Gulonolactone Addition to Human Hepatocellular Carcinoma Cells with Gene Transfer of Gulonolactone Oxidase Restores Ascorbate Biosynthesis and Reduces Hypoxia Inducible Factor 1


 and
and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
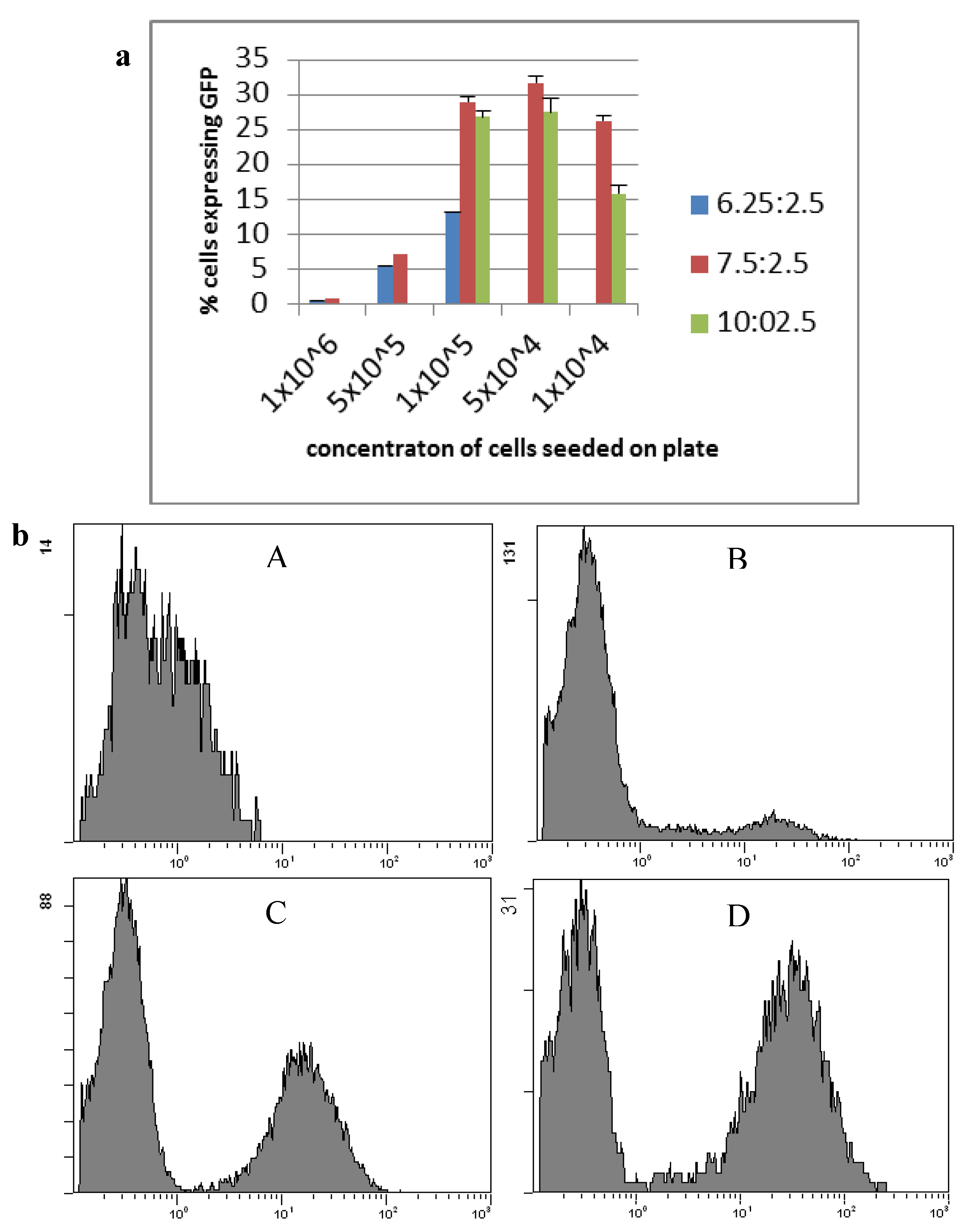
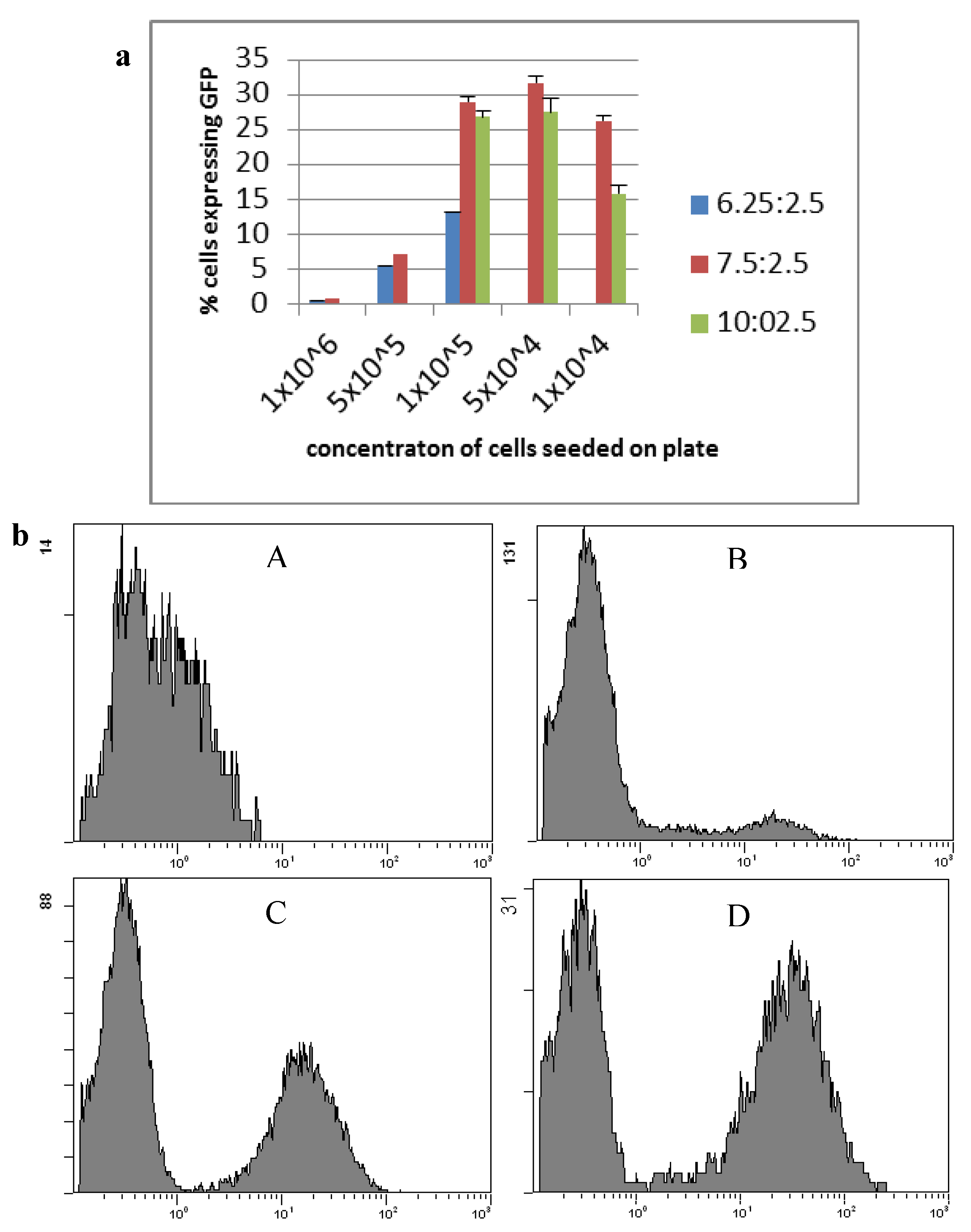
2.1. Transfection
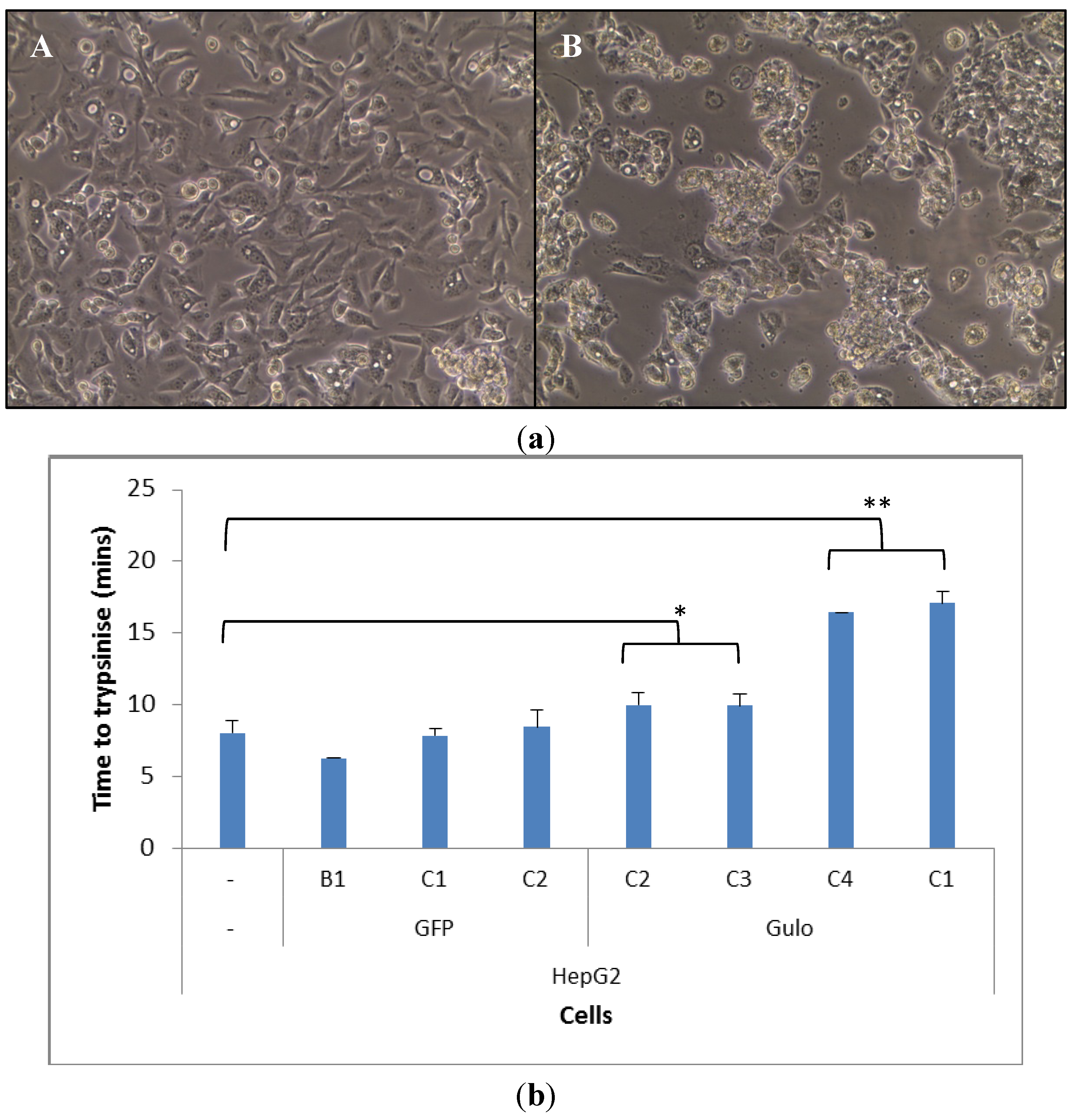
2.2. Cell Morphology
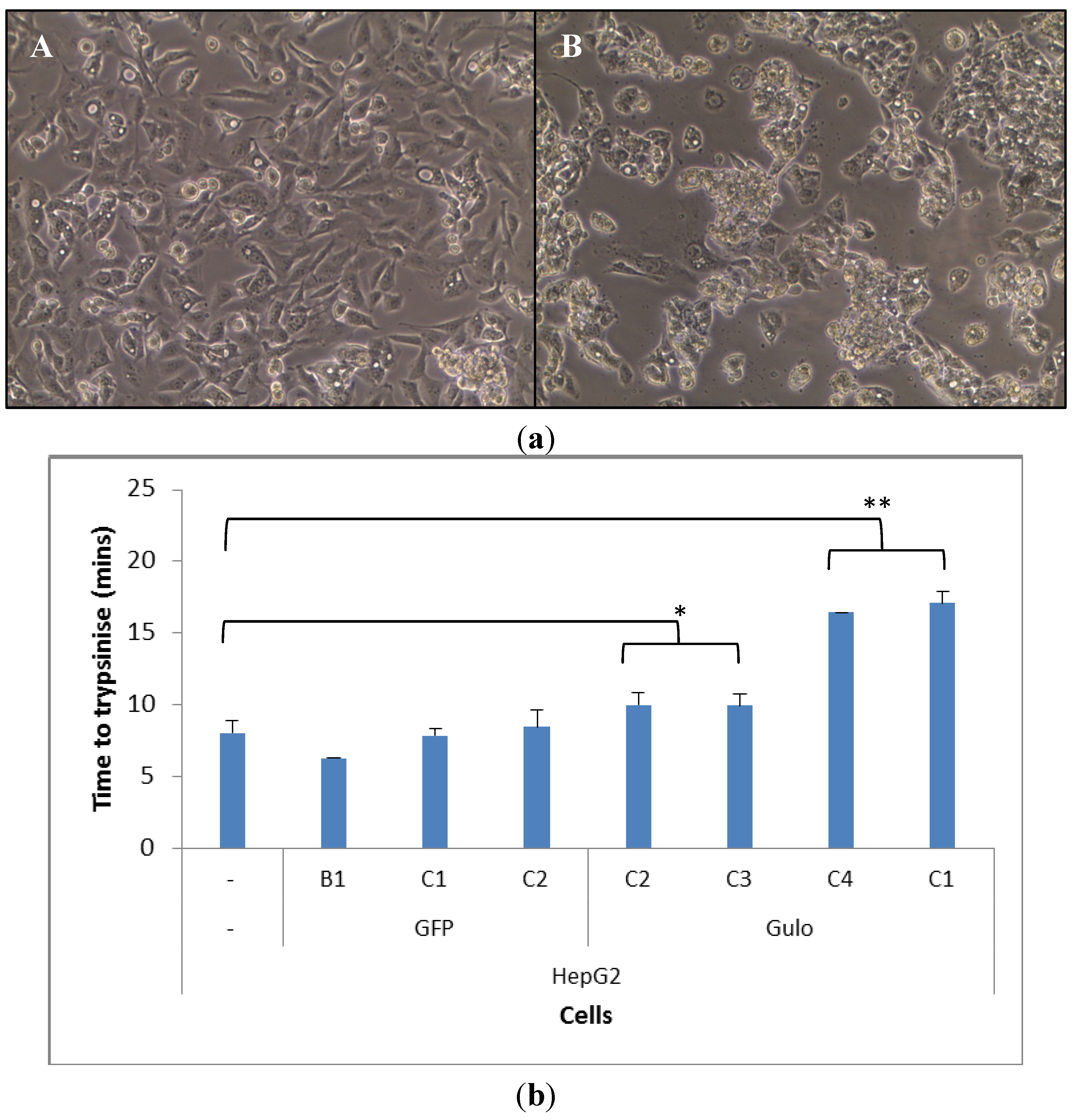
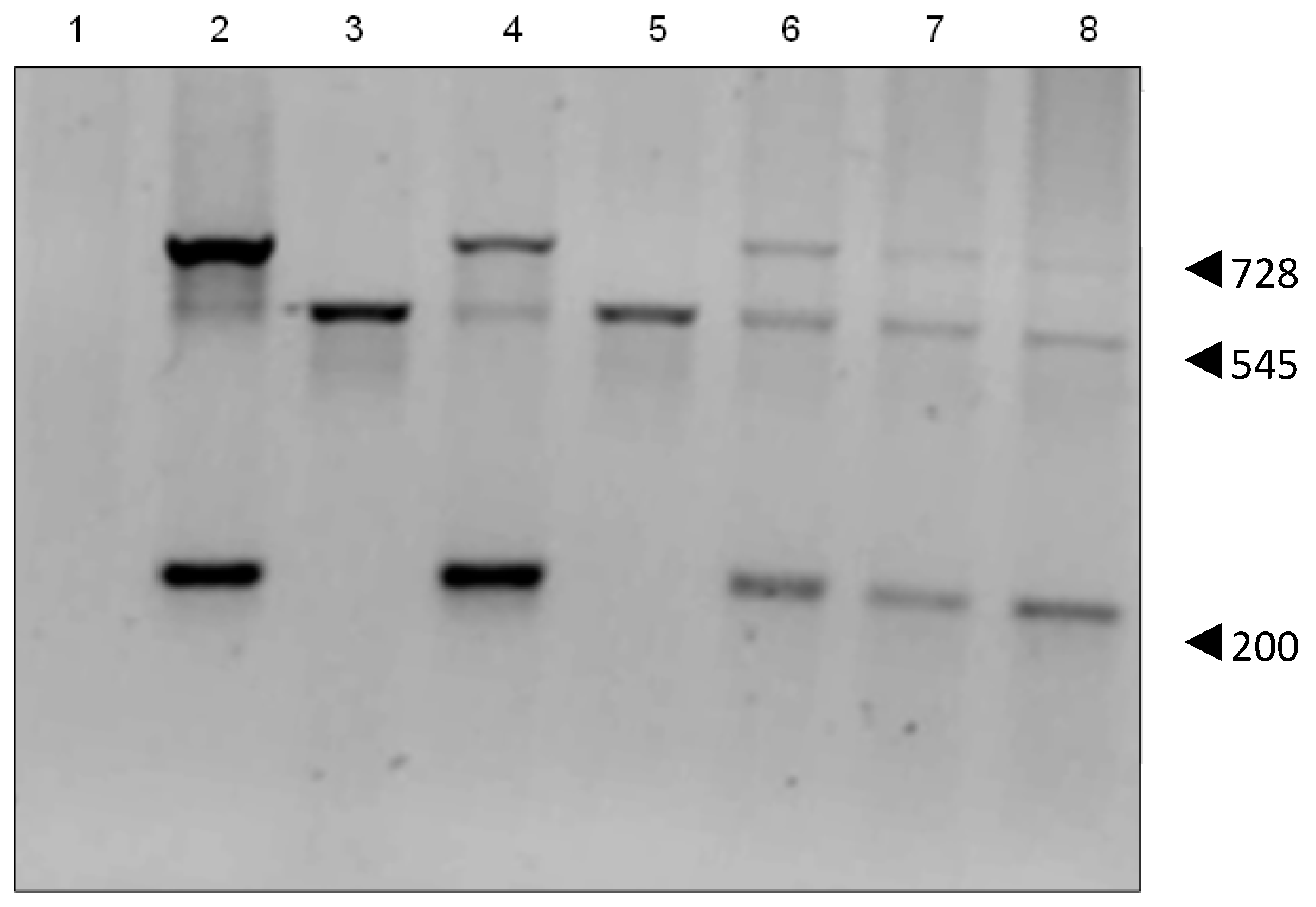
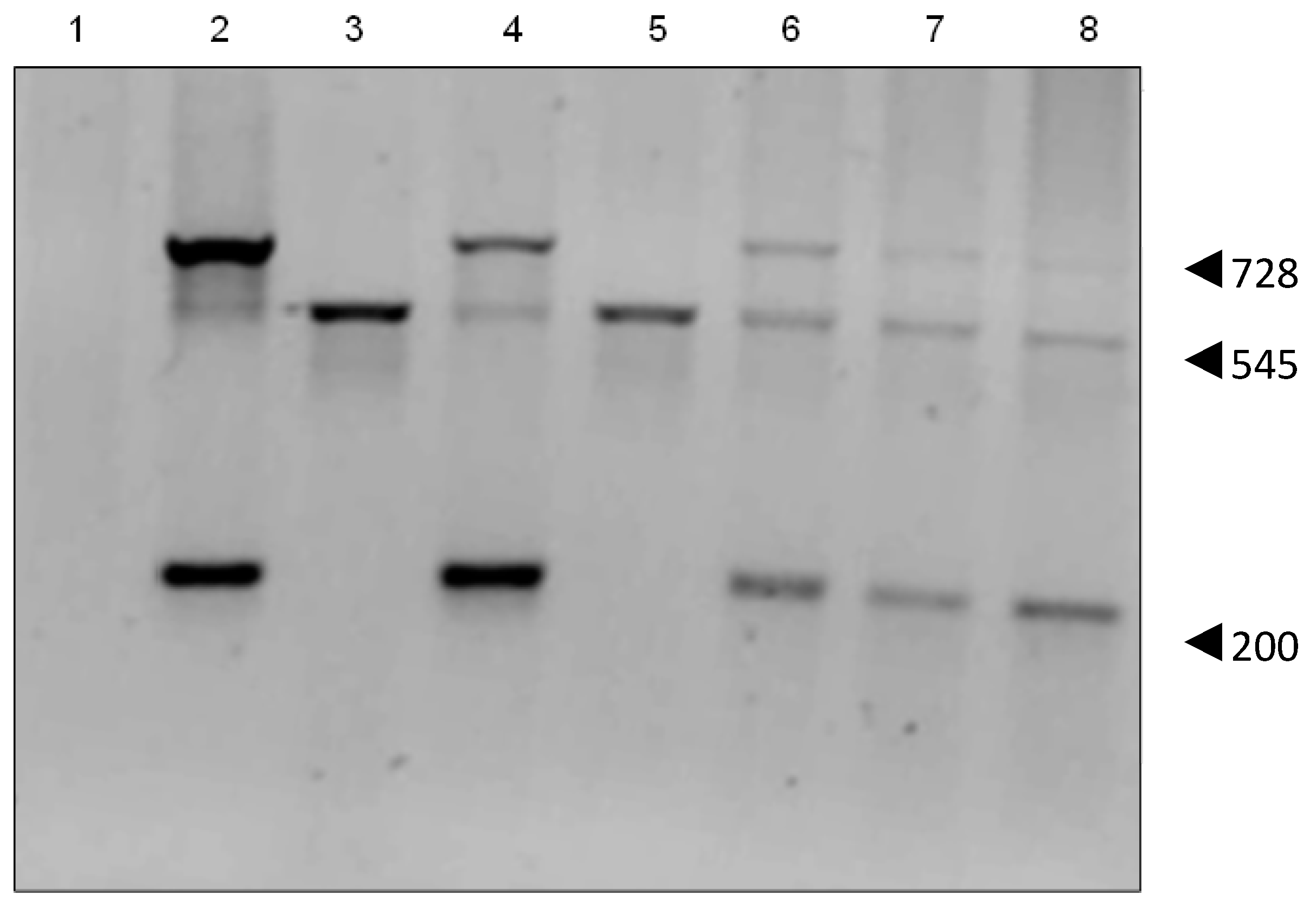
2.3. Genomic Insertion
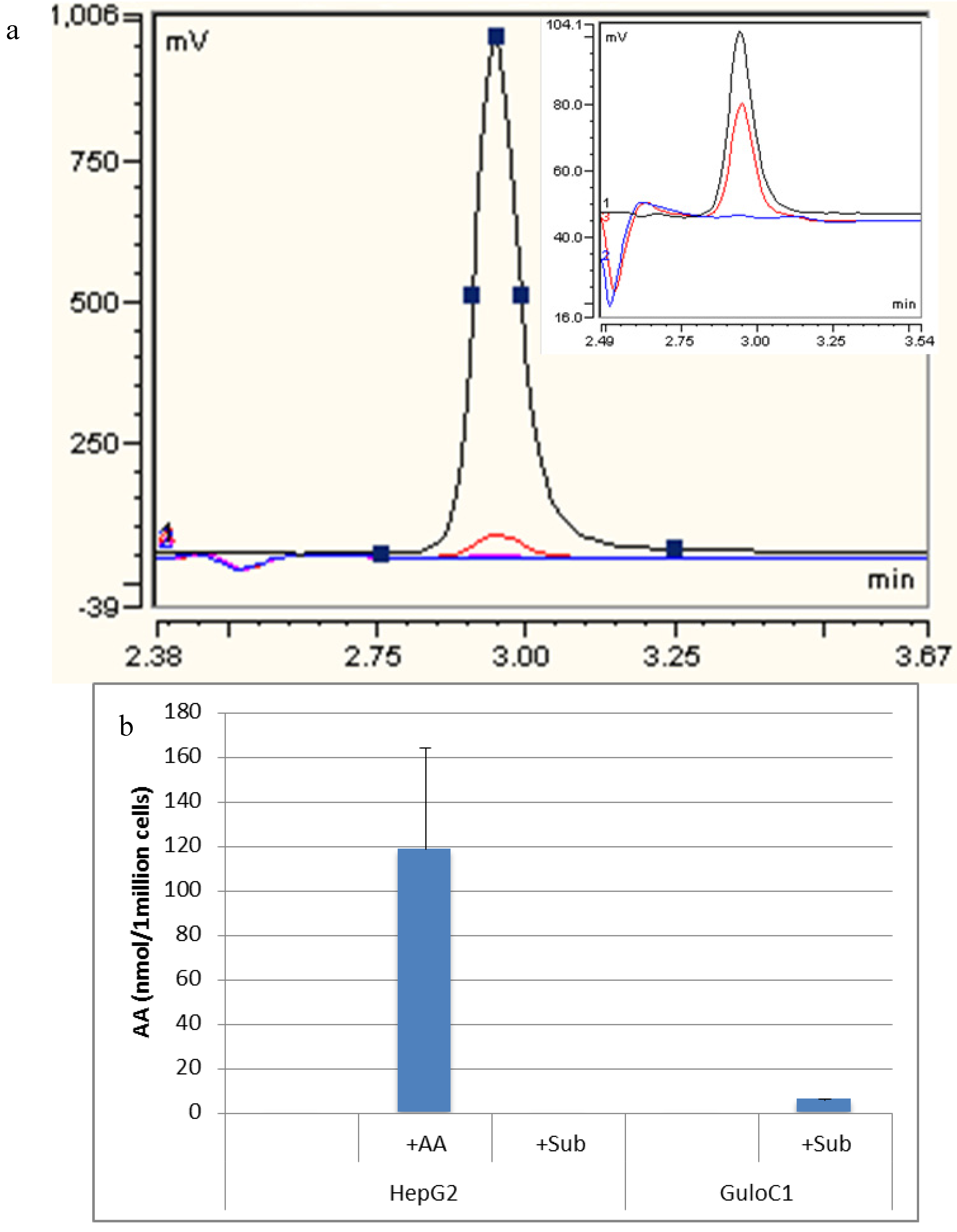
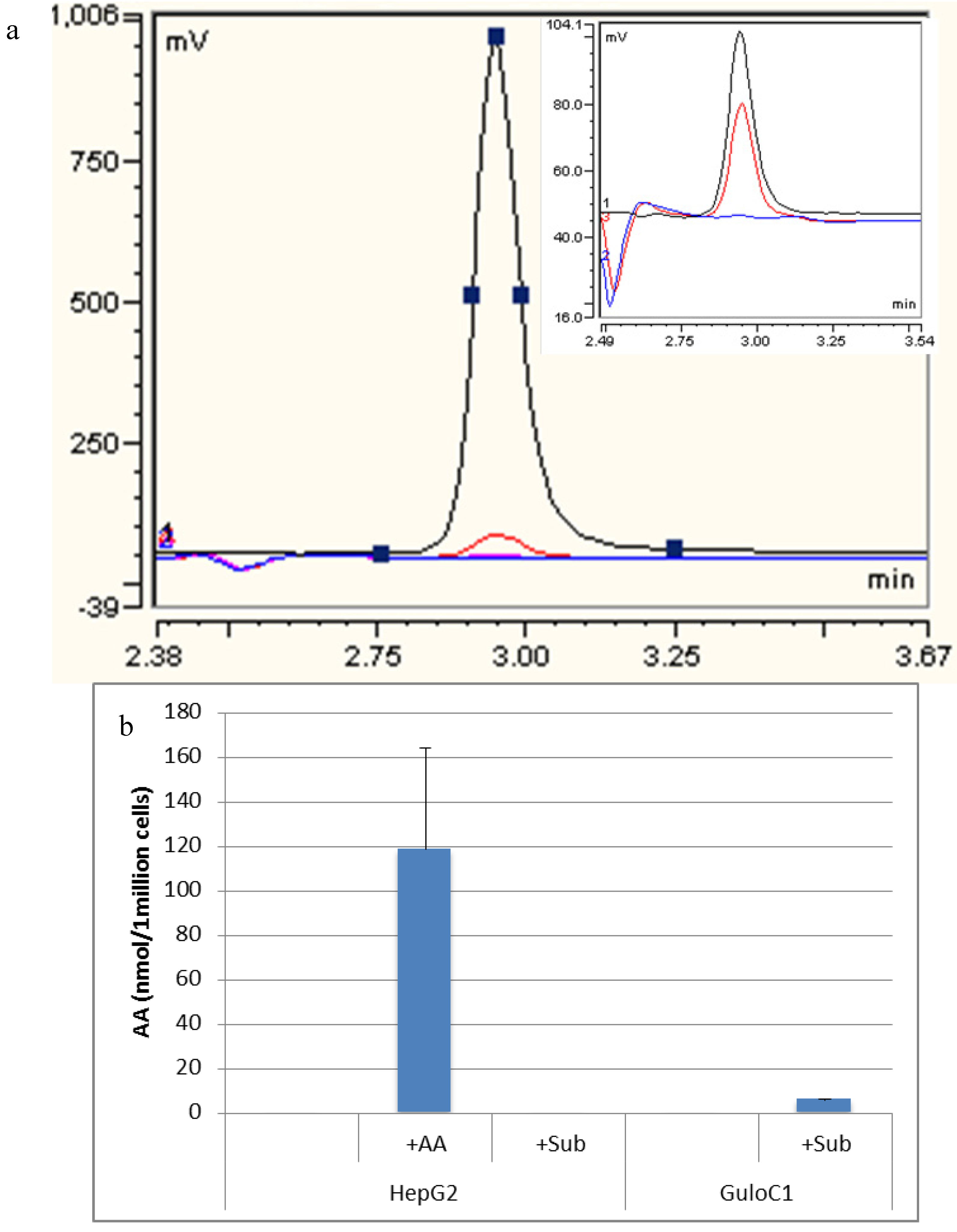
2.4. Ascorbate Synthesis
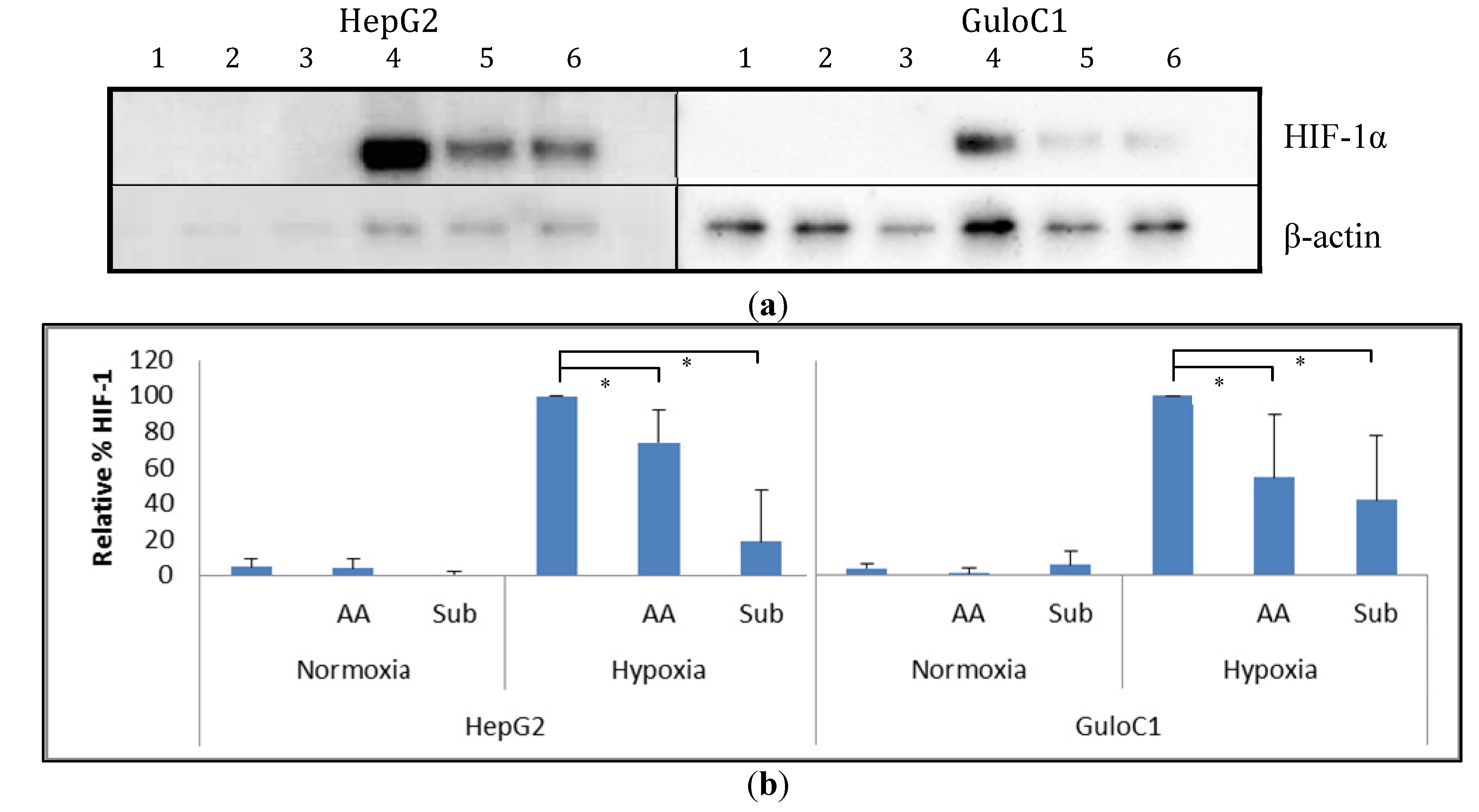
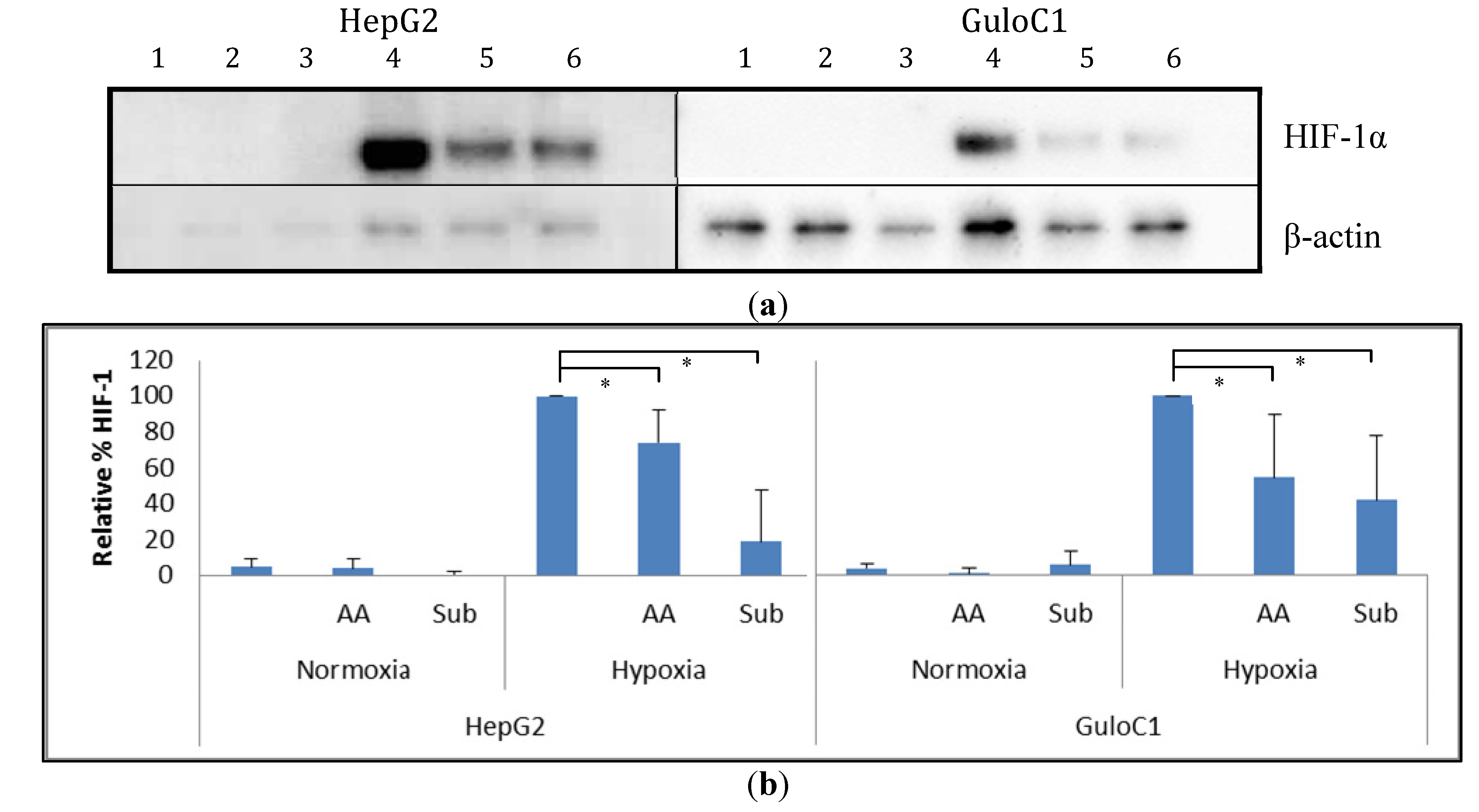
2.5. Hypoxia Inducible Factor 1 Levels
3. Experimental Section
3.1. Cells and Cell Culture
3.2. Hypoxic Conditions
3.3. Transfection
