
Combined Genetic and Transcriptional Study Unveils the Role of DGAT1 Gene Mutations in Congenital Diarrhea


Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Evaluation
2.2. Whole-Exome Sequencing and Analysis
2.3. RNA Sequencing and Data Analysis
2.4. Literature Review of Reported DGAT1 Variant Cases
3. Results
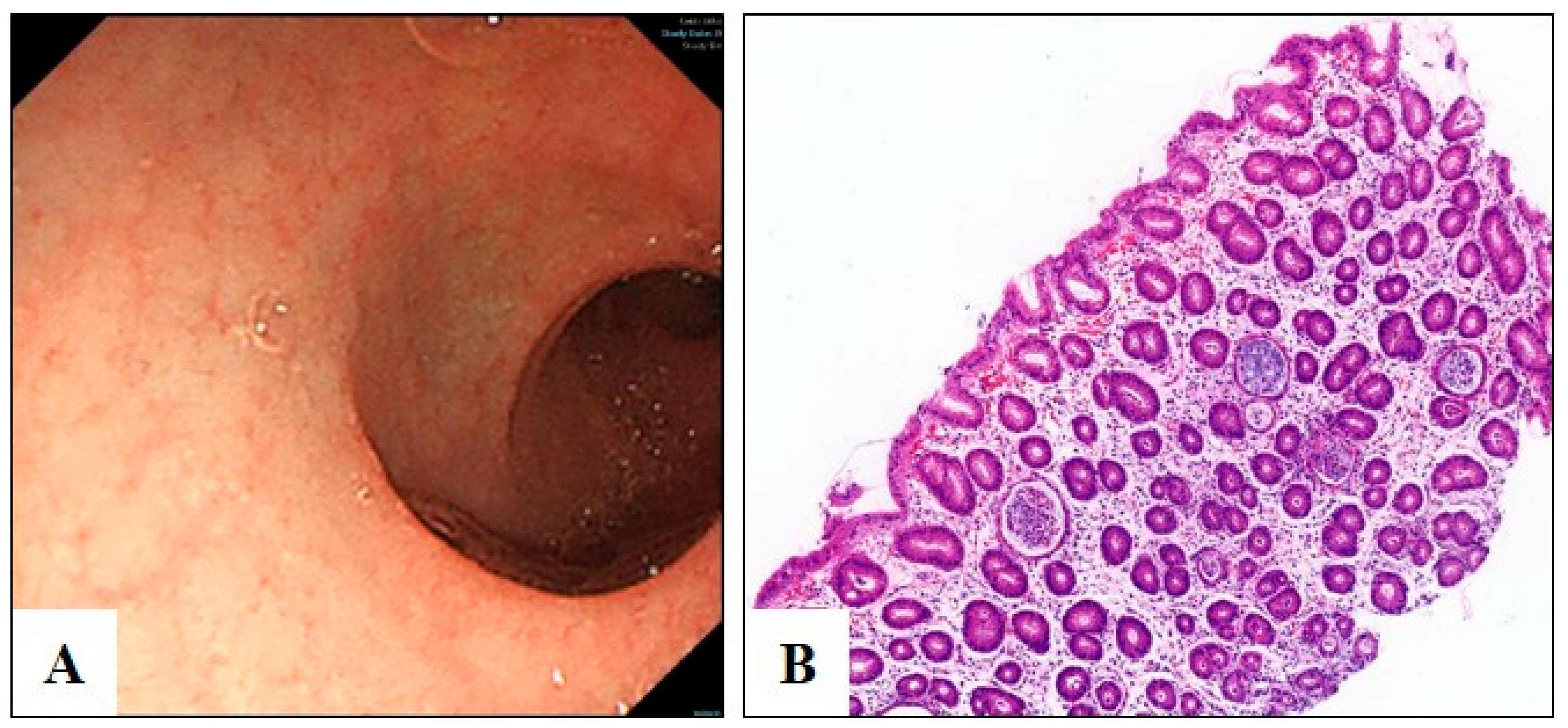
3.1. Clinical Diagnosis of Congenital Diarrhea
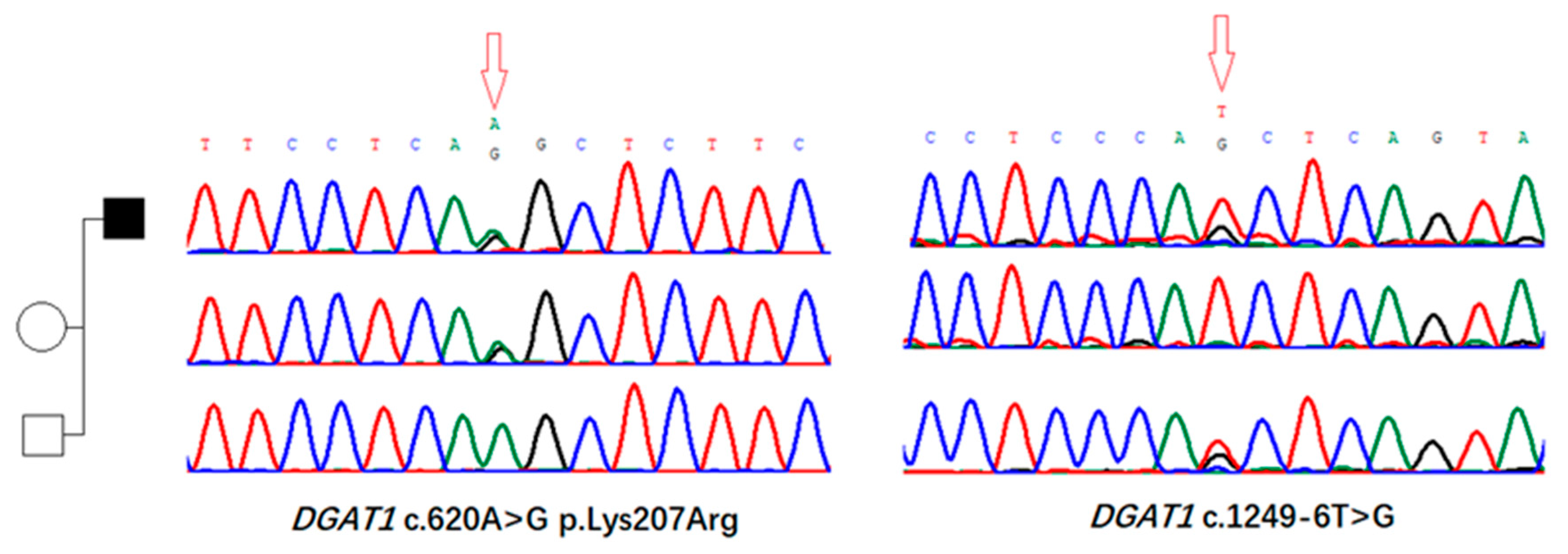
3.2. Identification of Genomic Variants in DGAT1 Gene
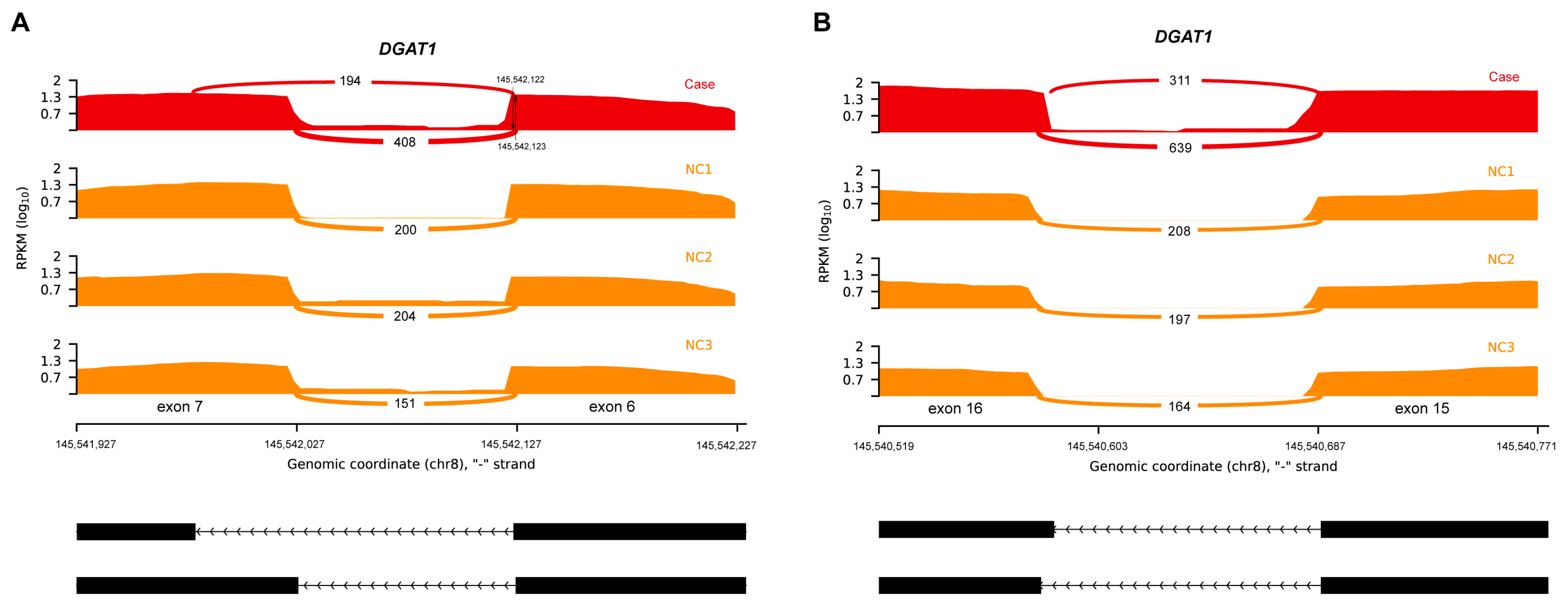
3.3. Aberrant Splicing Event Revealed by RNA Sequencing
3.4. Review of Reported DGAT1 Cases
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cases, S.; Smith, S.J.; Zheng, Y.W.; Myers, H.M.; Lear, S.R.; Sande, E.; Novak, S.; Collins, C.; Welch, C.B.; Lusis, A.J.; et al. Identification of a gene encoding an acyl CoA:diacylglycerol acyltransferase, a key enzyme in triacylglycerol synthesis. Proc. Natl. Acad. Sci. USA 1998, 95, 13018–13023. [Google Scholar] [CrossRef] [PubMed]
- Haas, J.T.; Winter, H.S.; Lim, E.; Kirby, A.; Blumenstiel, B.; DeFelice, M.; Gabriel, S.; Jalas, C.; Branski, D.; Grueter, C.A.; et al. DGAT1 mutation is linked to a congenital diarrheal disorder. J. Clin. Investig. 2012, 122, 4680–4684. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.-L.E.; Stone, S.J.; Koliwad, S.; Harris, C.; Farese, R.V., Jr. Thematic review series: Glycerolipids. DGAT enzyme and triacylglycerol biosynthesis. J. Lipid Res. 2008, 49, 2283–2301. [Google Scholar] [CrossRef] [PubMed]
