Restoration of DAP Kinase Tumor Suppressor Function: A Therapeutic Strategy to Selectively Induce Apoptosis in Cancer Cells Using Immunokinase Fusion Proteins


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
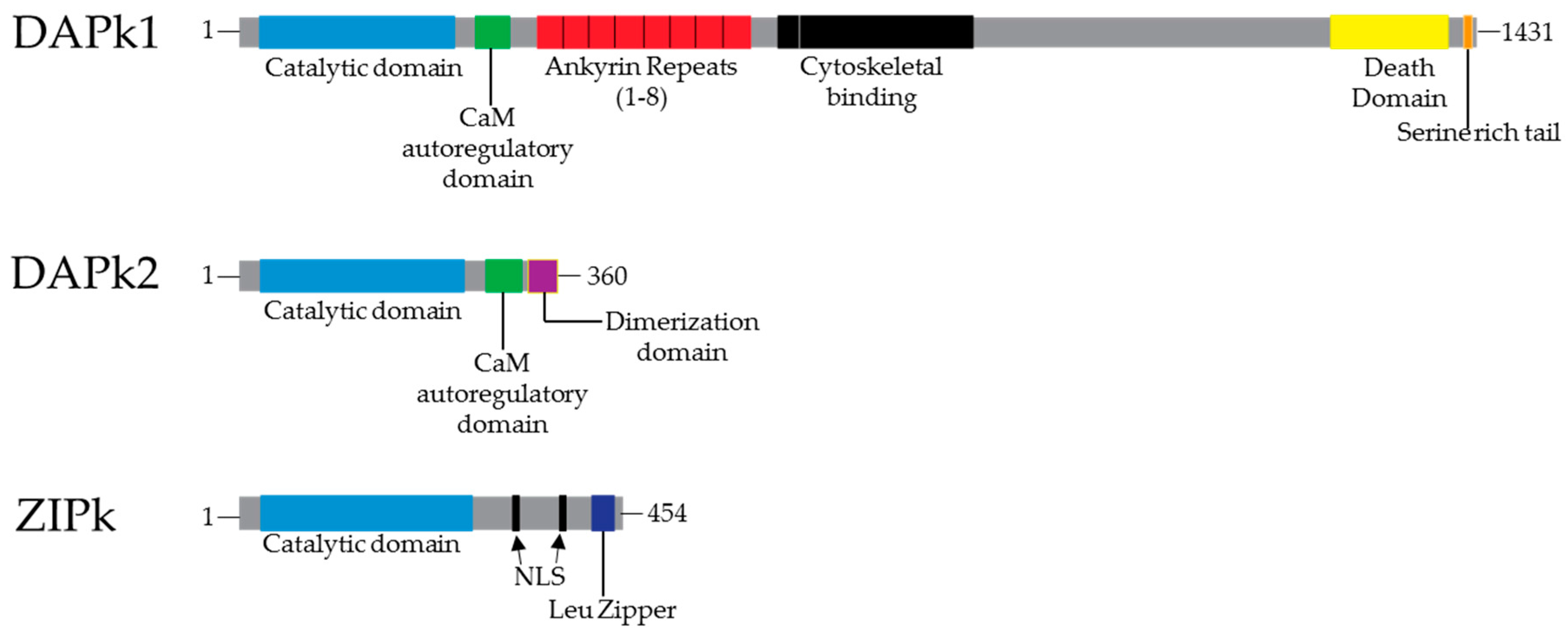
2. Death Associated Protein Kinases (DAPk)
3. Structure and Function
4. DAPk1 and DAPk2: Regulation and Signaling Pathway
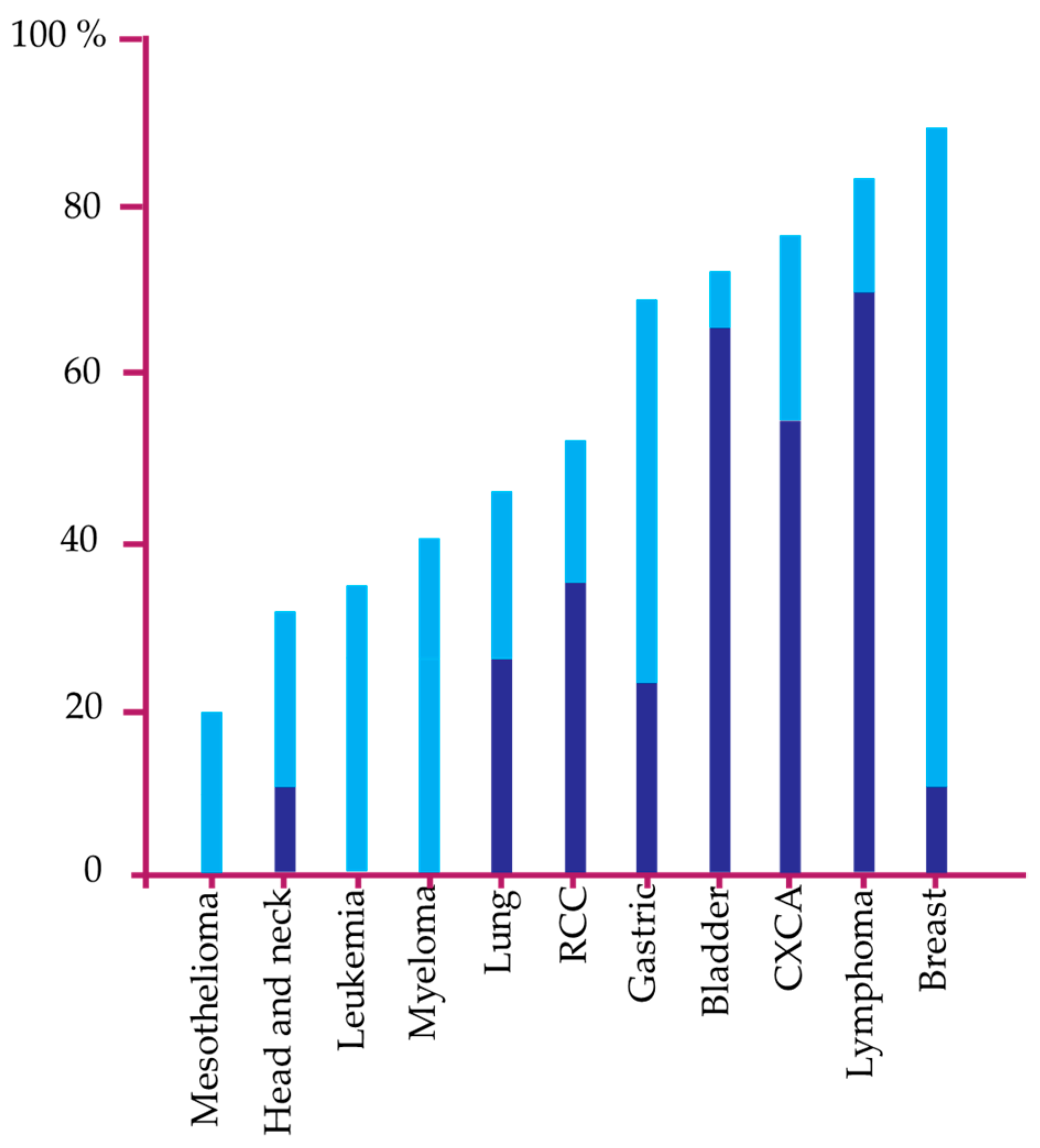
5. Loss and Restoration of DAPk Tumor Suppressor Function in Cancer
6. Immunokinase-Based Fusion Proteins
7. Restoring Apoptosis by Targeted Delivery of DAPk into Tumor Cells
7.1. DAPk1-Based Fusion Proteins for Chronic Lymphocytic Leukaemia
7.2. DAPk2-Based Fusion Proteins for Hodgkin’s Lymphoma
8. Conclusions
Conflicts of Interest
References
- Portt, L.; Norman, G.; Clapp, C.; Greenwood, M.; Greenwood, M.T. Anti-apoptosis and cell survival: A review. Biochim. Biophys. Acta 2011, 1813, 238–259. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Mallipeddi, R.; Wessagowit, V.; South, A.P.; Robson, A.M.; Orchard, G.E.; Eady, R.A.; McGrath, J.A. Reduced expression of insulin-like growth factor-binding protein-3 (IGFBP-3) in squamous cell carcinoma complicating recessive dystrophic epidermolysis bullosa. J. Investig. Dermatol. 2004, 122, 1302–1309. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, S.H.; Gores, G.J. Apoptosis in cancer: Cause and cure. Bioessays 2000, 22, 1007–1017. [Google Scholar] [CrossRef]
- Bates, S.; Phillips, A.C.; Clark, P.A.; Stott, F.; Peters, G.; Ludwig, R.L.; Vousden, K.H. p14ARF links the tumour suppressors RB and p53. Nature 1998, 395, 124–125. [Google Scholar] [CrossRef] [PubMed]
- Michie, A.M.; McCaig, A.M.; Nakagawa, R.; Vukovic, M. Death-associated protein kinase (DAPK) and signal transduction: Regulation in cancer. FEBS J. 2010, 277, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Laird, P.W. Oncogenic mechanisms mediated by DNA methylation. Mol. Med. Today 1997, 3, 223–229. [Google Scholar] [CrossRef]
- Baylin, S.B. Mechanisms underlying epigenetically mediated gene silencing in cancer. Sem. Cancer Biol. 2002, 12, 331–337. [Google Scholar] [CrossRef]
- Baylin, S.; Bestor, T.H. Altered methylation patterns in cancer cell genomes: Cause or consequence? Cancer Cell 2002, 1, 299–305. [Google Scholar] [CrossRef]
