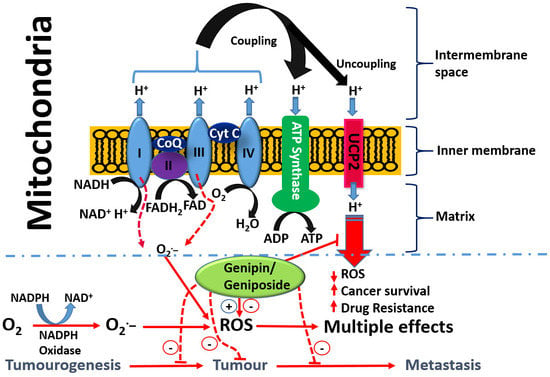
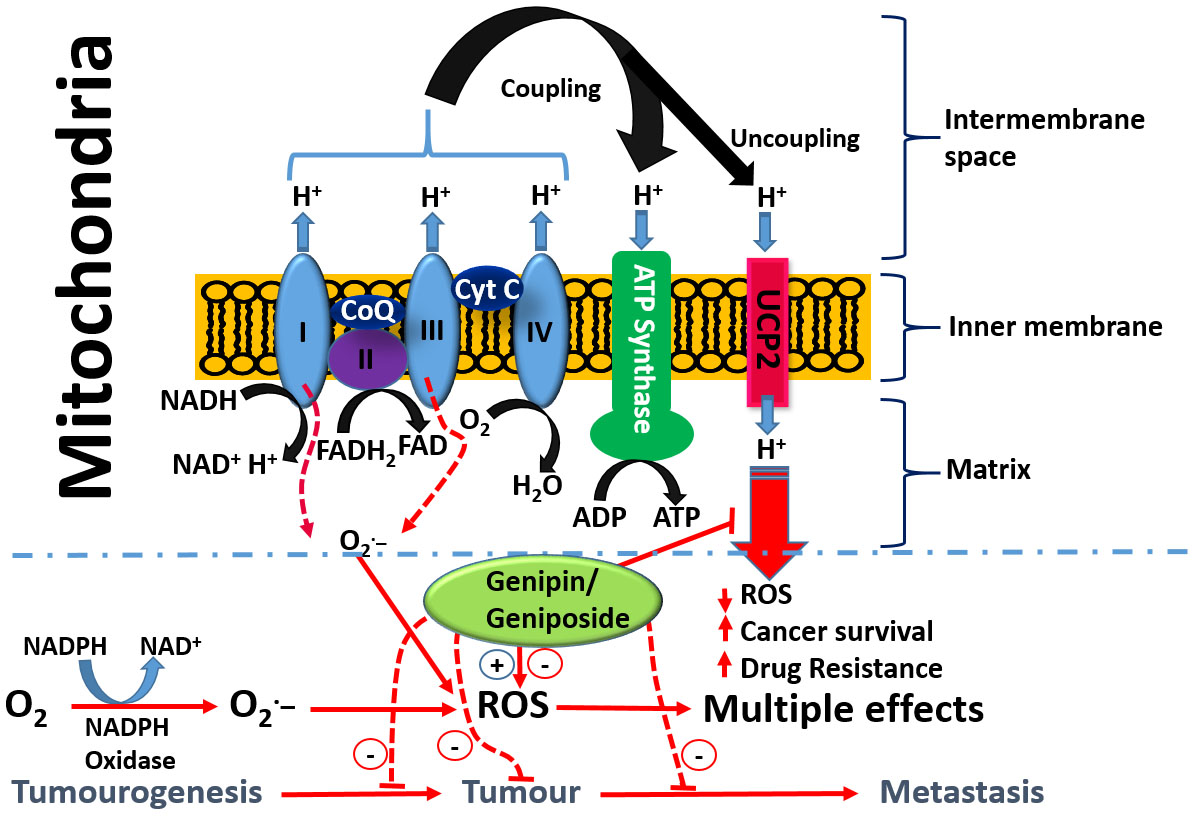{kind=link}
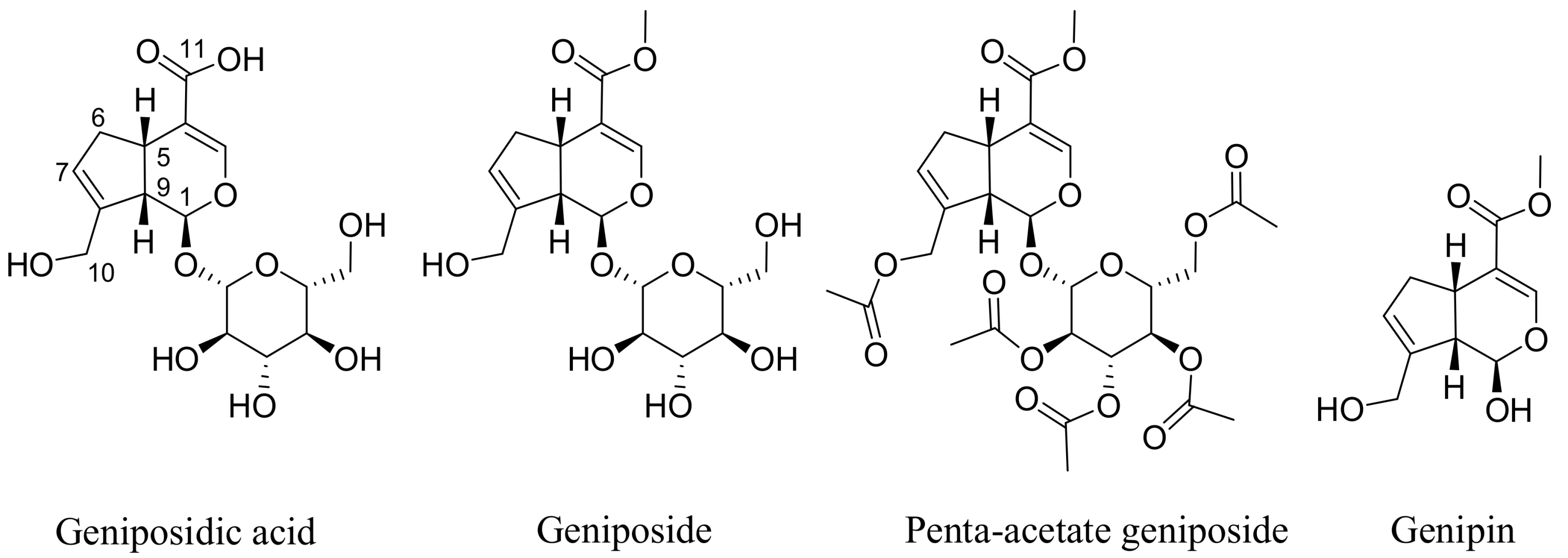{kind=link}
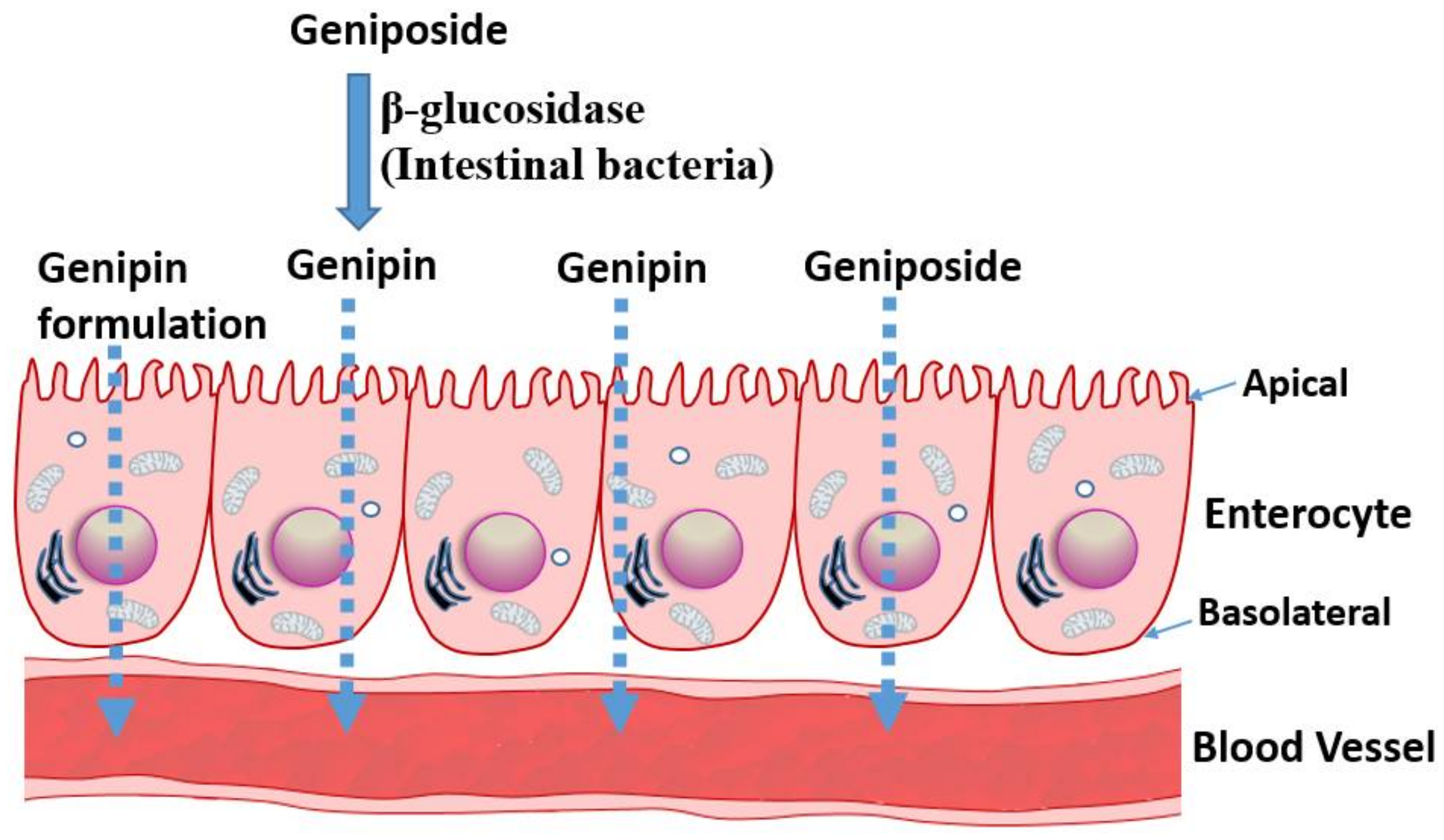{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Natural Sources of Geniposide and Genipin
3. Physicochemical Properties and Associated Pharmacokinetics Profile
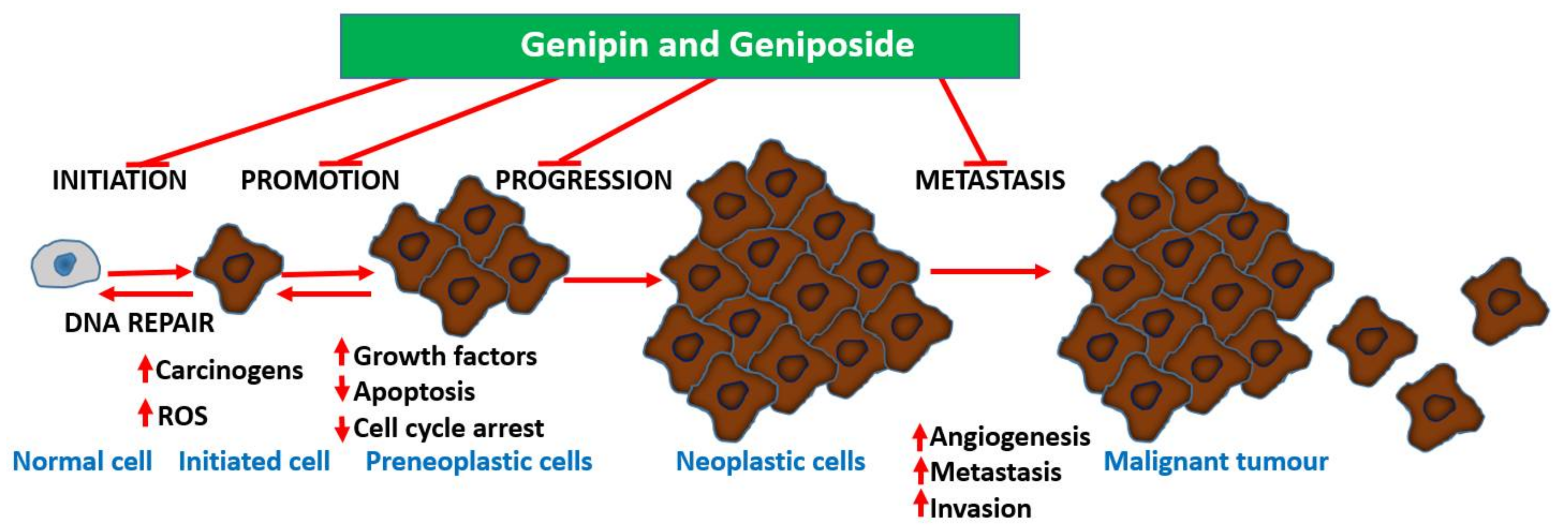
4. Anticancer Effects of Geniposide and Genipin
4.1. Direct Cytotoxic Effect on Cancer Cells
4.2. Effects on Carcinogenesis
4.3. Effects on Cancer Metastasis
