The Pathogenetic Role of the HGF/c-Met System in Papillary Carcinoma of the Thyroid


Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Papillary Carcinoma of the Thyroid and MET Expression
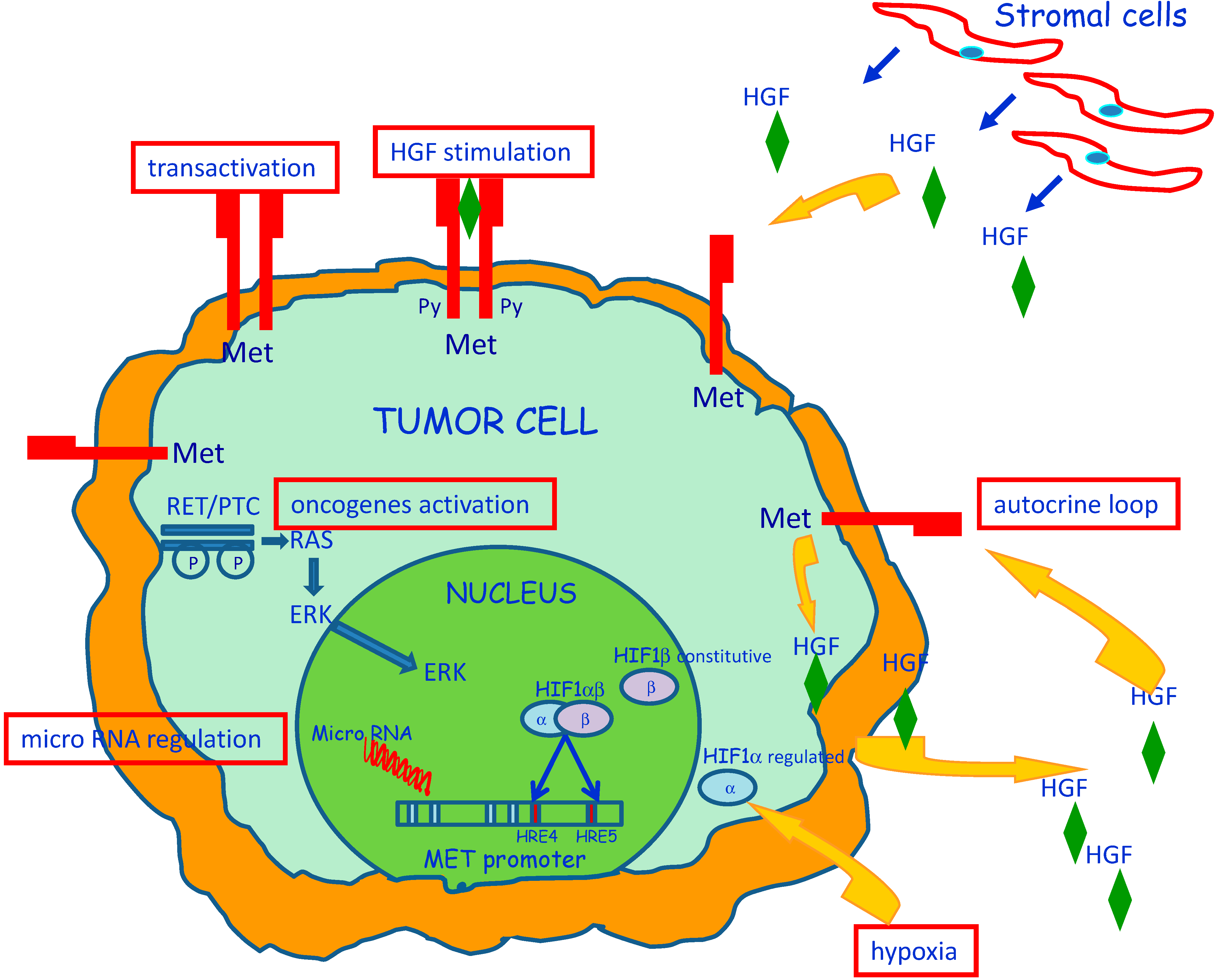
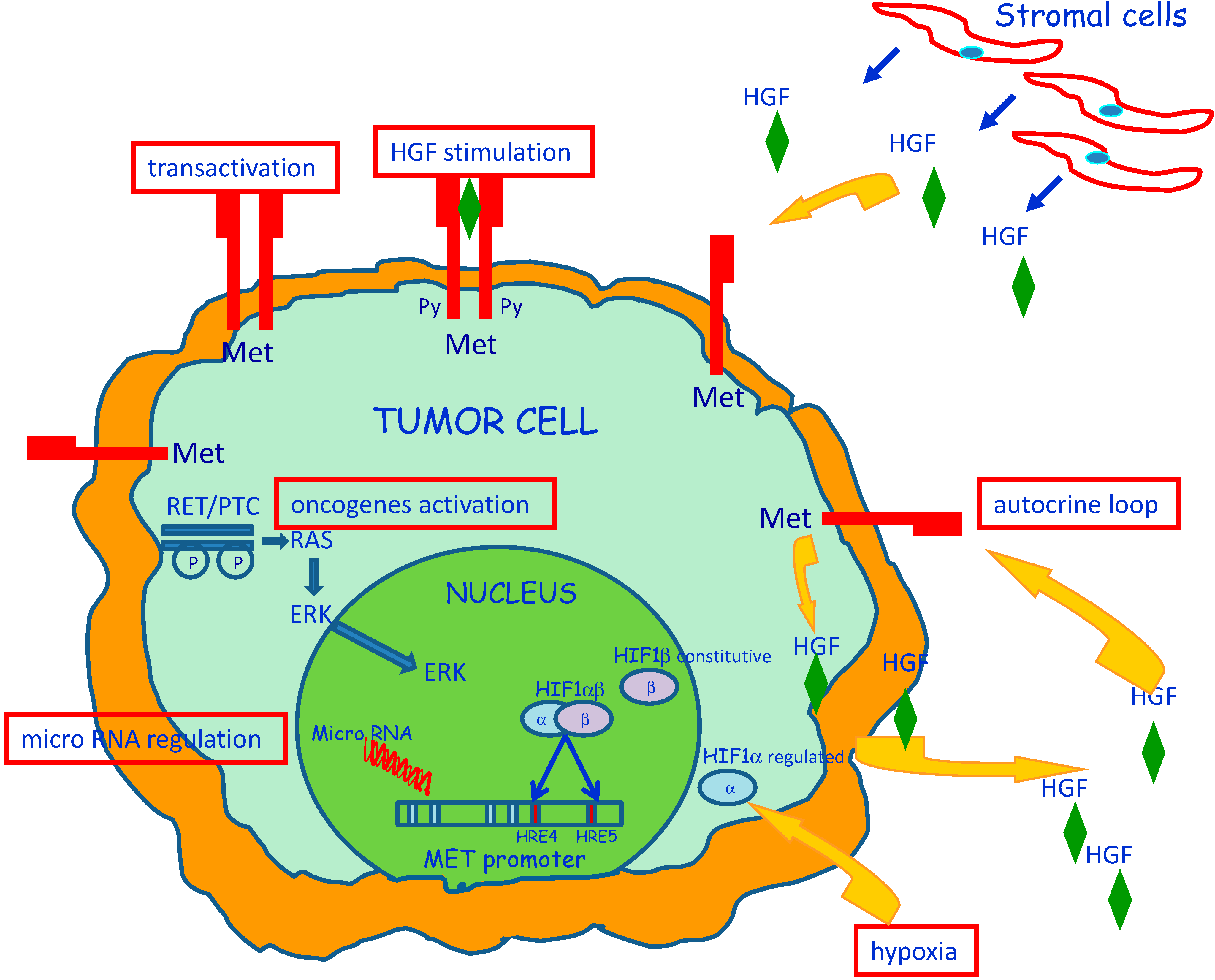
3. Molecular Mechanisms of MET Dysregulation in Papillary Carcinoma of the Thyroid (PTC)
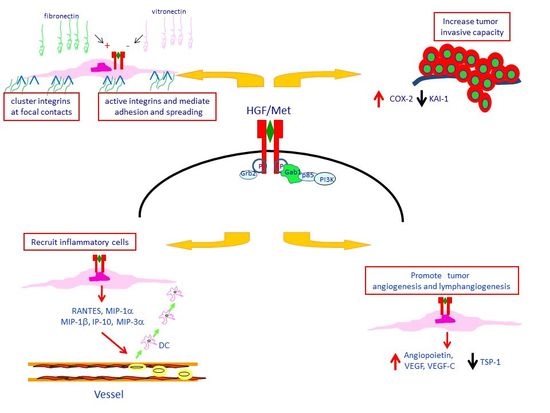
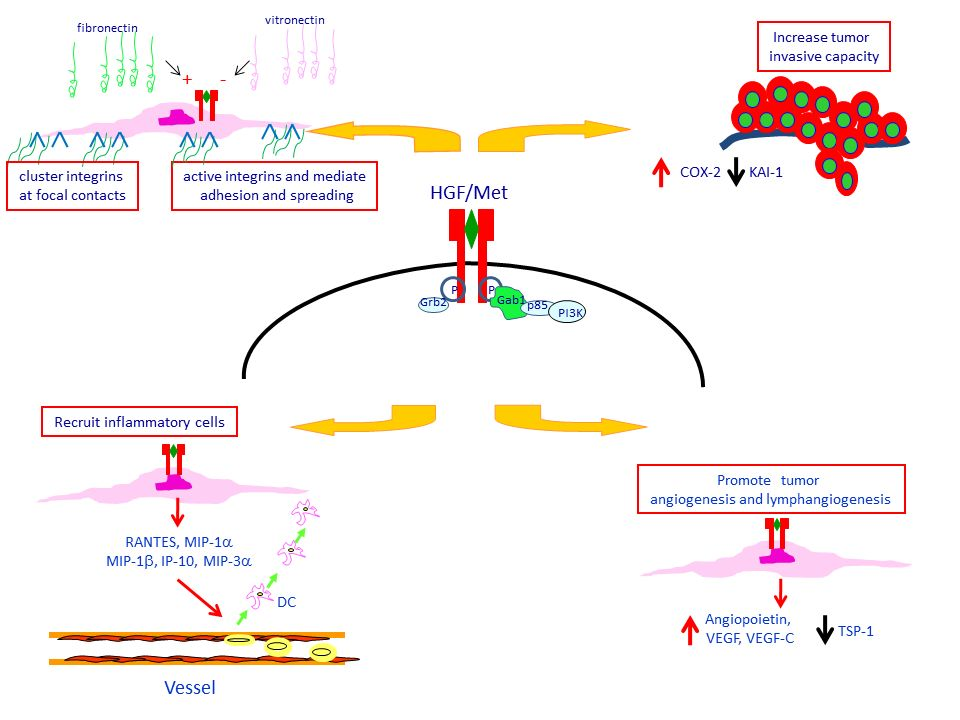
4. Met Protein Over-Expression Facilitates Invasiveness of PTC
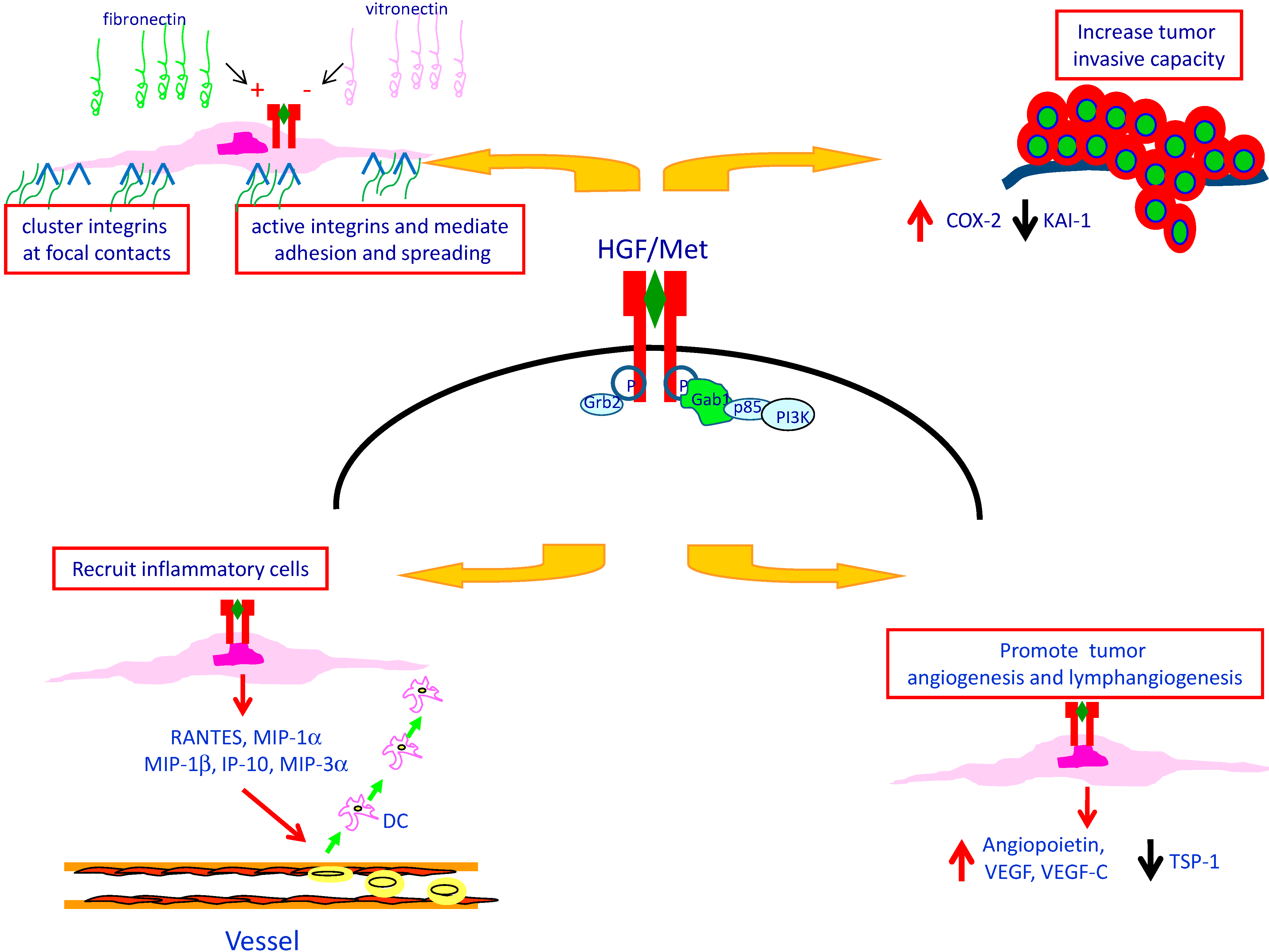
5. Met Protein Over-Expression May Influences the Tumor Microenvironment in PTC
6. Conclusions
Conflicts of Interest
References
- Rosai, J.; Carcangiu, M.L.; DeLellis, R.A. Tumors of the thyroid gland. Atlas Tumor Pathol. 1992, 10008339689. [Google Scholar]
- Kondo, T.; Ezzat, S.; Asa, S.L. Pathogenetic mechanisms in thyroid follicular-cell neoplasia. Nat. Rev. Cancer 2006, 6, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Scarpino, S.; Stoppacciaro, A.; Ballerini, F.; Marchesi, M.; Prat, M.; Stella, M.C.; Sozzani, S.; Allavena, P.; Mantovani, A.; Ruco, L.P. Papillary carcinoma of the thyroid: Hepatocyte growth factor (HGF) stimulates tumor cells to release chemokines active in recruiting dendritic cells. Am. J. Pathol. 2000, 156, 831–837. [Google Scholar] [CrossRef] [PubMed]
- Scarpino, S.; D’Alena, F.C.; di Napoli, A.; Ballarini, F.; Prat, M.; Ruco, L.P. Papillary carcinoma of the thyroid: Evidence for a role for hepatocyte growth factor (HGF) in promoting tumour angiogenesis. J. Pathol. 2003, 199, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Pierotti, M.A.; Vigneri, P.; Bongarzone, I. Rearrangements of RET and NTRK1 tyrosine kinase receptors in papillary thyroid carcinomas. Recent Results Cancer Res. 1998, 154, 237–247. [Google Scholar] [PubMed]