- Yao, R.; Yang, F.; Zhang, Q.; Yu, T.; Yu, Y.; Chang, G.; Wang, X. Clinical and molecular characterization of a patient with generalized arterial calcification of infancy caused by rare ABCC6 mutation. J. Pers. Med. 2023, 14, 54. [Google Scholar] [CrossRef] [PubMed]
- Talevich, E.; Shain, A.H.; Botton, T.; Bastian, B.C. CNVkit: Genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput. Biol. 2016, 12, e1004873. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Gu, W.; Luo, Y.; Lou, J.; Chen, J. DGAT1 mutations leading to delayed chronic diarrhoea: A case report. BMC Med. Genet. 2020, 21, 239. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Huang, Y.; Wang, Y.; Lu, J.; Wu, J.; Yu, Z. Phenotype and genotype of a cohort of Chinese children with early-onset protein-losing enteropathy. J. Pediatr. 2019, 208, 38–42.e3. [Google Scholar] [CrossRef] [PubMed]
- van Rijn, J.M.; Ardy, R.C.; Kuloğlu, Z.; Härter, B.; van Haaften-Visser, D.Y.; van der Doef, H.P.; van Hoesel, M.; Kansu, A.; van Vugt, A.H.; Thian, M.; et al. Intestinal failure and aberrant lipid metabolism in patients with DGAT1 deficiency. Gastroenterology 2018, 155, 130–143.e15. [Google Scholar] [CrossRef] [PubMed]