- Kögel, D.; Prehn, J.H.; Scheidtmann, K.H. The DAP kinase family of pro-apoptotic proteins: Novel players in the apoptotic game. Bioessays 2001, 23, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Zöchbauer-Müller, S.; Fong, K.M.; Virmani, A.K.; Geradts, J.; Gazdar, A.F.; Minna, J.D. Aberrant promoter methylation of multiple genes in non-small cell lung cancers. Cancer Res. 2001, 61, 249–255. [Google Scholar] [PubMed]
- Lehmann, U.; Celikkaya, G.; Hasemeier, B.; Länger, F.; Kreipe, H. Promoter hypermethylation of the death-associated protein kinase gene in breast cancer is associated with the invasive lobular subtype. Cancer Res. 2002, 62, 6634–6638. [Google Scholar] [PubMed]
- Kiriazis, A. Synthesis of Six-Membered Rings and Inhibitors of Protein Kinases. Ph.D. Thesis, University of Helsinki, Helsinki, Finland, 2012. [Google Scholar]
- Bialik, S.; Kimchi, A. DAP-kinase as a target for drug design in cancer and diseases associated with accelerated cell death. Sem. Cancer Biol. 2004, 14, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Hupp, T.R. Death-associated protein kinase (DAPK) and signal transduction. FEBS J. 2010, 277, 47. [Google Scholar] [CrossRef] [PubMed]
- Gade, P.; Manjegowda, S.B.; Nallar, S.C.; Maachani, U.B.; Cross, A.S.; Kalvakolanu, D.V. Regulation of the death-associated protein kinase 1 expression and autophagy via ATF6 requires apoptosis signal-regulating kinase 1. Mol. Cell. Biol. 2014, 34, 4033–4048. [Google Scholar] [CrossRef] [PubMed]
- Lukas, T.J. DAPK1. In Encyclopedia of Signaling Molecules; Choi, S., Ed.; Springer: New York, NY, USA, 2016; pp. 1–8. [Google Scholar]
- Deiss, L.P.; Feinstein, E.; Berissi, H.; Cohen, O.; Kimchi, A. Identification of a novel serine/threonine kinase and a novel 15-kD protein as potential mediators of the gamma interferon-induced cell death. Genes Dev. 1995, 9, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Ravanan, P.; Talwar, P. Death associated protein kinase 1 (DAPK1): A regulator of apoptosis and autophagy. Front. Mol. Neurosci. 2016, 9. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Hagberg, H.; Krishnamurthy, R.; Thornton, C.; Mallard, C. Death associated protein kinases: Molecular structure and brain injury. Int. J. Mol. Sci. 2013, 14, 13858–13872. [Google Scholar] [CrossRef] [PubMed]
- Inbal, B.; Shani, G.; Cohen, O.; Kissil, J.L.; Kimchi, A. Death-associated protein kinase-related protein 1, a novel serine/threonine kinase involved in apoptosis. Mol. Cell. Biol. 2000, 20, 1044–1054. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Atmanene, C.; Xu, Q.; Fouillen, L.; Van Dorsselaer, A.; Bonnet, D.; Marsol, C.; Hibert, M.; Sanglier-Cianferani, S.; Pigault, C. Homodimerization of the death-associated protein kinase catalytic domain: Development of a new small molecule fluorescent reporter. PLoS ONE 2010, 5, e14120. [Google Scholar] [CrossRef] [PubMed]