- Kimura, E.T.; Nikiforova, M.N.; Zhu, Z.; Knauf, J.A.; Nikiforov, Y.E.; Fagin, J.A. High prevalence of BRAF mutations in thyroid cancer. Cancer Res. 2003, 63, 1454–1457. [Google Scholar] [PubMed]
- Cohen, Y.; Xing, M.; Mambo, E.; Guo, Z.; Wu, G.; Trink, B.; Beller, U.; Westra, W.H.; Ladenson, P.W.; Sidransky, D. BRAF mutation in papillary thyroid carcinoma. J. Natl. Cancer Inst. 2003, 95, 625–627. [Google Scholar] [CrossRef] [PubMed]
- Soares, P.; Trovisco, V.; Rocha, A.S.; Lima, J.; Castro, P.; Preto, A.; Máximo, V.; Botelho, T.; Seruca, R.; Sobrinho-Simões, M. BRAF mutations and RET/PTC rearrangements are alternative events in the etiopathogenesis of PTC. Oncogene 2003, 22, 4578–4580. [Google Scholar] [CrossRef] [PubMed]
- Delellis, R.A.; Lloyd, R.V.; Heitz, P.U.; Eng, C. Pathology and Genetics of Tumors of Endocrine Organs; World Health Organization: Lyon, France, 2004. [Google Scholar]
- Huang, Y.; Prasad, M.; Lemon, W.J.; Hampel, H.; Wright, F.A.; Kornacker, K.; LiVolsi, V.; Frankel, W.; Kloos, R.T.; Eng, C.; et al. Gene expression in papillary thyroid carcinoma reveals highly consistent profiles. Proc. Natl. Acad. Sci. USA 2001, 98, 15044–15049. [Google Scholar] [CrossRef]
- Prat, M.; Narsimhan, R.; Crepaldi, T.; Nicotra, M.R.; Natali, P.G.; Comoglio, P.M. The receptor encoded by the human c-MET oncogene is expressed in hepatocytes, epithelial cells and solid tumors. Int. J. Cancer 1991, 49, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Ruco, L.P.; Ranalli, T.; Marzullo, A.; Bianco, P.; Prat, M.; Comoglio, P.M.; Baroni, C.D. Expression of Met protein in thyroid tumours. J. Pathol. 