5. Lessons on the Mechanisms of Action
5.1. Mechanisms Related to Cell Cycle Regulation
5.2. General Anti-Inflammatory Mechanisms
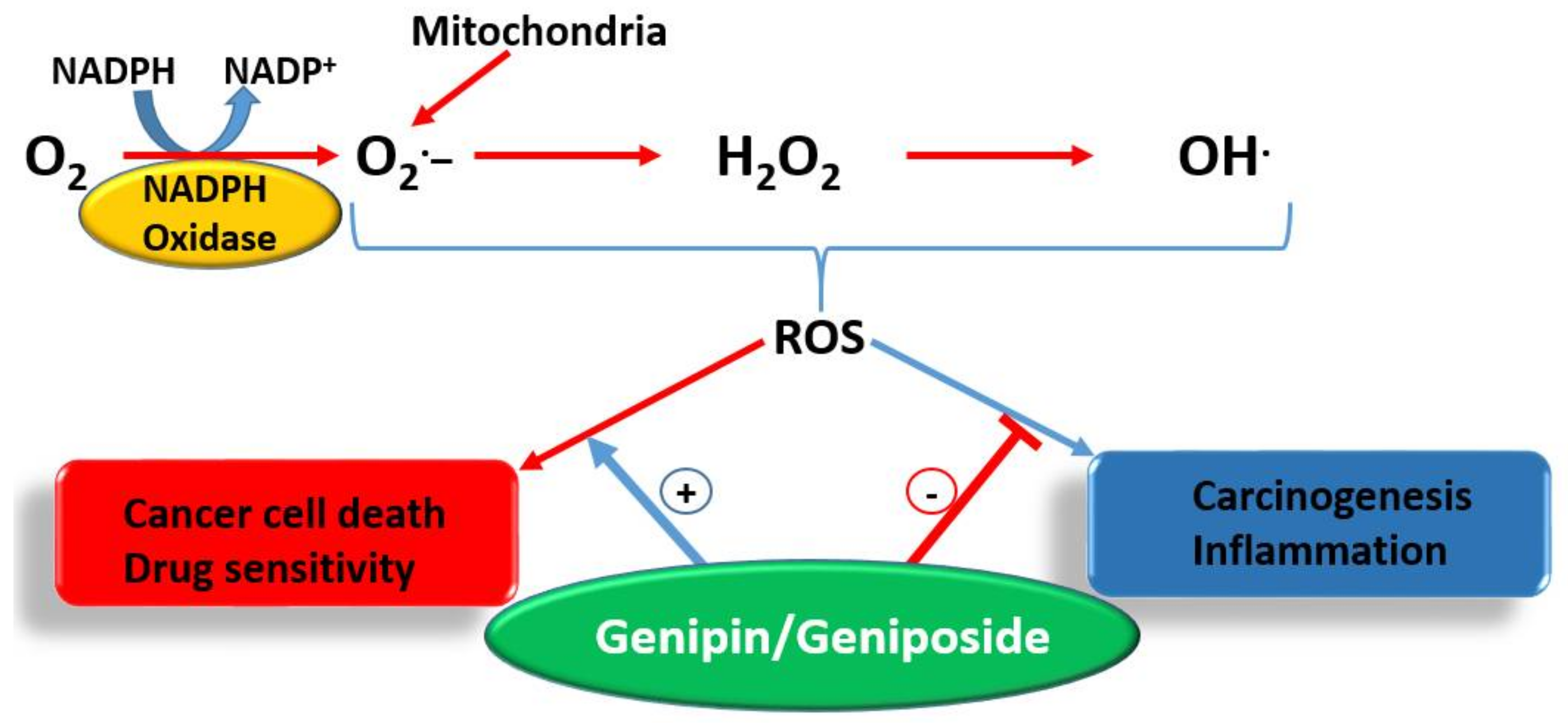
5.3. Cancer Cell Killing by Weaponizing Oxygen
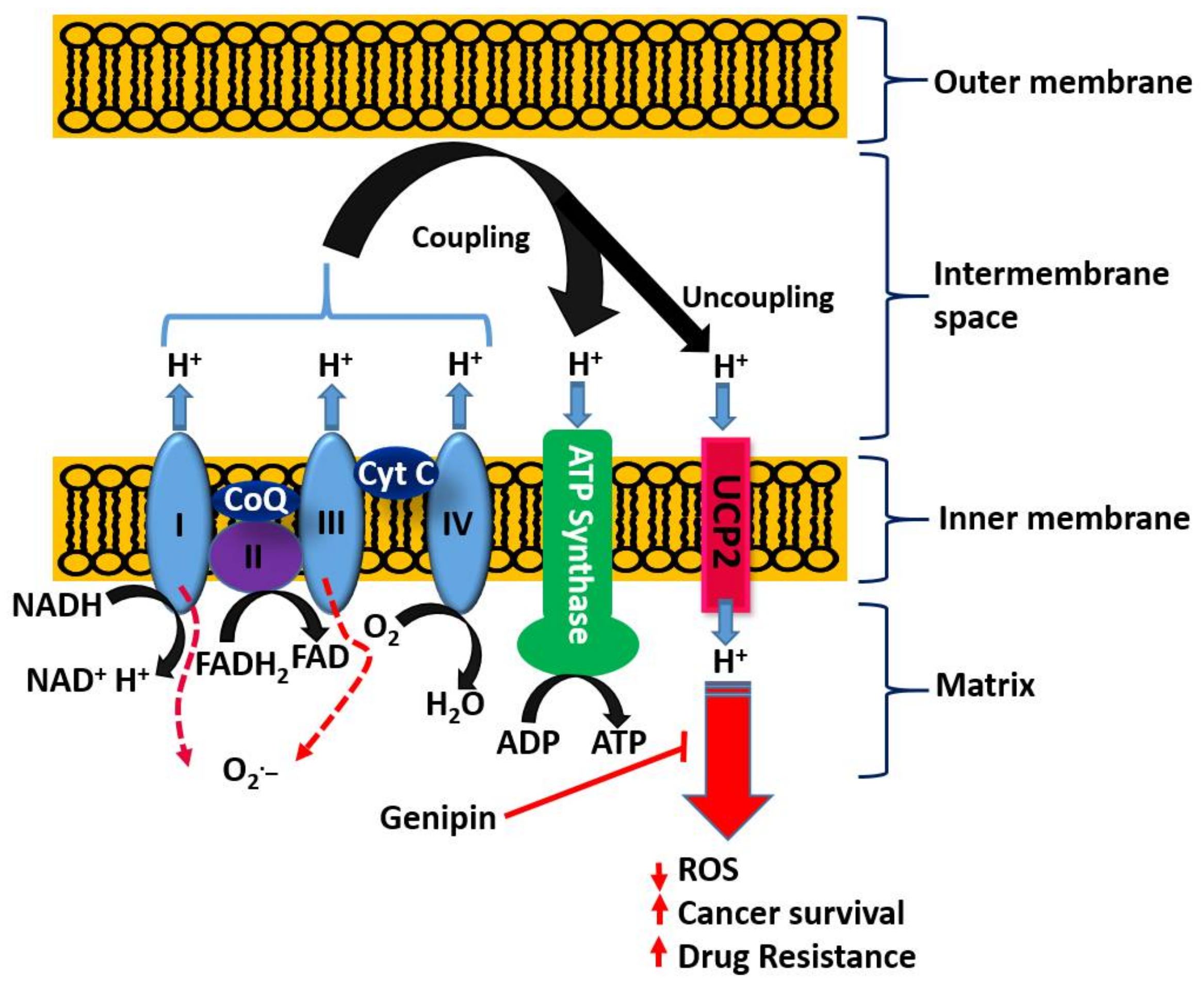
5.4. Emerging Role of the Mitochondrial Uncoupling Protein-2 (UCP2) in Cancer Biology and Chemotherapy
6. Drug Potentiation
7. General Summary and Conclusions
- Anticarcinogenic effect via antioxidant and anti-inflammatory (e.g., Nrf2, GPx induction) mechanisms.