- Shani, G.; Marash, L.; Gozuacik, D.; Bialik, S.; Teitelbaum, L.; Shohat, G.; Kimchi, A. Death-associated protein kinase phosphorylates ZIP kinase, forming a unique kinase hierarchy to activate its cell death functions. Mol. Cell. Biol. 2004, 24, 8611–8626. [Google Scholar] [CrossRef] [PubMed]
- Velentza, A.V.; Schumacher, A.M.; Weiss, C.; Egli, M.; Watterson, D.M. A protein kinase associated with apoptosis and tumor suppression structure, activity, and discovery of peptide substrates. J. Biol. Chem. 2001, 276, 38956–38965. [Google Scholar] [CrossRef] [PubMed]
- Shohat, G.; Spivak-Kroizman, T.; Cohen, O.; Bialik, S.; Shani, G.; Berrisi, H.; Eisenstein, M.; Kimchi, A. The pro-apoptotic function of death-associated protein kinase is controlled by a unique inhibitory autophosphorylation-based mechanism. J. Biol. Chem. 2001, 276, 47460–47467. [Google Scholar] [CrossRef] [PubMed]
- Shani, G.; Henis-Korenblit, S.; Jona, G.; Gileadi, O.; Eisenstein, M.; Ziv, T.; Admon, A.; Kimchi, A. Autophosphorylation restrains the apoptotic activity of DRP-1 kinase by controlling dimerization and calmodulin binding. EMBO J. 2001, 20, 1099–1113. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, R.; Bialik, S.; Kimchi, A. The DAPK family: A structure–function analysis. Apoptosis 2014, 19, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Wang, W.J.; Kuo, J.C.; Tsai, H.C.; Lin, J.R.; Chang, Z.F.; Chen, R.H. Bidirectional signals transduced by DAPK–ERK interaction promote the apoptotic effect of DAPK. EMBO J. 2005, 24, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Llambi, F.; Lourenço, F.C.; Gozuacik, D.; Guix, C.; Pays, L.; del Rio, G.; Kimchi, A.; Mehlen, P. The dependence receptor UNC5H2 mediates apoptosis through DAP-kinase. EMBO J. 2005, 24, 1192–1201. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.R.; Yuan, W.C.; Ho, H.C.; Chen, C.H.; Shih, H.M.; Chen, R.H. The Cullin 3 substrate adaptor KLHL20 mediates DAPK ubiquitination to control interferon responses. EMBO J. 2010, 29, 1748–1761. [Google Scholar] [CrossRef] [PubMed]
- Schlegel, C.; Fonseca, A.; Stöcker, S.; Georgiou, M.; Misterek, M.; Munro, C.; Carmo, C.; Seckl, M.; Costa-Pereira, A. DAPK2 is a novel modulator of TRAIL-induced apoptosis. Cell Death Differ. 2014, 21, 1780–1791. [Google Scholar] [CrossRef] [PubMed]