1996, 180, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Belfiore, A.; Gangemi, P.; Costantino, A.; Russo, G.; Santonocito, G.M.; Ippolito, O.; di Renzo, M.F.; Comoglio, P.; Fiumara, A.; Vigneri, R. Negative/low expression of the Met/hepatocyte growth factor receptor identifies papillary thyroid carcinomas with high risk of distant metastases. J. Clin. Endocrinol. Metab. 1997, 82, 2322–2328. [Google Scholar] [PubMed]
- Oyama, T.; Ichimura, E.; Sano, T.; Kashiwabara, K.; Fukuda, T.; Nakajima, T. c-Met expression of thyroid tissue with special reference to papillary carcinoma. Pathol. Int. 1998, 48, 763–768. [Google Scholar] [CrossRef] [PubMed]
- Trovato, M.; Villari, D.; Bartolone, L. Expression of the hepatocyte growth factor and c-Met in normal thyroid, nonneoplastic, and neoplastic nodules. Thyroid 1998, 8, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, R.M.; Vitarelli, E.; Barresi, G.; Trimarchi, F.; Benvenga, S.; Trovato, M. HGF/c-Met system pathways in benign and malignant histotypes of thyroid nodules: An immunohistochemical characterization. Histol. Histopathol. 2012, 27, 113–121. [Google Scholar] [PubMed]
- Zanetti, A.; Stoppacciaro, A.; Marzullo, A.; Ciabatta, M.; Fazioli, F.; Prat, M.; Comoglio, P.M.; Baroni, C.D.; Ruco, L.P. Expression of Met protein and and urokinase-type plasminogen activator receptor (uPA-R) in papillary carcinoma of the thyroid. J. Pathol. 1998, 186, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Giordano, S.; Ponzetto, C.; di Renzo, M.F.; Cooper, C.S.; Comoglio, P.M. Tyrosine kinase receptor indistinguishable from the c-Met protein. Nature 1989, 339, 155–156. [Google Scholar] [CrossRef] [PubMed]