3.4. PCR Analysis
3.5. Western Blot Analysis
3.6. Ascorbate and Gulonolactone Loading
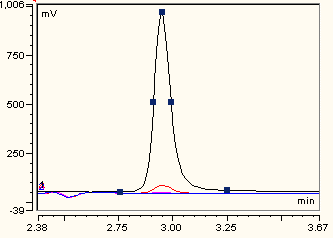
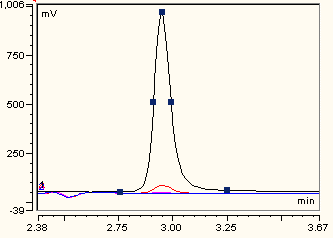
3.7. HPLC Analysis
3.8. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mandl, J.; Szarka, A.; Bánhegyi, G. Vitamin C: Update on physiology and pharmacology. Br. J. Pharmacol. 2009, 157, 1097–1110. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr.; Ratcliffe, P.J. Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef]
- Semenza, G.L. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 2010, 29, 625–634. [Google Scholar] [CrossRef]
- Maxwell, P.H.; Dachs, G.U.; Gleadle, J.M.; Nicholls, L.G.; Harris, A.L.; Stratford, I.J.; Hankinson, O.; Pugh, C.W.; Ratcliffe, P.J. Hypoxia-inducible factor-1 modulates gene expression in solid tumors and influences both angiogenesis and tumor growth. Proc. Natl. Acad. Sci. USA 1997, 94, 8104–8109. [Google Scholar] [CrossRef]