- Vetterkind, S.; Illenberger, S.; Kubicek, J.; Boosen, M.; Appel, S.; Naim, H.Y.; Scheidtmann, K.-H.; Preuss, U. Binding of Par-4 to the actin cytoskeleton is essential for Par-4/Dlk-mediated apoptosis. Exp. Cell Res. 2005, 305, 392–408. [Google Scholar] [CrossRef] [PubMed]
- Bialik, S.; Kimchi, A. The death-associated protein kinases: Structure, function, and beyond. Annu. Rev. Biochem. 2006, 75, 189–210. [Google Scholar] [CrossRef] [PubMed]
- Almeida, F.G.O.; Aquino, P.F.; Souza, A.D.L.; Souza, A.Q.L.; Vinhote, S.C.; Mac-Cormick, T.M.; Silva, M.S.M.; Chalub, S.R.S.; da Gama Fischer, J.S.; Carvalho, P.C. Colorectal cancer DNA methylation patterns from patients in Manaus, Brazil. Biol. Res. 2015, 48, 50. [Google Scholar] [CrossRef] [PubMed]
- Cohen, O.; Inbal, B.; Kissil, J.L.; Raveh, T.; Berissi, H.; Spivak-Kroizaman, T.; Feinstein, E.; Kimchi, A. DAP-kinase participates in TNF-α–and Fas-induced apoptosis and its function requires the death domain. J. Cell Biol. 1999, 146, 141–148. [Google Scholar] [PubMed]
- Jang, C.-W.; Chen, C.-H.; Chen, C.-C.; Chen, J.-y.; Su, Y.-H.; Chen, R.-H. TGF-β induces apoptosis through Smad-mediated expression of DAP-kinase. Nat. Cell Biol. 2002, 4, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Martoriati, A.; Doumont, G.; Alcalay, M.; Bellefroid, E.; Pelicci, P.G.; Marine, J.-C. DAPK1, encoding an activator of a p19ARF-p53-mediated apoptotic checkpoint, is a transcription target of p53. Oncogene 2005, 24, 1461–1466. [Google Scholar] [CrossRef] [PubMed]
- Britschgi, A.; Trinh, E.; Rizzi, M.; Jenal, M.; Ress, A.; Tobler, A.; Fey, M.; Helin, K.; Tschan, M. DAPK2 is a novel E2F1/KLF6 target gene involved in their proapoptotic function. Oncogene 2008, 27, 5706–5716. [Google Scholar] [CrossRef] [PubMed]
- Gozuacik, D.; Kimchi, A. DAPk protein family and cancer. Autophagy 2006, 2, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Raveh, T.; Droguett, G.; Horwitz, M.S.; DePinho, R.A.; Kimchi, A. DAP kinase activates a p19ARF/p53-mediated apoptotic checkpoint to suppress oncogenic transformation. Nat. Cell Biol. 2001, 3, 1–7. [Google Scholar] [PubMed]
- Wang, W.-J.; Kuo, J.-C.; Yao, C.-C.; Chen, R.-H. DAP-kinase induces apoptosis by suppressing integrin activity and disrupting matrix survival signals. J. Cell Biol. 2002, 159, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-Y.; Lee, Y.-R.; Chen, R.-H. The functions and regulations of DAPK in cancer metastasis. Apoptosis 2014, 19, 364. [Google Scholar] [CrossRef] [PubMed]