- Bottaro, D.P.; Rubin, J.S.; Faletto, D.L.; Chan, A.M.; Kmiecik, T.E.; vande Woude, G.F.; Aaronson, S.A. Identification of the hepatocyte growth factor receptor as the c-Met proto-oncogene product. Science 1991, 251, 802–804. [Google Scholar] [CrossRef] [PubMed]
- Naldini, L.; Vigna, E.; Narsimhan, R.P.; Gaudino, G.; Zarnegar, R.; Michalopoulos, G.K.; Comoglio, P.M. Hepatocyte growth factor (HGF) stimulates the tyrosine kinase activity of the receptor encoded by the proto-oncogene c-Met. Oncogene 1991, 6, 501–504. [Google Scholar] [PubMed]
- Gonzatti-Haces, M.; Seth, A.; Park, M.; Copeland, T.; Oroszlan, S.; vande Woude, G.F. Characterization of the TPR–Met oncogene p65 and the met proto-oncogene p140 protein-tyrosine kinases. Biochemistry 1988, 85, 21–25. [Google Scholar]
- Di Renzo, M.F.; Olivero, M.; Ferro, S. Over-expression of the c-Met/HGF receptor gene in human thyroid carcinomas. Oncogene 1992, 7, 2549–2553. [Google Scholar] [PubMed]
- Migliore, C.; Petrelli, A.; Ghiso, E.; Corso, S.; Capparuccia, L.; Eramo, A.; Comoglio, P.M.; Giordano, S. MicroRNAs impair Met-mediated invasive growth. Cancer Res. 2008, 68, 10128–10136. [Google Scholar] [CrossRef] [PubMed]
- Webb, C.P.; Taylor, G.A.; Jeffers, M. Evidence for a role of Met-HGF/SF during Ras-mediated tumorigenesis/metastasis. Oncogene 1998, 17, 2019–2025. [Google Scholar] [CrossRef] [PubMed]