- Targeting specific enzymes (e.g., GGT, MMPs) involved in carcinogenesis.
- Modulation of signal transduction pathways (e.g., MAPK such as JNK, p38, and ERK; PI3K, Akt, JAK1, etc.) involved in cell proliferation, inflammation, and cell death.
- Suppression of the production and function of proinflammatory cytokines (such as IL-1, IL-6, and TNF-α) and other proteins (iNOS).
- Modulation of various transcription factors (Egr1, NF-κB, AP-1, p21, STAT3) involved in inflammation and cancer biology and of transcriptional modulators such as SMAD2.
- Upregulation of genes/proteins that promote cell death and downregulation of survival genes/proteins; p53, Bcl-2, Bcl-xL, survivin, c-Myc, Bax, etc. are classical examples.
- Enhancement od ROS formation both by the NADPH oxidase and UCP2 pathways (see Figure 5).
- Triggering of cell cycle arrest (G1/S phase or G2/M phase) by modulating cyclin-dependent kinases.
- Mechanism related to topoisomerase I poisoning for cytotoxicity and downregulation of P-glycoprotein that allow drug potentiation and/or combination therapy.
- Activation of procaspases (e.g., procaspase-8 and 9) and caspases including the final apoptosis executioner, caspase 3.
Conflicts of Interest
Abbreviations
| AP-1 | Activator protein-1 |
| AFB1 | Aflatoxin B1 |
| Akt | Protein kinase B (PKB) |
| αSMA | α-Smooth muscle actin |
| ALT | Alanine aminotransferase |
| AST | Aspartate aminotransferase |
| BAK-1 | BCL2 Antagonist/Killer 1 |
| Bax | BCL2-associated X |
| Bcl-2 | B cell lymphoma gene 2 |
| Cdk | Cyclin-dependent kinase |
| c-Fos | C-Jun, c-Myc, and c-Src; proto-oncogenes |
| EBV | Epstein-Barr virus |
| ECM | Extracellular matrix |
| Egr1 | Early growth response-1 |
| ERK1/2 | Extracellular signal-regulated kinases 1 and 2 |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| GGT | γ-Glutamyl transpeptidase |
| GPx | Glutathione peroxidase |
| GSH | Glutathione |
| GST | Glutathione-S-transferase |
| γ-GT | γ-Glutamyltranspeptidase |
| HO-1 | Haem oxygenase-1 |
| H2O2 | Hydrogen peroxide |
| IFN-γ | Interferon-γ |
| IL-1 | Interleukin 1 |
| IL-6 | Interleukin 6 |
| iNOS | Inducible nitric oxide synthase |
| JAK1 | Janus kinase-1 |
| JNK | C-Jun-NH2-kinase |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen-activated protein kinase |
| MEK1/2 | Mitogen-activated protein kinase kinase 1 and 2 |
| MLK3 | Mixed lineage kinase-3 |
| MMP-2 | Matrix metalloproteinase-2 |
| MT1-MMP | Membrane type I matrix metalloproteinase |
| mTOR | Mechanistic target of rapamycin |
| NF-κB | Nuclear factor kappa B |
| NGF | Nerve growth factor |
| NO | Nitric oxide |
| p21 | Cyclin-dependent kinase inhibitor p21 |
| PKCδ | Protein kinase Cδ |
| PI3K | Phosphoinositide 3-kinase |
| Rb | Retinoblastoma |
| ROS | Reactive oxygen species |
| SHP-1 | c-Src homology 2 domain-containing phosphatase-1 |
| SMAD2 | Mothers against decapentaplegic homolog 2 also known as SMAD family member 2 |
| SMase | Sphingomyelinase |
| STAT3 | Signal-transducer-and-activator-of-transcription-3 |
| TGFβ1 | Transforming growth factor β1 |
| TIMP-1 | Tissue inhibitor of matrix metalloproteinase-1 |
| TNF-α | Tumour necrosis factor-α TPA, 12-O-Tetradecanoylphorbol-13-acetate |
| VEGF | Vascular endothelial growth factor |
References
- Mukhtar, E.; Adhami, V.M.; Mukhtar, H. Targeting microtubules by natural agents for cancer therapy. Mol. Cancer Ther. 2014, 13, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Ivano, A.; Andrey, L.; Richard, A.K.; Gerry, M.; Alexey, V.A. Polypharmacology of approved anticancer drugs. Curr. Drug Targets 2017, 18, 534–543. [Google Scholar] [CrossRef]
- Ke, X.; Shen, L. Molecular targeted therapy of cancer: The progress and future prospect. Front. Lab. Med. 2017, 1, 69–75. [Google Scholar] [CrossRef]
- Corraliza-Gorjón, I.; Somovilla-Crespo, B.; Santamaria, S.; Garcia-Sanz, J.A.; Kremer, L. New strategies using antibody combinations to increase cancer treatment effectiveness. Front. Immunol. 2017, 8, 1804. [Google Scholar] [CrossRef] [PubMed]
- Butler, T.; Maravent, S.; Boisselle, J.; Valdes, J.; Fellner, C. A review of 2014 cancer drug approvals, with a look at 2015 and beyond. Pharm. Ther. 2015, 40, 191–205. [Google Scholar] [PubMed]
- CenterWatch. FDA Approved Drugs. Available online: https://www.centerwatch.com/drug-information/fda-approved-drugs/ (accessed on 25 January 2018).
- Davis, C.; Gurpinar, E.; Pinto, A. Availability of evidence of benefits on overall survival and quality of life of cancer drugs approved by European Medicines Agency: Retrospective cohort study of drug approvals 2009–13. BMJ 2017, 359, j4530. [Google Scholar] [CrossRef] [PubMed]
- WHO. Cancer: Fact Sheet. Available online: http://www.who.int/mediacentre/factsheets/fs297/en/ (accessed on 25 January 2018).