- Gozuacik, D.; Bialik, S.; Raveh, T.; Mitou, G.; Shohat, G.; Sabanay, H.; Mizushima, N.; Yoshimori, T.; Kimchi, A. DAP-kinase is a mediator of endoplasmic reticulum stress-induced caspase activation and autophagic cell death. Cell Death Differ. 2008, 15, 1875–1886. [Google Scholar] [CrossRef] [PubMed]
- Zalckvar, E.; Berissi, H.; Eisenstein, M.; Kimchi, A. Phosphorylation of Beclin 1 by DAP-kinase promotes autophagy by weakening its interactions with Bcl-2 and Bcl-XL. Autophagy 2009, 5, 720–722. [Google Scholar] [CrossRef] [PubMed]
- Bialik, S.; Kimchi, A. Lethal weapons: DAP-kinase, autophagy and cell death: DAP-kinase regulates autophagy. Curr. Opin. Cell Biol. 2010, 22, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Geering, B.; Stoeckle, C.; Rožman, S.; Oberson, K.; Benarafa, C.; Simon, H.-U. DAPK2 positively regulates motility of neutrophils and eosinophils in response to intermediary chemoattractants. J. Leukoc. Biol. 2014, 95, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Lilienthal, N.; Lohmann, G.; Crispatzu, G.; Vasyutina, E.; Zittrich, S.; Mayer, P.; Herling, C.D.; Tur, M.K.; Hallek, M.; Pfitzer, G.; et al. A Novel Recombinant Anti-CD22 Immunokinase Delivers Proapoptotic Activity of Death-Associated Protein Kinase (DAPK) and Mediates Cytotoxicity in Neoplastic B Cells. Mol. Cancer Ther. 2016, 15, 971–984. [Google Scholar] [CrossRef] [PubMed]
- Inbal, B.; Bialik, S.; Sabanay, I.; Shani, G.; Kimchi, A. DAP kinase and DRP-1 mediate membrane blebbing and the formation of autophagic vesicles during programmed cell death. J. Cell Biol. 2002, 157, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhu, M.; Zhang, X.; Cheng, D.; Ma, X. Clinical significance of DAPK promoter hypermethylation in lung cancer: A meta-analysis. Drug Design Dev. Ther. 2015, 9, 1785. [Google Scholar]
- Martinez-Glez, V.; Franco-Hernandez, C.; Gonzalez-Gomez, P.; Isla, A.; de Campos, J.; Vaquero, J.; Gutierrez, M.; Casartelli, C.; Rey, J. DAPK1 promoter hypermethylaiton in brain metastases and peripheral blood. Neoplasma 2007, 54, 123. [Google Scholar] [PubMed]