- Gambarotta, G.; Boccaccio, C.; Giordano, S.; Andŏ, M.; Stella, M.C.; Comoglio, P.M. Ets up-regulates MET transcription. Oncogene 1996, 13, 1911–1917. [Google Scholar] [PubMed]
- Ivan, M.; Bond, J.A.; Prat, M.; Comoglio, P.M.; Wynford-Thomas, D. Activated RAS and RET oncogenes induce over-expression of c-Met (hepatocyte growth factor receptor) in human thyroid epithelial cells. Oncogene 1997, 14, 2417–2423. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, T.; Suzuki, S.; Mashiko, M.; Ohtake, T.; Endo, Y.; Takebayashi, Y.; Sekikawa, K.; Hagiwara, K.; Takenoshita, S. BRAF mutations in papillary carcinomas of the thyroid. Oncogene 2003, 22, 6455–6457. [Google Scholar] [CrossRef] [PubMed]
- Scarpino, S.; di Napoli, A.; Rapazzotti-Onelli, M.; Pilozzi, E.; Ruco, L. Papillary carcinoma of the thyroid: Methylation is not involved in the regulation of MET expression. Br. J. Cancer 2004, 91, 703–706. [Google Scholar] [PubMed]
- Minna, E.; Romeo, P.; de Cecco, L.; Dugo, M.; Cassinelli, G.; Pilotti, S.; Degl’Innocenti, D.; Lanzi, C.; Casalini, P.; Pierotti, M.; et al. miR-199a-3p displays tumor suppressor functions in papillary thyroid carcinoma. Oncotarget 2014, 15, 2513–2528. [Google Scholar]
- Pallante, P.; Visone, R.; Ferracin, M.; Ferraro, A.; Berlingieri, M.T.; Troncone, G.; Chiappetta, G.; Liu, C.G.; Santoro, M.; Negrini, M.; et al. MicroRNA deregulation in human thyroid papillary carcinomas. Endocr. Relat. Cancer 2006, 13, 497–508. [Google Scholar] [CrossRef]
- Boccaccio, C.; Gaudino, G.; Gambarotta, G.; Galimi, F.; Comoglio, P.M. Hepatocyte growth factor (HGF) receptor expression is inducible and is part of the delayed-early response to HGF. J. Biol. Chem. 1994, 269, 12846–12851. [Google Scholar] [PubMed]