- Shan, M.; Yu, S.; Yan, H.; Guo, S.; Xiao, W.; Wang, Z.; Zhang, L.; Ding, A.; Wu, Q.; Li, S.F.Y. A Review on the phytochemistry, pharmacology, pharmacokinetics and toxicology of geniposide, a natural product. Molecules 2017, 22, 1689. [Google Scholar] [CrossRef] [PubMed]
- Inouye, H.; SaitoTaguchi, S.; Endo, T. New iridoid glucosides gardenoside and geniposide from Gardenia jasminoides. Tetrahedron Lett. 1969, 28, 2347–2350. [Google Scholar] [CrossRef]
- Endo, T.; Taguchi, H. The constituents of Gardenia jasminoides: Geniposide and genipin gentiobioside. Chem. Pharm. Bull. 1973, 21, 2684–2688. [Google Scholar] [CrossRef]
- Habtemariam, S. Iridoids and other monoterpenes in the Alzheimer’s brain: Recent development and future prospects. Molecules 2018, 23, 117. [Google Scholar] [CrossRef] [PubMed]
- Habtemariam, S. Antidiabetic potential of monoterpenes: A case of small molecules punching above their weight. Int. J. Mol. Sci. 2018, 19, 4. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Wu, X.; Li, L.; Chen, Y.; Lu, T.; Li, W.; Cai, B.; Yin, W. Quality control of Gardeniae Fructus by HPLC-PDA fingerprint coupled with chemometric methods. J. Chromatogr. Sci. 2015, 53, 1685–16594. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Du, S.; Lu, Y.; Rao, X. Studies on O/W partition coefficient and absorption kinetics of geniposide in fructus gardeniae extract in rat intestine. Zhongguo Zhong Yao Za Zhi 2009, 34, 1840–1844. [Google Scholar] [PubMed]
- Yang, M.; Chen, X.Y.; Zhang, H.Y.; Wang, J.M.; Lu, Q.; Song, W. Pharmacokinetics of geniposide through 4 routes of administration. Chin. J. New Drugs 2010, 19, 746–750. [Google Scholar]
- Yu, D.; Zhang, Y.; Guo, L.W.; Zhang, Q.C.; Zhu, H.X. Study on the absorption mechanism of geniposide in the Chinese formula Huang-Lian-Jie-Du-Tang in rats. AAPS PharmSciTech 2017, 18, 1382–1392. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.J.; Kim, I.S.; Kim, D.H.; Yoo, H.H. Liquid chromatography-tandem mass spectrometric determination of geniposide in rat plasma and its pharmacokinetic application. Bull. Korean Chem. Soc. 2013, 34, 2760–2764. [Google Scholar] [CrossRef]
- Li, Y.; Cai, W.; Cai, Q.; Che, Y.Y.; Zhao, B.S.; Zhang, J.Y. Comprehensive characterization of the in vitro and in vivo metabolites of geniposide in rats using ultra-high-performance liquid chromatography coupled with linear ion trap-Orbitrap mass spectrometer. Xenobiotica 2016, 46, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Chen, X.; Du, S.; Wu, Q.; Yao, Z.; Zhai, Y. The in situ and in vivo study on enhancing effect of borneol in nasal absorption of geniposide in rats. Arch. Pharm. Res. 2010, 33, 691–696. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Du, S.Y.; Chen, X.L.; Wu, Q.; Song, X.; Xu, B.; Zhai, Y.S. Enhancing effect of natural borneol on the absorption of geniposide in rat via intranasal administration. J. Zhejiang Univ. Sci. B 2011, 12, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Du, S.; Bai, J.; Li, P.; Wen, R.; Zhao, X. Bioavailability and brain-targeting of geniposide in gardenia-borneol co-compound by different administration routes in mice. Int. J. Mol. Sci. 2012, 13, 14127–14135. [Google Scholar] [CrossRef] [PubMed]
- Akao, T.; Kobayashi, K.; Aburada, M. Enzymatic studies on the animal and intestinal bacterial metabolism of geniposide. Biol. Pharm. Bull. 1994, 17, 1573–1576. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Akao, T. Relation of intestinal bacteria to pharmacological effects of glycosides. Biosci. Microflora 1997, 16, 1–7. [Google Scholar] [CrossRef]
- Yim, J.S.; Kim, Y.S.; Moon, S.K.; Cho, K.H.; Bae, H.S.; Kim, J.J.; Park, E.K.; Kim, D.H. Metabolic activities of ginsenoside Rb1, baicalin, glycyrrhizin and geniposide to their bioactive compounds by human intestinal microflora. Biol. Pharm. Bull. 2004, 27, 1580–1583. [Google Scholar] [CrossRef] [PubMed]
- Habtemariam, S.; Belai, A. Natural therapies of the inflammatory bowel disease: The case of rutin and its aglycone, quercetin. Mini-Rev. Med. Chem. 2018, 18, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Kim, S.Y.; Park, S.Y.; Han, M.J. Metabolism of quercitrin by human intestinal bacteria and its relation to some biological activities. Biol. Pharm. Bull. 1999, 22, 749–751. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Yang, J.; Qian, D.; Guo, J.; Shang, E.-X.; Duan, J.-A.; Xu, J. Rapid screening and identification of metabolites of quercitrin produced by the human intestinal bacteria using ultra performance liquid chromatography/quadrupole-time-of-flight mass spectrometry. Arch. Pharm. Res. 2014, 37, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.J.; Khanal, T.; Kim, H.G.; Lee, D.H.; Yeo, H.K.; Lee, Y.S.; Ahn, Y.T.; Kim, D.H.; Jeong, H.G.; Jeong, T.C. Role of metabolism by human intestinal microflora in geniposide-induced toxicity in HepG2 cells. Arch. Pharm. Res. 2012, 35, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.J.; Kim, I.S.; Kim, D.-H.; Yoo, H.H. Effects of intestinal microbiota on the bioavailability of geniposide in Rats. J. Agric. Food Chem. 2014, 62, 9632–9636. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Lee, C.-J.; Ma, J.Y. Enhancement of active compound, genipin, from Gardeniae fructus using immobilized glycosyl hydrolase family 3 β-glucosidase from Lactobacillus antri. AMB Express 2017, 7, 64. [Google Scholar] [CrossRef] [PubMed]
- Ku, S.; You, H.J.; Park, M.S.; Ji, G.E. Whole-cell biocatalysis for producing ginsenoside Rd from Rb1 using Lactobacillus rhamnosus GG. J. Microbiol. Biotechnol. 2016, 26, 1206–1215. [Google Scholar] [CrossRef] [PubMed]
- Xiudong, X.; Ying, W.; Xiaoli, L.; Ying, L.; Jianzhong, Z. Soymilk residue (okara) as a natural immobilization carrier for Lactobacillus plantarum cells enhances soymilk fermentation, glucosidic isoflavone bioconversion, and cell survival under simulated gastric and intestinal conditions. Peer J. 2016, 4, e2701. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Xu, H.; Wang, X.; Liu, Z. Lactobacillus raises in vitro anticancer effect of geniposide in HSC-3 human oral squamous cell carcinoma cells. Exp. Ther. Med. 2017, 14, 4586–4594. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.R.; Jeong, Y.A.; Na, Y.J.; Park, S.H.; Jo, M.J.; Kim, J.L.; Jeong, S.; Lee, S.Y.; Kim, H.J.; Oh, S.C.; et al. Genipin suppresses colorectal cancer cells by inhibiting the Sonic Hedgehog pathway. Oncotarget 2017, 8, 101952–101964. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, R.; MoYung, K.C.; Zhao, Y.J.; Poon, K. Mechanism for the temporal potentiation of genipin to the cytotoxicity of cisplatin in colon cancer cells. Int. J. Med. Sci. 2016, 13, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Dando, I.; Pacchiana, R.; Pozza, E.D.; Cataldo, I.; Bruno, S.; Conti, P.; Cordani, M.; Grimaldi, A.; Butera, G.; Caraglia, M.; et al. UCP2 inhibition induces ROS/Akt/mTOR axis: Role of GAPDH nuclear translocation in genipin/everolimus anticancer synergism. Free Radic. Biol. Med. 2017, 113, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.; Kim, J.M.; Kim, S.J.; Shim, S.H.; Ha, C.H.; Chang, H.I. Induction of apoptosis by genipin inhibits cell proliferation in AGS human gastric cancer cells via Egr1/p21 signaling pathway. Bioorg. Med. Chem. Lett. 2015, 25, 4191–4196. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Ko, H.; Kim, S.J.; Shim, S.H.; Ha, C.H.; Chang, H.I. Chemopreventive properties of genipin on AGS cell line via induction of JNK/Nrf2/ARE signaling pathway. J. Biochem. Mol. Toxicol. 2016, 30, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Lee, D.U.; Jeong, C.S. Gardenia jasminoides Ellis ethanol extract and its constituents reduce the risks of gastritis and reverse gastric lesions in rats. Food Chem. Toxicol. 2009, 47, 1127–1131. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yao, J.; Luo, Y.; Han, Y.; Wang, Z.; Du, L. P38 MAP kinase mediates apoptosis after genipin treatment in non-small-cell lung cancer H1299 cells via a mitochondrial apoptotic cascade. J. Pharmacol. Sci. 2013, 121, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Feng, Q.; Xu, W.; Li, X.; Kang, Z.; Ren, Y.; Du, L. Genipin induced apoptosis associated with activation of the c-Jun NH2-terminal kinase and p53 protein in HeLa cells. Biol. Pharm. Bull. 2010, 33, 1343–1348. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.; Kim, C.; Kim, S.M.; Kim, W.S.; Choi, S.H.; Chang, I.M.; Ahn, K.S. The hydrolyzed products of iridoid glycoside with β-glucosidase treatment exert anti-proliferative effects through suppression of STAT3 activation and STAT3-regulated gene products in several human cancer cells. Pharm. Biol. 2012, 50, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.Y.; Kim, B.C. Mixed lineage kinase 3 connects reactive oxygen species to c-Jun NH2-terminal kinase-induced mitochondrial apoptosis in genipin-treated PC3 human prostate cancer cells. Biochem. Biophys. Res. Commun. 2007, 362, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.C.; Kim, H.G.; Lee, S.A.; Lim, S.; Park, E.H.; Kim, S.J.; Lim, C.J. Genipin-induced apoptosis in hepatoma cells is mediated by reactive oxygen species/c-Jun NH2-terminal kinase-dependent activation of mitochondrial pathway. Biochem. Pharmacol. 