- Dachs, G.U.; Tozer, G.M. Hypoxia modulated gene expression: Angiogenesis, metastasis and therapeutic exploitation. Eur. J. Cancer 2000, 36, 1649–1660. [Google Scholar] [CrossRef]
- Giaccia, A.; Siim, B.G.; Johnson, R.S. HIF-1 as a target for drug development. Nat. Rev. Drug Discov. 2003, 2, 803–811. [Google Scholar] [CrossRef]
- Kuiper, C.; Molenaar, I.G.; Dachs, G.U.; Currie, M.J.; Sykes, P.H.; Vissers, M.C. Low ascorbate levels are associated with increased hypoxia-inducible factor-1 activity and an aggressive tumor phenotype in endometrial cancer. Cancer Res. 2010, 70, 5749–5758. [Google Scholar] [CrossRef]
- Kuiper, C.; Dachs, G.U.; Munn, D.; Currie, M.J.; Robinson, B.A.; Pearson, J.F.; Vissers, M.C.M. Increased tumor ascorbate is associated with extended disease-free survival and decreased hypoxia-inducible factor-1 activation in human colorectal cancer. Front. Oncol. 2014, 4, 1–10. [Google Scholar]
- Vissers, M.C.; Gunningham, S.P.; Morrison, M.J.; Dachs, G.U.; Currie, M.J. Modulation of hypoxia-inducible factor-1 alpha in cultured primary cells by intracellular ascorbate. Free Radic. Biol. Med. 2007, 42, 765–772. [Google Scholar] [CrossRef]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef]
- Gilkes, D.M.; Bajpai, S.; Wong, C.C.; Chaturvedi, P.; Hubbi, M.E.; Wirtz, D.; Semenza, G.L. Procollagen lysyl hydroxylase 2 is essential for hypoxia-induced breast cancer metastasis. Mol. Cancer Res. 2013, 11, 456–466. [Google Scholar] [CrossRef]
- Linster, C.L.; van Schaftingen, E. Vitamin C. Biosynthesis, recycling and degradation in mammals. FEBS J. 2007, 274, 1–22. [Google Scholar]