- Pennacchietti, S.; Michieli, P.; Galluzzo, M.; Mazzone, M.; Giordano, S.; Comoglio, P.M. Hypoxia promotes invasive growth by transcriptional activation of the MET protooncogene. Cancer Cell 2003, 3, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Scarpino, S.; d’Alena, F.C.; di Napoli, A.; Pasquini, A.; Marzullo, A.; Ruco, L. Increased expression of Met protein is associated with up-regulation of hypoxia inducible factor-1 (HIF-1) in tumour cells in papillary carcinoma of the thyroid. J. Pathol. 2004, 202, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Ponzetto, C.; Bardelli, A.; Zhen, Z.; Maina, F.; dalla Zonca, P.; Giordano, S.; Graziani, A.; Panayotou, G.; Comoglio, P.M. A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell 1994, 77, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Pelicci, G.; Giordano, S.; Zhen, Z.; Salcini, A.E.; Lanfrancone, L.; Bardelli, A.; Panayotou, G.; Waterfield, M.D.; Ponzetto, C. The motogenic and mitogenic responses to HGF are amplified by the Shc adaptor protein. Oncogene 1995, 10, 1631–1638. [Google Scholar] [PubMed]
- Weidner, K.M.; di Cesare, S.; Sachs, M.; Brinkmann, V.; Behrens, J.; Birchmeier, W. Interaction between Gab1 and the c-Met receptor tyrosine kinase is responsible for epithelial morphogenesis. Nature 1996, 384, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Nusrat, A.; Parkos, C.A.; Bacarra, A.E. Hepatocyte growthfactor/scatter factor effects on epithelia. Regulation of intercellular junctions in transformed and non-transformed cell lines, basolateral polarization of c-met receptor in transformed and natural intestinal epithelia, and induction of rapid wound repair in a transformed model epithelium. J. Clin. Investig. 1994, 93, 2056–2065. [Google Scholar] [CrossRef] [PubMed]