2005, 70, 1398–1407. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zhu, M.; Tsao, S.W.; Man, K.; Zhang, Z.; Feng, Y. Up-regulation of TIMP-1 by genipin inhibits MMP-2 activities and suppresses the metastatic potential of human hepatocellular carcinoma. PLoS ONE 2012, 7, e46318. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S.; Jeong, C.S.; Moon, A. Genipin, a constituent of Gardenia jasminoides Ellis, induces apoptosis and inhibits invasion in MDA-MB-231 breast cancer cells. Oncol. Rep. 2012, 27, 567–572. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Feng, Q.; Cao, H.L.; Xu, W.; Li, X.R.; Ren, Y.Q.; Du, L.F. Apoptosis induced by genipin in human leukemia K562 cells: Involvement of c-Jun N-terminal kinase in G2/M arrest. Acta Pharmacol. Sin. 2011, 32, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Ahn, K.S.; Jeong, S.J.; Jung, J.H.; Kwon, T.R.; Rhee, Y.H.; Kim, S.H.; Kim, S.Y.; Yoon, H.J.; Zhu, S.; et al. Signal transducer and activator of transcription 3 pathway mediates genipin-induced apoptosis in U266 multiple myeloma cells. J. Cell Biochem. 2011, 112, 1552–1562. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J.; Adjeitey, C.N.; Harper, M.E. Genipin-induced inhibition of uncoupling protein-2 sensitizes drug-resistant cancer cells to cytotoxic agents. PLoS ONE 2010, 5, e13289. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.C.; Tseng, T.H.; Lee, M.J.; Hsu, J.D.; Wang, C.J. Induction of apoptosis by penta-acetyl geniposide in rat C6 glioma cells. Chem. Biol. Interact. 2002, 141, 243–257. [Google Scholar] [CrossRef]
- Peng, C.H.; Huang, C.N.; Hsu, S.P.; Wang, C.J. Penta-acetyl geniposide-induced apoptosis involving transcription of NGF/p75 via MAPK-mediated AP-1 activation in C6 glioma cells. Toxicology 2007, 238, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.H.; Huang, C.N.; Hsu, S.P.; Wang, C.J. Penta-acetyl geniposide induce apoptosis in C6 glioma cells by modulating the activation of neutral sphingomyelinase-induced p75 nerve growth factor receptor and protein kinase Cdelta pathway. Mol. Pharmacol. 2006, 70, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.H.; Huang, C.N.; Wang, C.J. The anti-tumor effect and mechanisms of action of penta-acetyl geniposide. Curr. Cancer Drug Targets 2005, 5, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.H.; Tseng, T.H.; Liu, J.Y.; Huang, C.N.; Hsu, S.P.; Wang, C.J. Penta-acetyl geniposide-induced C6 glioma cell apoptosis was associated with the activation of protein kinase C-δ. Chem.-Biol. Interact. 2004, 147, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.J.; Chu, C.Y.; Tseng, T.H.; Lin, J.K. Penta-acetyl geniposide inhibits the growth and development of C6 glioma cells in rats. Cancer Lett. 1993, 70, 113–118. [Google Scholar] [CrossRef]
- Wang, C.J.; Tseng, T.H.; Lin, J.K. Penta-acetyl geniposide: Isolation, identification and primary effect on C6 glioma cell in vitro. Anticancer Res. 1992, 12, 911–916. [Google Scholar] [PubMed]
- Chow, J.-M.; Shen, S.-C.; Huan, S.K.; Lin, H.-Y.; Chen, Y.-C. Quercetin, but not rutin and quercitrin, prevention of H2O2-induced apoptosis via anti-oxidant activity and heme oxygenase 1 gene expression in macrophages. Biochem. Pharmacol. 2005, 69, 1839–1851. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.H.; Shen, S.-C.; Hsu, C.-S.; Chen, Y.-C. Mitochondrial-dependent, reactive oxygen species-independent apoptosis by myricetin: Role of protein kinase C, cytochrome c, and caspase cascade. Biochem. Pharmacol. 2005, 69, 913–927. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-C.; Chou, F.-P.; Huang, H.-P.; Hsu, J.-D.; Wang, C.-J. Inhibition of cell cycle progression by penta-acetyl geniposide in rat C6 glioma cells. Toxicol. Appl. Pharmacol. 2004, 198, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.Y.; Yang, J.J.; Lin, S.Y.; Lin, C.C. Comparisons of geniposidic acid and geniposide on antitumor and radioprotection after sublethal irradiation. Cancer Lett. 1997, 113, 31–37. [Google Scholar] [CrossRef]
- Liesmann, J.E.; Cook, J.A.; Lipschultz, C.; Teague, D.; Fisher, J.; Mitchell, J.B. Cytotoxic studies of paclitaxel (Taxol) in human tumour cell lines. Br. J. Cancer 1993, 68, 1104–1109. [Google Scholar] [CrossRef]
- Habtemariam, S.; Varghese, G.K. A Novel Diterpene Skeleton: Identification of a highly aromatic, cytotoxic and antioxidant 5-methyl-10-demethyl-abietane-type diterpene from Premna serratifolia. Phytother. Res. 2015, 29, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Medina, A.; Peña-Rodríguezb, L.M.; May-Pat, F.; Karagianisd, G.; Waterman, P.G.; Mallet, A.I.; Habtemariam, S. Identification of sakurasosaponin as a cytotoxic principle from Jacquinia flammea. Nat. Prod. Commun. 2010, 5, 365–368. [Google Scholar] [PubMed]
- Habtemariam, S. Cytotoxicity and immunosuppressive activity of withanolides from Discopodium penninervium. Planta Med. 1997, 63, 15–17. [Google Scholar] [CrossRef] [PubMed]
- Habtemariam, S. Cytotoxicity of diterpenes from Premna schimperi and Premna oligotricha. Planta Med. 1995, 6, 368–369. [Google Scholar] [CrossRef] [PubMed]
- Habtemariam, S. Cytotoxic and cytostatic activity of erlangerins from Commiphora erlangeriana. Toxicon 2003, 41, 723–727. [Google Scholar] [CrossRef]
- Nakayachi, T.; Yasumoto, E.; Nakano, K.; Morshed, S.R.; Hashimoto, K.; Kikuchi, H.; Nishikawa, H.; Kawase, M.; Sakagami, H. Structure-activity relationships of alpha, beta-unsaturated ketones as assessed by their cytotoxicity against oral tumor cells. Anticancer Res. 2004, 24, 737–742. [Google Scholar] [PubMed]
- Yu, S.-X.; Du, C.-T.; Chen, W.; Lei, Q.-Q.; Li, N.; Qi, S.; Zhang, X.-J.; Hu, G.-Q.; Deng, X.-M.; Han, W.-Y.; et al. Genipin inhibits NLRP3 and NLRC4 inflammasome activation via autophagy suppression. Sci. Rep. 2015, 5, 17935. [Google Scholar] [CrossRef] [PubMed]
- Habtemariam, S.; Gray, A.I.; Halbert, G.W.; Waterman, P.G. A novel antibacterial diterpene from Premna schimperi. Planta Med. 1990, 56, 187–189. [Google Scholar] [CrossRef] [PubMed]
- Habtemariam, S.; Gray, A.I.; Waterman, P.G. Antibacterial diterpenes from the aerial parts of Premna oligotricha. Planta Med. 1992, 58, 109–110. [Google Scholar] [CrossRef] [PubMed]
- Habtemariam, S.; Gray, A.I.; Waterman, P.G. A new antibacterial sesquiterpene from Premna oligotricha. J. Nat. Prod. 1993, 56, 140–143. [Google Scholar] [CrossRef] [PubMed]
- Kouam, S.F.; Ngouonpe, A.W.; Bullach, A.; Lamshöft, M.; Kuigoua, G.M.; Spiteller, M. Monoterpenes with antibacterial activities from a Cameroonian medicinal plant Canthium Multiflorum (Rubiaceae). Fitoterapia 2013, 91, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Amslinger, S. The tunable functionality of α,β-unsaturated carbonyl compounds enables their differential application in biological systems. ChemMedChem 2010, 5, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Hsu, J.D.; Wang, C.J. Inhibition of 12-O-tetradecanoylphorbol-13-acetate-caused tumor promotion in benzo(a)pyrene initiated CD-1 mouse skin by geniposide. Anticancer Res. 1995, 15, 411–416. [Google Scholar] [PubMed]
- Wang, S.W.; Lai, C.Y.; Wang, C.J. Inhibitory effect of geneposide on aflatoxin B1-induced DNA repair synthesisi in primary cultured rat hepatocytes. Cancer Lett. 1992, 65, 133–137. [Google Scholar] [CrossRef]
- Tseng, T.H.; Chu, C.Y.; Wang, C.J. Inhibition of penta-acetyl geniposide on AFB1-induced genotoxicity in C3H10T1/2 cells. Cancer Lett. 1992, 62, 233–242. [Google Scholar] [CrossRef]
- Wang, C.J.; Wang, S.W.; Lin, J.K. Suppressive effect of geniposide on the hepatotoxicity and hepatic DNA binding of aflatoxin B1 in rats. Cancer Lett. 1991, 60, 95–102. [Google Scholar] [CrossRef]
- Lin, Y.L.; Hsu, J.D.; Chou, F.P.; Lee, M.J.; Shiow, S.J.; Wang, C.J. Suppressive effect of penta-acetyl geniposide on the development of g-glutamyl transpeptidase foci-induced by aflatoxin B1 in rats. Chem. Biol. Interact. 2000, 128, 115–126. [Google Scholar] [CrossRef]
- Han, L.; Hiratake, J.; Kamiyama, A.; Sakata, K. Design, synthesis, and evaluation of gamma-phosphono diester analogues of glutamate as highly potent inhibitors and active site probes of gamma-glutamyl transpeptidase. Biochemistry 2007, 46, 1432–1447. [Google Scholar] [CrossRef] [PubMed]
- Wickham, S.; Regan, N.; West, M.B.; Thai, J.; Cook, P.F.; Terzyan, S.S.; Li, P.K.; Hanigan, M.H. Inhibition of human γ-glutamyl transpeptidase: Development of more potent, physiologically relevant, uncompetitive inhibitors. Biochem. J. 2013, 450, 547–557. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yamamoto, S.; Watanabe, B.; Hiratake, J.; Tanaka, R.; Ohkita, M.; Matsumura, Y. Preventive effect of GGsTop, a novel and selective γ-glutamyl transpeptidase inhibitor, on ischemia/reperfusion-induced renal injury in rats. J. Pharmacol. Exp. Ther. 2011, 339, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Hanigan, M.H. Gamma-Glutamyl Transpeptidase: Redox regulation and drug resistance. Adv. Cancer Res. 2014, 122, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Stark, A.A. Oxidative metabolism of glutathione by gamma-glutamyl transpeptidase and peroxisome proliferation: The relevance to hepatocarcinogenesis. A hypothesis. Mutagenesis 1991, 6, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Ding, L.; Zhen, Y.; Lin, P.; Zhu, Y.; Chen, Y.; Hu, X. Progress in studies on the antimutagenicity and anticarcinogenicity of green tea epicatechins. Chin. Med. Sci. J. 1991, 6, 233–238. [Google Scholar] [PubMed]