- Huang, Y.; Chen, L.; Guo, L.; Hupp, T.R.; Lin, Y. Evaluating DAPK as a therapeutic target. Apoptosis 2014, 19, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Benderska, N.; Schneider-Stock, R. Transcription control of DAPK. Apoptosis 2014, 19, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-Y.; Lin, Y.-M.; Chung, H.-C.; Lang, Y.-D.; Lin, C.-J.; Huang, J.; Wang, W.-C.; Lin, F.-M.; Chen, Z.; Huang, H.-D. miR-103/107 promote metastasis of colorectal cancer by targeting the metastasis suppressors DAPK and KLF4. Cancer Res. 2012, 72, 3631–3641. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, K.-I.; Oda, Y.; Saito, T.; Yamamoto, H.; Takahira, T.; Tamiya, S.; Iwamoto, Y.; Tsuneyoshi, M. Death-associated protein kinase (DAP kinase) alteration in soft tissue leiomyosarcoma: Promoter methylation or homozygous deletion is associated with a loss of DAP kinase expression. Hum. Pathol. 2004, 35, 1266–1271. [Google Scholar] [CrossRef] [PubMed]
- Raveh, T.; Kimchi, A. DAP kinase—A proapoptotic gene that functions as a tumor suppressor. Exp. Cell Res. 2001, 264, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-J.; Kuo, J.-C.; Ku, W.; Lee, Y.-R.; Lin, F.-C.; Chang, Y.-L.; Lin, Y.-M.; Chen, C.-H.; Huang, Y.-P.; Chiang, M.-J. The tumor suppressor DAPK is reciprocally regulated by tyrosine kinase Src and phosphatase LAR. Mol. Cell 2007, 27, 701–716. [Google Scholar] [CrossRef] [PubMed]
- Tur, M.K.; Neef, I.; Jost, E.; Galm, O.; Jäger, G.; Stöcker, M.; Ribbert, M.; Osieka, R.; Klinge, U.; Barth, S. Targeted restoration of down-regulated DAPK2 tumor suppressor activity induces apoptosis in Hodgkin lymphoma cells. J. Immunother. 2009, 32, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Hackanson, B.; Bennett, K.L.; Brena, R.M.; Jiang, J.; Claus, R.; Chen, S.-S.; Blagitko-Dorfs, N.; Maharry, K.; Whitman, S.P.; Schmittgen, T.D. Epigenetic modification of CCAAT/enhancer binding protein α expression in acute myeloid leukemia. Cancer Res. 2008, 68, 3142–3151. [Google Scholar] [CrossRef] [PubMed]
- Inbal, B.; Cohen, O.; Polak-Charcon, S.; Kopolovic, J.; Vadai, E.; Eisenbach, L.; Kimchi, A. DAP kinase links the control of apoptosis to metastasis. Nature 1997, 390, 180–184. [Google Scholar] [PubMed]
- Gozuacik, D.; Kimchi, A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene 2004, 23, 2891–2906. [Google Scholar] [CrossRef] [PubMed]