- Gabbay, K.H.; Bohren, K.M.; Morello, R.; Bertin, T.; Liu, J.; Vogel, P. Ascorbate synthesis pathway: Dual role of ascorbate in bone homeostasis. J. Biol. Chem. 2010, 285, 19510–19520. [Google Scholar]
- Corti, A.; Casini, A.F.; Pompella, A. Cellular pathways for transport and efflux of ascorbate and dehydroascorbate. Arch. Biochem. Biophys. 2010, 500, 107–115. [Google Scholar] [CrossRef]
- Ha, M.N.; Graham, F.L.; D’Souza, C.K.; Muller, W.J.; Igdoura, S.A.; Schellhorn, H.E. Functional rescue of vitamin C synthesis deficiency in human cells using adenoviral-based expression of murine l-gulono-gamma-lactone oxidase. Genomics 2004, 83, 482–492. [Google Scholar] [CrossRef]
- Li, Y.; Shi, C.X.; Mossman, K.L.; Rosenfeld, J.; Boo, Y.C.; Schellhorn, H.E. Restoration of vitamin C synthesis in transgenic Gulo−/− mice by helper-dependent adenovirus-based expression of gulonolactone oxidase. Hum. Gene Ther. 2008, 19, 1349–1358. [Google Scholar] [CrossRef]