- Son, M.; Lee, M.; Ryu, E.; Moon, A.; Jeong, C.S.; Jung, Y.W.; Park, G.H.; Sung, G.H.; Cho, H.; Kang, H. Genipin as a novel chemical activator of EBV lytic cycle. J. Microbiol. 2015, 53, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Mui, U.N.; Haley, C.T.; Tyring, S.K. Viral Oncology: Molecular biology and pathogenesis. J. Clin. Med. 2017, 6, E111. [Google Scholar] [CrossRef] [PubMed]
- Yasunaga, J.I.; Matsuoka, M. Oncogenic spiral by infectious pathogens: Cooperation of multiple factors in cancer development. Cancer Sci. 2018, 109, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.; Jung, S.W.; Lee, S.; Son, K.; Park, G.H.; Jung, J.W.; Shin, Y.S.; Seo, T.; Cho, H.; Kang, H. Genipin enhances Kaposi’s Sarcoma-associated herpesvirus genome maintenance. PLoS ONE 2016, 11, e0163693. [Google Scholar] [CrossRef]
- Zhang, Y.; Yao, J.; Qi, X.; Liu, X.; Lu, X.; Feng, G. Geniposide demonstrates anti-inflammatory and antiviral activity against pandemic A/Jiangsu/1/2009 (H1N1) influenza virus infection in vitro and in vivo. Antivir. Ther. 2017, 22, 599–611. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.J.; Lai, C.C.; Lai, C.H.; Sue, S.C.; Lin, C.W.; Hung, C.H.; Lin, T.H.; Hsu, W.Y.; Huang, S.M.; Hung, Y.L.; et al. Inhibition of enterovirus 71 infections and viral IRES activity by Fructus gardeniae and geniposide. Eur. J. Med. Chem. 2013, 62, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.P.; Shih, Y.W.; Wu, C.H.; Lai, P.J.; Hung, C.N.; Wang, C.J. Inhibitory effect of penta-acetyl geniposide on C6 glioma cells metastasis by inhibiting matrix metalloproteinase-2 expression involved in both the PI3K and ERK signaling pathways. Chem. Biol. Interact. 2009, 181, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Jiang, R.; Zhu, X.; Zhang, X.; Zhan, Z. Genipin inhibits TNF-α-induced vascular smooth muscle cell proliferation and migration via induction of HO-1. PLoS ONE 2013, 8, e74826. [Google Scholar] [CrossRef] [PubMed]
- Kitano, A.; Saika, S.; Yamanaka, O.; Ikeda, K.; Reinach, P.S.; Nakajima, Y.; Okada, Y.; Shirai, K.; Ohnishi, Y. Genipin suppresses subconjunctival fibroblast migration, proliferation and myofibroblast transdifferentiation. Ophthalmic Res. 2006, 38, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, S.; Reis, C.A.; Pinho, S.S. Cadherins glycans in cancer: Sweet players in a bitter process. Trends Cancer 2016, 2, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Daniele, A.; Abbate, I.; Oakley, C.; Casamassima, P.; Savino, E.; Casamassima, A.; Sciortino, G.; Fazio, V.; Gadaleta-Caldarola, G.; Giotta, F.; et al. Clinical and prognostic role of matrix metalloproteinase-2, -9 and their inhibitors in breast cancer and liver diseases: A review. Int. J. Biochem. Cell Biol. 2016, 77, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Merchant, N.; Nagaraju, G.P.; Rajitha, B.; Lammata, S.; Jella, K.K.; Buchwald, Z.S.; Lakka, S.S.; Ali, A.N. Matrix metalloproteinases: Their functional role in lung cancer. Carcinogenesis 2017, 38, 766–780. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, T.; Seiki, M. Integrated functions of membrane-type 1 matrix metalloproteinase in regulating cancer malignancy: Beyond a proteinase. Cancer Sci. 2017, 108, 1095–1100. [Google Scholar] [CrossRef] [PubMed]
- Ci, Y.; Qiao, J.; Han, M. Molecular mechanisms and metabolomics of natural polyphenols interfering with breast cancer metastasis. Molecules 2016, 21, E1634. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.-J.; Lee, S.; Shin, K.-H.; Kim, B.-C.; Lim, C.-J.; Park, E.-H. Geniposide, an anti-angiogenic compound from the fruits of Gardenia jasminoides. Planta Med. 2004, 70, 467–469. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B.; Sethi, G.; Ahn, K.S.; Sandur, S.K.; Pandey, M.K.; Kunnumakkara, A.B.; Sung, B.; Ichikawa, H. Targeting signal-transducer-and-activator-of-transcription-3 for prevention and therapy of cancer: Modern target but ancient solution. Ann. N. Y. Acad. Sci. 2006, 1091, 151–169. [Google Scholar] [CrossRef] [PubMed]
- Cafferkey, C.; Chau, I. Novel STAT 3 inhibitors for treating gastric cancer. Expert Opin. Investig. Drugs 2016, 25, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Radha, G.; Raghavan, S.C. BCL2: A promising cancer therapeutic target. Biochim. Biophys. Acta 2017, 1868, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Cory, S.; Adams, J.M. The Bcl2 family: Regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2002, 2, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Rosse, T.; Olivier, R.; Monney, L.; Rager, M.; Conus, S.; Fellay, I.; Jansen, B.; Borner, C. Bcl-2 prolongs cell survival after Bax-induced release of cytochrome c. Nature 1998, 391, 496–499. [Google Scholar] [CrossRef] [PubMed]
- Moll, U.M.; Wolff, S.; Speidel, D.; Deppert, W. Transcription-independent pro-apoptotic functions of p53. Curr. Opin. Cell Biol. 2005, 17, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Speidel, D. Transcription-independent p53 apoptosis: An alternative route to death. Trends Cell Biol. 2010, 20, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Vaseva, A.V.; Moll, U.M. The mitochondrial p53 pathway. Biochim. Biophys. Acta 2009, 1787, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Lavin, M.F.; Gueven, N. The complexity of p53 stabilization and activation. Cell Death Differ. 2006, 13, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Liu, Q. Cell cycle regulation in treatment of breast cancer. Adv. Exp. Med. Biol. 2017, 1026, 251–270. [Google Scholar] [CrossRef] [PubMed]
- De Groot, A.F.; Kuijpers, C.J.; Kroep, J.R. CDK4/6 inhibition in early and metastatic breast cancer: A review, Cancer Treat. Rev. 2017, 60, 130–138. [Google Scholar] [CrossRef]
- Vijayaraghavan, S.; Moulder, S.; Keyomarsi, K.; Layman, R.M. Inhibiting CDK in cancer therapy: Current evidence and future directions. Target Oncol. 2018, 13, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Casimiro, M.C.; Velasco-Velázquez, M.; Aguirre-Alvarado, C.; Pestell, R.G. Overview of cyclins D1 function in cancer and the CDK inhibitor landscape: Past and present. Expert Opin. Investig. Drugs 2014, 23, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Blattner, C.; Sparks, A.; Lane, D. Transcription factor E2F-1 is upregulated in response to DNA damage in a manner analogous to that of p53. Mol. Cell Biol. 1999, 19, 3704–3713. [Google Scholar] [CrossRef] [PubMed]
- Engeland, K. Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ. 2018, 25, 114–132. [Google Scholar] [CrossRef] [PubMed]
- Gálvez, M.; Martín-Cordero, C.; Ayuso, M.J. Iridoids as DNA topoisomerase I poisons. J. Enzym. Inhib. Med. Chem. 2005, 20, 389–392. [Google Scholar] [CrossRef] [PubMed]
- Kuraishy, A.; Karin, M.; Grivennikov, S.I. Tumor promotion via injury- and death-induced inflammation. Immunity 2011, 35, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Mao, R.; Yang, J. NF-κB and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein Cell 2013, 4, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Ben-Neriah, Y.; Karin, M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat. Immunol. 2011, 12, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.M.; Chua, W.; Charles, K.A.; Clarke, S.J. Inflammation and cancer: Causes and consequences. Clin. Pharmacol. Ther. 2010, 87, 504–508. [Google Scholar] [CrossRef] [PubMed]
- Crusz, S.M.; Balkwill, F.R. Inflammation and cancer: Advances and new agents. Nat. Rev. Clin. Oncol. 2015, 12, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Mukhtar, H.; Elmets, C.A. Photocarcinogenesis: Mechanisms, models and human health implications. Photochem. Photobiol. 1996, 63, 356–357. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.Y.; He, Y.-Y. Ultraviolet radiation-induced non-melanoma skin cancer: Regulation of DNA damage repair and inflammation. Genes Dis. 2014, 1, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Moore, P.S.; Chang, Y. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat. Rev. Cancer 2010, 10, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.-Z. Inflammation fires up cancer metastasis. Sem. Cancer Biol. 2017, 47, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, S.; Goto, T.; Mikami, K.; Miura, K.; Ohshima, S.; Yoneyama, K.; Sato, M.; Shibuya, T.; Watanabe, D.; Kataoka, E.; et al. Genipin prevents fulminant hepatic failure resulting in reduction of lethality through the suppression of TNF-α production. Hepatol. Res. 2005, 33, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Shoda, J.; Kano, M.; Suzuki, S.; Ohtake, N.; Yamamoto, M.; Takahashi, H.; Utsunomiya, H.; Oda, K.; Sato, K.; et al. Inchikoto, a herbal medicine, and its ingredients dually exert Mrp2/MRP2-mediated choleresis and Nrf2-mediated antioxidative action in rat livers. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G1450–G1463. [Google Scholar] [CrossRef] [PubMed]