- Ventura, A.; Kirsch, D.G.; McLaughlin, M.E.; Tuveson, D.A.; Grimm, J.; Lintault, L.; Newman, J.; Reczek, E.E.; Weissleder, R.; Jacks, T. Restoration of p53 function leads to tumour regression in vivo. Nature 2007, 445, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; El-Deiry, W.S. Restoration of p53 to limit tumor growth. Curr. Opin. Oncol. 2008, 20, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Tur, M.K.; Neef, I.; Jager, G.; Teubner, A.; Stocker, M.; Melmer, G.; Barth, S. Immunokinases, a novel class of immunotherapeutics for targeted cancer therapy. Curr. Pharm. Des. 2009, 15, 2693–2699. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg-Lerner, A.; Kimchi, A. DAPk silencing by DNA methylation conveys resistance to anti EGFR drugs in lung cancer cells. Cell Cycle 2012, 11, 2051. [Google Scholar] [CrossRef] [PubMed]
- Kang, G.H.; Shim, Y.-H.; Jung, H.-Y.; Kim, W.H.; Ro, J.Y.; Rhyu, M.-G. CpG island methylation in premalignant stages of gastric carcinoma. Cancer Res. 2001, 61, 2847–2851. [Google Scholar] [PubMed]
- Sanchez-Cespedes, M.; Esteller, M.; Wu, L.; Nawroz-Danish, H.; Yoo, G.H.; Koch, W.M.; Jen, J.; Herman, J.G.; Sidransky, D. Gene promoter hypermethylation in tumors and serum of head and neck cancer patients. Cancer Res. 2000, 60, 892–895. [Google Scholar] [PubMed]
- Fischer, J.R.; Ohnmacht, U.; Rieger, N.; Zemaitis, M.; Stoffregen, C.; Kostrzewa, M.; Buchholz, E.; Manegold, C.; Lahm, H. Promoter methylation of RASSF1A, RARβ and DAPK predict poor prognosis of patients with malignant mesothelioma. Lung Cancer 2006, 54, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.-P.; Zhang, B.; Zhang, X.-Q.; Chen, M.; Cao, J.; Liu, W.-J. Promoter hypermethylation of multiple genes in early gastric adenocarcinoma and precancerous lesions. Hum. Pathol. 2009, 40, 1534–1542. [Google Scholar] [CrossRef] [PubMed]
- Yuregir, O.O.; Yurtcu, E.; Kizilkilic, E.; Kocer, N.; Ozdogu, H.; Sahin, F. Detecting methylation patterns of p16, MGMT, DAPK and E-cadherin genes in multiple myeloma patients. Int. J. Lab. Hematol. 2010, 32, 142–149. [Google Scholar] [CrossRef] [PubMed]
- De Schutter, H.; Geeraerts, H.; Verbeken, E.; Nuyts, S. Promoter methylation of TIMP3 and CDH1 predicts better outcome in head and neck squamous cell carcinoma treated by radiotherapy only. Oncol. Rep. 2009, 21, 507–513. [Google Scholar] [PubMed]
- Leung, R.C.-Y.; Liu, S.S.; Chan, K.Y.-K.; Tam, K.-F.; Chan, K.-L.; Wong, L.-C.; Ngan, H.Y.-S. Promoter methylation of death-associated protein kinase and its role in irradiation response in cervical cancer. Oncol. Rep. 2008, 19, 1339–1345. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Iida, S.; Uetake, H.; Takagi, Y.; Yamashita, T.; Inokuchi, M.; Yamada, H.; Kojima, K.; Sugihara, K. Methylated TMS1 and DAPK genes predict prognosis and response to chemotherapy in gastric cancer. Int. J. Cancer 2008, 122, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Christoph, F.; Kempkensteffen, C.; Weikert, S.; Köllermann, J.; Krause, H.; Miller, K.; Schostak, M.; Schrader, M. Methylation of tumour suppressor genes APAF-1 and DAPK-1 and in vitro effects of demethylating agents in bladder and kidney cancer. Br. J. Cancer 2006, 95, 1701–1707. [Google Scholar] [CrossRef] [PubMed]
- Hervouet, E.; Cheray, M.; Vallette, F.M.; Cartron, P.-F. DNA methylation and apoptosis resistance in cancer cells. Cells 2013, 2, 545–573. [Google Scholar] [CrossRef] [PubMed]
- Barth, S.; Gattenlöhner, S.; Tur, M.K. Immunokinases. In Fusion Protein Technologies for Biopharmaceuticals: Applications and Challenges; Wiley: Hoboken, NJ, USA, 2013; pp. 329–336. [Google Scholar]
- Reddy, A.N.; Jiang, W.W.; Kim, M.; Benoit, N.; Taylor, R.; Clinger, J.; Sidransky, D.; Califano, J.A. Death-associated protein kinase promoter hypermethylation in normal human lymphocytes. Cancer Res. 2003, 63, 7694–7698. [Google Scholar] [PubMed]




© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tur, M.K.; Daramola, A.K.; Gattenlöhner, S.; Herling, M.; Chetty, S.; Barth, S. Restoration of DAP Kinase Tumor Suppressor Function: A Therapeutic Strategy to Selectively Induce Apoptosis in Cancer Cells Using Immunokinase Fusion Proteins. Biomedicines 2017, 5, 59. https://doi.org/10.3390/biomedicines5040059
Tur MK, Daramola AK, Gattenlöhner S, Herling M, Chetty S, Barth S. Restoration of DAP Kinase Tumor Suppressor Function: A Therapeutic Strategy to Selectively Induce Apoptosis in Cancer Cells Using Immunokinase Fusion Proteins. Biomedicines. 2017; 5(4):59. https://doi.org/10.3390/biomedicines5040059
Chicago/Turabian StyleTur, Mehmet Kemal, Adebukola K. Daramola, Stefan Gattenlöhner, Marco Herling, Shivan Chetty, and Stefan Barth. 2017. "Restoration of DAP Kinase Tumor Suppressor Function: A Therapeutic Strategy to Selectively Induce Apoptosis in Cancer Cells Using Immunokinase Fusion Proteins" Biomedicines 5, no. 4: 59. https://doi.org/10.3390/biomedicines5040059
APA StyleTur, M. K., Daramola, A. K., Gattenlöhner, S., Herling, M., Chetty, S., & Barth, S. (2017). Restoration of DAP Kinase Tumor Suppressor Function: A Therapeutic Strategy to Selectively Induce Apoptosis in Cancer Cells Using Immunokinase Fusion Proteins. Biomedicines, 5(4), 59. https://doi.org/10.3390/biomedicines5040059