- Nishikimi, M.; Fukuyama, R.; Minoshima, S.; Shimizu, N.; Yagi, K. Cloning and chromosomal mapping of the human nonfunctional gene for l-gulono-gamma-lactone oxidase, the enzyme for l-ascorbic acid biosynthesis missing in man. J. Biol. Chem. 1994, 269, 13685–13688. [Google Scholar]
- Murine gulonolactone oxidase-encoding gene. Available online: http://www.ncbi.nlm.nih.gov/nucleotide/372099096?report=gbwithparts&from=65987566&to=66009047&RID=AYZAPXT201R (accessed on 17 December 2013).
- Karp, S.; Ciambra, C.M.; Miklean, S. High-performance liquid chromatographic post-column reaction system for the electrochemical detection of ascorbic acid and dehydroascorbic acid. J. Chromatogr. 1990, 504, 434–440. [Google Scholar] [CrossRef]
- Philips, N.; Dulaj, L.; Upadhya, T. Cancer cell growth and extracellular matrix remodeling mechanism of ascorbate; beneficial modulation by P. leucotomos. Anticancer Res. 2009, 29, 3233–3238. [Google Scholar]
- Flashman, E.; Davies, S.L.; Yeoh, K.K.; Schofield, C.J. Investigating the dependence of the hypoxia-inducible factor hydroxylases (factor inhibiting HIF and prolyl hydroxylase domain 2) on ascorbate and other reducing agents. Biochem. J. 2010, 427, 135–142. [Google Scholar] [CrossRef]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Flett, T.; Campbell, E.J.; Phillips, E.; Vissers, M.C.M.; Dachs, G.U. Gulonolactone Addition to Human Hepatocellular Carcinoma Cells with Gene Transfer of Gulonolactone Oxidase Restores Ascorbate Biosynthesis and Reduces Hypoxia Inducible Factor 1. Biomedicines 2014, 2, 98-109. https://doi.org/10.3390/biomedicines2010098
Flett T, Campbell EJ, Phillips E, Vissers MCM, Dachs GU. Gulonolactone Addition to Human Hepatocellular Carcinoma Cells with Gene Transfer of Gulonolactone Oxidase Restores Ascorbate Biosynthesis and Reduces Hypoxia Inducible Factor 1. Biomedicines. 2014; 2(1):98-109. https://doi.org/10.3390/biomedicines2010098
Chicago/Turabian StyleFlett, Teresa, Elizabeth J. Campbell, Elisabeth Phillips, Margreet C. M. Vissers, and Gabi U. Dachs. 2014. "Gulonolactone Addition to Human Hepatocellular Carcinoma Cells with Gene Transfer of Gulonolactone Oxidase Restores Ascorbate Biosynthesis and Reduces Hypoxia Inducible Factor 1" Biomedicines 2, no. 1: 98-109. https://doi.org/10.3390/biomedicines2010098
APA StyleFlett, T., Campbell, E. J., Phillips, E., Vissers, M. C. M., & Dachs, G. U. (2014). Gulonolactone Addition to Human Hepatocellular Carcinoma Cells with Gene Transfer of Gulonolactone Oxidase Restores Ascorbate Biosynthesis and Reduces Hypoxia Inducible Factor 1. Biomedicines, 2(1), 98-109. https://doi.org/10.3390/biomedicines2010098