- Lv, C.; Wang, L.; Liu, X.; Cong, X.; Yan, S.S.; Wang, Y.; Zhang, W. Geniposide attenuates oligomeric Aβ(1-42)-induced inflammatory response by targeting RAGE-dependent signaling in BV2 cells. Curr. Alzheimer Res. 2014, 11, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Lv, C.; Wang, L.; Liu, X.; Yan, S.; Yan, S.S.; Wang, Y.; Zhang, W. Multi-faced neuroprotective effects of geniposide depending on the RAGE-mediated signaling in an Alzheimer mouse model. Neuropharmacology 2015, 89, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Sohn, Y.A.; Hwang, I.Y.; Lee, S.Y.; Cho, H.S.; Jeong, C.S. Protective effects of genipin on gastrointestinal disorders. Biol. Pharm. Bull. 2017, 40, 151–154. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, Z.; Li, Y.; Shen, P.; Li, S.; Lu, X.; Liu, J.; Cao, Y.; Liu, B.; Fu, Y.; Zhang, N. Administration of geniposide ameliorates dextran sulfate sodium-induced colitis in mice via inhibition of inflammation and mucosal damage. Int. Immunopharmacol. 2017, 49, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Li, W.; Li, X.; Li, F.; Zhang, L.; Wang, B.; Huang, G.; Guo, X.; Wan, L.; Liu, Y.; et al. Geniposide attenuates inflammatory response by suppressing P2Y14 receptor and downstream ERK1/2 signaling pathway in oxygen and glucose deprivation-induced brain microvascular endothelial cells. J. Ethnopharmacol. 2016, 185, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.-J.; Song, Y.S.; Kim, H.-J.; Lee, Y.H.; Hong, S.M.; Kim, S.J.; Jin, C.; Lim, C.J.; Park, E.H. Antiinflammatory effects of genipin, an active principle of gardenia. Eur. J. Pharmacol. 2004, 495, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.J.; Lim, K.H.; Jung, H.J.; Park, E.H. Anti-inflammatory evaluation of gardenia extract, geniposide and genipin. J. Ethnopharmacol. 2006, 103, 496–500. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Kondo, K.; Ikeda, Y.; Umemura, K. Antithrombotic effect of geniposide and genipin in the mouse thrombosis model. Planta Med. 2001, 67, 807–810. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Y.; Liu, H.; Yang, M.; Wei, S.F. Antithrombotic activities of aqueous extract from Gardenia jasminoides and its main constituent. Pharm. Biol. 2013, 51, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Kuo, W.H.; Wang, C.J.; Young, S.C.; Sun, Y.C.; Chen, Y.J.; Chou, F.P. Differential induction of the expression of GST subunits by geniposide in rat hepatocytes. Pharmacology 2004, 70, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Lau, F.T.K.; Pak, R.C.K. Effects of Gardenia jasminoides and geniposides on hepatic drug-metabolizing enzyme activity implications for α-nephthylisothiocyanate-induced hepatotoxicity. Asia Pacific J. Pharmacol. 1986, 1, 91–98. [Google Scholar]
- Liu, J.; Yin, F.; Zheng, X.; Jing, J.; Hu, Y. Geniposide, a novel agonist for GLP-1 receptor, prevents PC12 cells from oxidative damage via MAP kinase pathway. Neurochem. Int. 2007, 51, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.H.; Yin, F.; Guo, L.X.; Deng, X.H.; Hu, Y.H. Neuroprotection of geniposide against hydrogen peroxide induced PC12 cells injury: Involvement of PI3 kinase signal pathway. Acta Pharmacol. Sin. 2009, 30, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.T.; Li, X.F.; Li, W.X.; Yang, Y.; Huang, C.; Meng, X.M.; Zhang, L.; Li, J. Geniposide alleviates inflammation by suppressing MeCP2 in mice with carbon tetrachloride-induced acute liver injury and LPS-treated THP-1 cells. Int. Immunopharmacol. 2015, 29, 739–747. [Google Scholar] [CrossRef] [PubMed]
- Sakaida, I.; Tsuchiya, M.; Kawaguchi, K.; Kimura, T.; Terai, S.; Okita, K. Herbal medicine Inchinko- to (TJ-135) prevents liver fibrosis and enzyme-altered lesions in rat liver cirrhosis induced by a choline deficient l-amino acid-defined diet. J. Hepatol. 2003, 38, 762–769. [Google Scholar] [CrossRef]
- Yamamoto, M.; Ogawa, K.; Morota, M.; Fukuda, K.; Komatsu, Y. The herbal medicine Inchin-ko-to inhibits liver cell apoptosis induced by transforming growth factor beta 1. Hepatology 1996, 23, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Miura, N.; Ohtake, N.; Amagaya, S.; Ishige, A.; Sasaki, H.; Komatsu, Y.; Fukuda, K.; Ito, T.; Terasawa, K. Genipin, a metabolite derived from the herbal medicine Inchin-ko-to, and suppression of Fas-induced lethal liver apoptosis in mice. Gastroenterology 2000, 118, 380–389. [Google Scholar] [CrossRef]
- Saha, S.K.; Lee, S.B.; Won, J.; Choi, H.Y.; Kim, K.; Yang, G.M.; Dayem, A.A.; Cho, S.G. Correlation between oxidative stress, nutrition, and cancer initiation. Int. J Mol. Sci. 2017, 18, E1544. [Google Scholar] [CrossRef] [PubMed]
- Kruk, J.; Aboul-Enein, H.Y. Reactive oxygen and nitrogen species in carcinogenesis: Implications of oxidative stress on the progression and development of several cancer types. Mini Rev. Med. Chem. 2017, 17, 904–919. [Google Scholar] [CrossRef] [PubMed]
- Mahalingaiah, P.K.; Singh, K.P. Chronic oxidative stress increases growth and tumorigenic potential of MCF-7 breast cancer cells. PLoS ONE 2014, 9, e87371. [Google Scholar] [CrossRef] [PubMed]
- McGowan, J.V.; Chung, R.; Maulik, A.; Piotrowska, I.; Walker, J.M.; Yellon, D.M. Anthracycline chemotherapy and cardiotoxicity. Cardiovasc. Drugs Ther. 2017, 31, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Ueda, U.; Nakamura, H.; Masutani, H.; Sasada, T.; Takabayashi, A.; Yamaoka, Y.; Yodoi, J. Baicalin induces apoptosis via mitochondrial pathway as prooxidant. Mol. Immunol. 2002, 38, 781–791. [Google Scholar] [CrossRef]
- Galati, G.; Sabzevari, O.; Wilson, J.X.; O’Brien, P.J. Prooxidant activity and cellular effects of the phenoxyl radicals of dietary flavonoids and other polyphenolics. Toxicology 2002, 177, 91–104. [Google Scholar] [CrossRef]
- Atsumi, T.; Fujisawa, S.; Tonosaki, K. A comparative study of the antioxidant/prooxidant activities of eugenol and isoeugenol with various concentrations and oxidation conditions. Toxicol. In Vitro 2005, 19, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-C.; Kim, J.; Park, J.-K.; Chung, G.-H.; Jang, Y.S. The antioxidant, rather than prooxidant, activities of quercetin on normal cells: Quercetin protects mouse thymocytes from glucose oxidase-mediated apoptosis. Exp. Cell Res. 2003, 291, 386–397. [Google Scholar] [CrossRef]
- Murias, M.; Jäger, W.; Handler, N.; Erker, T.; Horvath, Z.; Szekeres, T.; Nohl, H.; Gille, L. Antioxidant, prooxidant and cytotoxic activity of hydroxylated resveratrol analogues: Structure–activity relationship. Biochem. Pharmacol. 2005, 69, 903–912. [Google Scholar] [CrossRef] [PubMed]
- Sahu, S.C.; Ruggles, D.I.; O’Donnell, M.W. Prooxidant activity and toxicity of nordihydroguaiaretic acid in clone-9 rat hepatocyte cultures. Food Chem. Toxicol. 2006, 44, 1751–1757. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, M.; Haneda, M.O.; Naruse, M.; Htay, H.H.; Tsubouchi, R.; Qiao, S.L.; Li, W.H.; Murakami, K.; Yokochi, T. Prooxidant activity of curcumin: Copper-dependent formation of 8-hydroxy-2′-deoxyguanosine in DNA and induction of apoptotic cell death. Toxicol. In Vitro 2004, 18, 783–789. [Google Scholar] [CrossRef] [PubMed]
- Habtemariam, S. Methyl-3-O-methyl gallate and gallic acid from the leaves of Peltiphyllum peltatum: Isolation and comparative antioxidant, prooxidant, and cytotoxic effects in neuronal cells. J. Med. Food 2011, 14, 1412–1418. [Google Scholar] [CrossRef] [PubMed]
- Habtemariam, S. Knipholone anthrone from Kniphofia foliosa induces a rapid onset of necrotic cell death in cancer cells. Fitoterapia 2010, 81, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- Habtemariam, S.; Dagne, E. Differential cytotoxic and prooxidnant activity of knipholone and knipholone anthrone. Planta Med. 2009, 75, 885. [Google Scholar] [CrossRef]
- Habtemariam, S.; Dagne, E. Comparative antioxidant, prooxidant and cytotoxic activity of sigmoidin A and eriodictyol. Planta Med. 2010, 76, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Habtemariam, S. Flavonoids as inhibitors or enhancers of the cytotoxicity of tumor necrosis factor-α in L-929 tumor cells. J. Nat Prod. 1997, 60, 775–778. [Google Scholar] [CrossRef] [PubMed]
- Kawanishi, S.; Oikawa, S.; Murata, M. Evaluation for safety of antioxidant chemopreventive agents. Antioxid. Redox Signal. 2005, 7, 1728–1739. [Google Scholar] [CrossRef] [PubMed]
- Solovieva, E.M.; Soloviev, V.V.; Akatov, V.S. Vitamin B12b increases the cytotoxicity of short-time exposure to ascorbic acid, inducing oxidative burst and iron-dependent DNA damage. Eur. J. Pharmacol. 2007, 566, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-W.; Lee, S.-M. Antioxidant and prooxidant properties of ascorbic acid on hepatic dysfunction induced by cold ischemia/reperfusion. Eur. J. Pharmacol. 2008, 580, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Lenaz, G. The mitochondrial production of reactive oxygen species: Mechanisms and implications in human pathology. IUBMB Life 2001, 52, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Habtemariam, S. Methodology for rapid Isolation of moringin: Potential anticancer compound from the seeds of Moringa stenopetala. Pharm. Anal. Acta 2017, 8, 558. [Google Scholar] [CrossRef]