- Tamagnone, L.; Comoglio, P.M. Control of invasive growth by hepatocyte growth factor (HGF) and related scatter factors. Cytokine Growth Factor Rev. 1997, 8, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Trusolino, L.; Serini, G.; Cecchini, G. Growth factor dependent activation of αVβ3 integrin in normal epithelial cells: Implications for tumor invasion. J. Cell Biol. 1998, 142, 1145–1156. [Google Scholar]
- Trusolino, L.; Cavassa, S.; Angelini, P.; Ando, M.; Bertotti, A.; Comoglio, P.M. Boccaccio HGF/scatter factor selectively promotes cell invasion by increasing integrin avidity. FASEB J. 2000, 11, 1629–1640. [Google Scholar] [CrossRef]
- Wang, R.; Kobayashi, R.; Bishop, J.M. Cellular adherence elicits ligand-independent activation of the Met cell-surface receptor. Proc. Natl. Acad. Sci. USA 1996, 93, 8425–8430. [Google Scholar] [CrossRef] [PubMed]
- Scarpino, S.; Stoppacciaro, A.; Colarossi, C.; Cancellario, F.; Marzullo, A.; Marchesi, M.; Biffoni, M.; Comoglio, P.M.; Prat, M.; Ruco, L.P. Hepatocyte growth factor (HGF) stimulates tumour invasiveness in papillary carcinoma of the thyroid. J. Pathol. 1999, 189, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Scarpino, S.; Giustiniani, M.C.; Ballarini, F.; Prat, M.; Mainiero, F.; Santoni, A.; Stoppacciaro, A.; Ruco, L. Papillary Carcinoma of the thyroid: The HGF-induced migratory response of tumor cells is regulated by extracellular matrix composition. Unpublished work.