- Sreedhar, A.; Zhao, Y. Uncoupling protein 2 and metabolic diseases. Mitochondrion 2017, 34, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Baffy, G. Uncoupling protein-2 and cancer. Mitochondrion 2010, 10, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Berthiaume, E.; Derdak, Z.; Konkin, T.A.; Resnick, M.B.; Wands, J.R.; Baffy, G. Increased expression of uncoupling protein-2 in cholangiocarcinoma cells may confer resistance to apoptosis. Hepatology 2004, 40, 372A–373A. [Google Scholar]
- Derdak, Z.; Mark, N.M.; Beldi, G.; Robson, S.C.; Wands, J.R.; Baffy, G. The mitochondrial uncoupling protein-2 promotes chemoresistance in cancer cells. Cancer Res. 2008, 68, 2813–2819. [Google Scholar] [CrossRef] [PubMed]
- Collins, P.; Jones, C.; Choudhury, S.; Damelin, L.; Hodgson, H. Increased expression of uncoupling protein 2 in HepG2 cells attenuates oxidative damage and apoptosis. Liver Int. 2005, 25, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J.; Harper, M.E. Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radic. Biol. Med. 2011, 51, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- Ayyasamy, V.; Owens, K.M.; Desouki, M.M.; Liang, P.; Bakin, A.; Thangaraj, K.; Buchsbaum, D.J.; LoBuglio, A.F.; Singh, K.K. Cellular model of Warburg effect identifies tumor promoting function of UCP2 in breast cancer and its suppression by genipin. PLoS ONE 2011, 6, e24792. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yang, Y.; Hou, J.; Ding, Y.; Zhang, T.; Zhang, Y.; Wang, J.; Shi, C.; Fu, W.; Cai, Z. The hydroxyl at position C1 of genipin is the active inhibitory group that affects mitochondrial uncoupling protein 2 in Panc-1 cells. PLoS ONE 2016, 11, e0147026. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Lee, J.H.; Jung, K.H.; Park, J.W.; Moon, S.H.; Choe, Y.S.; Lee, K.H. Molecular mechanism of (18)F-FDG uptake reduction induced by genipin in T47D cancer cell and role of uncoupling protein-2 in cancer cell glucose metabolism. Nucl. Med. Biol. 2016, 43, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.L.; Gu, J.; Zhang, Y.C.; Wang, N.; Zhu, Z.H.; Yang, Q.T.; Liu, M.; Xia, J.F. Inhibitory effect of Genipin on uncoupling protein-2 and energy metabolism of androgen-independent prostate cancer cells. Zhonghua Nan Ke Xue. 2015, 21, 973–976. [Google Scholar] [PubMed]
- Board, M.; Lopez, C.; van den Bos, C.; Callaghan, R.; Clarke, K.; Carr, C. Acetoacetate is a more efficient energy-yielding substrate for human mesenchymal stem cells than glucose and generates fewer reactive oxygen species. Int. J. Biochem. Cell Biol. 2017, 88, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Brandi, J.; Cecconi, D.; Cordani, M.; Torrens-Mas, M.; Pacchiana, R.; Dalla Pozza, E.; Butera, G.; Manfredi, M.; Marengo, E.; Oliver, J.; et al. The antioxidant uncoupling protein 2 stimulates hnRNPA2/B1, GLUT1 and PKM2 expression and sensitizes pancreas cancer cells to glycolysis inhibition. Free Radic. Biol. Med. 2016, 101, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Dadak, S.; Beall, C.; Vlachaki-Walker, J.M.; Soutar, M.P.M.; McCrimmon, R.J.; Ashford, M.L.J. Oleate induces KATP channel-dependent hyperpolarization in mouse hypothalamic glucose-excited neurons without altering cellular energy charge. Neuroscience 2017, 346, 29–42. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ge, H.; Zhang, F.; Duan, P.; Zhu, N.; Zhang, J.; Ye, F.; Shan, D.; Chen, H.; Lu, X.; Zhu, C.; et al. Mitochondrial Uuncoupling protein 2 in human cumulus cells is associated with regulating autophagy and apoptosis, maintaining gap junction integrity and progesterone synthesis. Mol. Cell Endocrinol. 2017, 443, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Jiang, Q.; Wang, Y.; Li, W.; Geng, M.; Han, Z.; Chen, X. The anti-proliferative effects of oleanolic acid on A7r5 cells-Role of UCP2 and downstream FGF-2/p53/TSP-1. Cell Biol. Int. 2017, 41, 1296–1306. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Fukuda, T.; Wada, T.; Kawanishi, M.; Tasaka, R.; Yasui, T.; Sumi, T. UCP2 expression may represent a predictive marker of neoadjuvant chemotherapy effectiveness for locally advanced uterine cervical cancer. Oncol. Lett. 2017, 14, 951–957. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lou, J.; Wang, Y.; Wang, X.; Jiang, Y. Uncoupling protein 2 regulates palmitic acid-induced hepatoma cell autophagy. Biomed. Res. Int. 2014, 2014, 810401. [Google Scholar] [CrossRef] [PubMed]
- Muller, S.; Klingbeil, S.M.; Sandica, A.; Jaster, R. Uncoupling protein 2 deficiency reduces proliferative capacity of murine pancreatic stellate cells. Hepatobiliary Pancreat. Dis. Int. 2016, 15, 647–654. [Google Scholar] [CrossRef]
- Rajanbabu, V.; Galam, L.; Fukumoto, J.; Enciso, J.; Tadikonda, P.; Lane, T.N.; Bandyopadhyay, S.; Parthasarathy, P.T.; Cho, Y.; Cho, S.H.; et al. Genipin suppresses NLRP3 inflammasome activation through uncoupling protein-2. Cell Immunol. 2015, 297, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.; Zhang, F.; Shan, D.; Chen, H.; Wang, X.; Ling, C.; Xi, H.; Huang, J.; Zhu, C.; Lv, J. Effects of mitochondrial uncoupling protein 2 inhibition by genipin in human cumulus cells. Biomed. Res. Int. 2015, 2015, 323246. [Google Scholar] [CrossRef] [PubMed]
- Dando, I.; Fiorini, C.; Pozza, E.D.; Padroni, C.; Costanzo, C.; Palmieri, M.; Donadelli, M. UCP2 inhibition triggers ROS-dependent nuclear translocation of GAPDH and autophagic cell death in pancreatic adenocarcinoma cells. Biochim. Biophys. Acta 2013, 1833, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.L.; Tang, W.X.; Tang, X.H.; Qin, W.; Gong, M. Downregulation of uncoupling protein-2 by genipin exacerbates diabetes-induced kidney proximal tubular cells apoptosis. Ren. Fail. 2014, 36, 1298–1303. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Yang, D.; Li, D.; Tan, Y.; Tang, B.; Yang, Y. Inhibition of uncoupling protein 2 with genipin exacerbates palmitate-induced hepatic steatosis. Lipids Health Dis. 2012, 11, 154. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhao, J.; Zhang, X. Inhibition of uncoupling protein 2 by genipin reduces insulin-stimulated glucose uptake in 3T3-L1 adipocytes. Arch. Biochem. Biophys. 2009, 486, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Y.; Parton, L.E.; Ye, C.P.; Krauss, S.; Shen, R.; Lin, C.T.; Porco, J.A.J.; Lowell, B.B. Genipin inhibits UCP2-mediated proton leak and acutely reverses obesity- and high glucose-induced beta cell dysfunction in isolated pancreatic islets. Cell Metab. 2006, 3, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Pons, D.G.; Nadal-Serrano, M.; Torrens-Mas, M.; Valle, A.; Oliver, J.; Roca, P. UCP2 inhibition sensitizes breast cancer cells to therapeutic agents by increasing oxidative stress. Free Radic. Biol. Med. 2015, 86, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Dalla Pozza, E.; Fiorini, C.; Dando, I.; Menegazzi, M.; Sgarbossa, A.; Costanzo, C.; Palmieri, M.; Donadelli, M. Role of mitochondrial uncoupling protein 2 in cancer cell resistance to gemcitabine. Biochim. Biophys. Acta 2012, 1823, 1856–1863. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.F.; Zhang, X.N.; Huang, Z.X.; Zhang, Y.; Zhou, Z.Y. Geniposide reverses multidrug resistance in vitro and in vivo by inhibiting the efflux function and expression of P-glycoprotein. Exp. Ther. Med. 2017, 13, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Su, W.P.; Chang, J.Y.; Kuo, C.C.; Su, W.C. Enhanced antitumor activity of rapamycin and genipin, a UCP-2 inhibitor, in lung cancer. Exp. Clin Ther. 2016, 76 (Suppl. 14), 1308. [Google Scholar] [CrossRef]
- Mahgoub, E.; Kumaraswamy, S.M.; Kader, K.H.; Venkataraman, B.; Ojha, S.; Adeghate, E.; Rajesh, M. Genipin attenuates cisplatin-induced nephrotoxicity by counteracting oxidative stress, inflammation, and apoptosis. Biomed. Pharmacother. 2017, 93, 1083–1097. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Habtemariam, S.; Lentini, G. Plant-Derived Anticancer Agents: Lessons from the Pharmacology of Geniposide and Its Aglycone, Genipin. Biomedicines 2018, 6, 39. https://doi.org/10.3390/biomedicines6020039
Habtemariam S, Lentini G. Plant-Derived Anticancer Agents: Lessons from the Pharmacology of Geniposide and Its Aglycone, Genipin. Biomedicines. 2018; 6(2):39. https://doi.org/10.3390/biomedicines6020039
Chicago/Turabian StyleHabtemariam, Solomon, and Giovanni Lentini. 2018. "Plant-Derived Anticancer Agents: Lessons from the Pharmacology of Geniposide and Its Aglycone, Genipin" Biomedicines 6, no. 2: 39. https://doi.org/10.3390/biomedicines6020039
APA StyleHabtemariam, S., & Lentini, G. (2018). Plant-Derived Anticancer Agents: Lessons from the Pharmacology of Geniposide and Its Aglycone, Genipin. Biomedicines, 6(2), 39. https://doi.org/10.3390/biomedicines6020039