- Takano, T.; Matsuzuka, F.; Sumizaki, H.; Kuma, K.; Amino, N. Rapid detection of specific messenger RNAs in thyroid carcinomas by reverse transcription-PCR with degenerate primers: Specific expression of oncofetal fibronectin messenger RNA in papillary carcinoma. Cancer Res. 1997, 57, 3792–3797. [Google Scholar] [PubMed]
- Scarpino, S.; Stoppacciaro, A.; Pellegrini, C.; Marzullo, A.; Zardi, L.; Tartaglia, F.; Viale, G.; Ruco, L.P. Expression of EDA/EDB isoforms of fibronectin in papillary carcinoma of the thyroid. J. Pathol. 1999, 188, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Mazhar, D.; Gillmore, R. COX and cancer. QJ Med. 2005, 98, 711–718. [Google Scholar] [CrossRef]
- Scarpino, S.; Duranti, E.; Stoppacciaro, A.; Pilozzi, E.; Natoli, G.; Sciacchitano, S.; Luciani, E.; Ruco, L. COX-2 is induced by HGF stimulation in Met-positive thyroid papillary carcinoma cells and is involved in tumour invasiveness. J. Pathol. 2009, 218, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Scarpino, S.; Duranti, E.; Giglio, S.; di Napoli, A.; Galafate, D.; del Bufalo, D.; Desideri, M.; Socciarelli, F.; Stoppacciaro, A.; Ruco, L. High expression of COX-2 and low expression of KAI-1/CD82 are associated with increased tumor invasiveness. Thyroid 2013, 23, 1127–1137. [Google Scholar] [CrossRef] [PubMed]
- Petrelli, A.; Valabrega, G. Multitarget drugs: The present and the future of cancer therapy. Expert Opin. Pharmacother. 2009, 10, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Carcangiu, M.L.; Zampi, G.; Pupi, A.; Castagnoli, A.; Rosai, J. Papillary carcinoma of the thyroid. A clinicopathologic study of 241 cases treated at the University of Florence, Italy. Cancer 1985, 55, 805–828. [Google Scholar] [CrossRef] [PubMed]
- Kashima, K.; Yokoyama, S.; Noguchi, S.; Murakami, N.; Yamashita, H.; Watanabe, S.; Uchino, S.; Toda, M.; Sasaki, A.; Daa, T.; et al. Chronic thyroiditis as a favorable prognostic factor in papillary thyroid carcinoma. Thyroid 1998, 8, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Loh, K.C.; Greenspan, F.S.; Dong, F.; Miller, T.R.; Yeo, P.P. Influence of lymphocytic thyroiditis on the prognostic outcome of patients with papillary thyroid carcinoma. J. Clin. Endocrinol. Metab. 1999, 84, 458–463. [Google Scholar] [CrossRef] [PubMed]
- Schaffler, A.; Palitzsch, K.D.; Seiffarth, C.; Hohne, H.M.; Riedhammer, F.J.; Hofstadter, F.; Scholmerich, J.; Ruschoff, J. Coexistent thyroiditis is associated with lower tumour stage in thyroid carcinoma. Eur. J. Clin. Investig. 1998, 28, 838–844. [Google Scholar] [CrossRef]
- Murray, P.J.; Thomas, A. Wynn Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruco, L.; Scarpino, S. The Pathogenetic Role of the HGF/c-Met System in Papillary Carcinoma of the Thyroid. Biomedicines 2014, 2, 263-274. https://doi.org/10.3390/biomedicines2040263
Ruco L, Scarpino S. The Pathogenetic Role of the HGF/c-Met System in Papillary Carcinoma of the Thyroid. Biomedicines. 2014; 2(4):263-274. https://doi.org/10.3390/biomedicines2040263
Chicago/Turabian StyleRuco, Luigi, and Stefania Scarpino. 2014. "The Pathogenetic Role of the HGF/c-Met System in Papillary Carcinoma of the Thyroid" Biomedicines 2, no. 4: 263-274. https://doi.org/10.3390/biomedicines2040263
APA StyleRuco, L., & Scarpino, S. (2014). The Pathogenetic Role of the HGF/c-Met System in Papillary Carcinoma of the Thyroid. Biomedicines, 2(4), 263-274. https://doi.org/10.3390/biomedicines2040263