- Gluchowski, N.L.; Chitraju, C.; Picoraro, J.A.; Mejhert, N.; Pinto, S.; Xin, W.; Kamin, D.S.; Winter, H.S.; Chung, W.K.; Walther, T.C.; et al. Identification and characterization of a novel DGAT1 missense mutation associated with congenital diarrhea. J. Lipid Res. 2017, 58, 1230–1237. [Google Scholar] [CrossRef] [PubMed]
- Schlegel, C.; Lapierre, L.A.; Weis, V.G.; Williams, J.A.; Kaji, I.; Pinzon-Guzman, C.; Prasad, N.; Boone, B.; Jones, A.; Correa, H.; et al. Reversible deficits in apical transporter trafficking associated with deficiency in diacylglycerol acyltransferase. Traffic 2018, 19, 879–892. [Google Scholar] [CrossRef] [PubMed]
- Stephen, J.; Vilboux, T.; Haberman, Y.; Pri-Chen, H.; Pode-Shakked, B.; Mazaheri, S.; Marek-Yagel, D.; Barel, O.; Di Segni, A.; Eyal, E.; et al. Congenital protein losing enteropathy: An inborn error of lipid metabolism due to DGAT1 mutations. Eur. J. Hum. Genet. 2016, 24, 1268–1273. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Dsouza, N.R.; Zarate, Y.A.; Lombardo, R.; Hopkin, R.; Linehan, A.R.; Simpson, J.; McCarrier, J.; Agre, K.E.; Gavrilova, R.H.; et al. Genetic variants in DGAT1 cause diverse clinical presentations of malnutrition through a specific molecular mechanism. Eur. J. Med. Genet. 2020, 63, 103817. [Google Scholar] [CrossRef] [PubMed]
- Ratchford, T.L.; Kirby, A.J.; Pinz, H.; Patel, D.R. Congenital diarrhea from DGAT1 mutation leading to electrolyte derangements, protein-losing enteropathy, and rickets. J. Pediatr. Gastroenterol. Nutr. 2018, 66, e82–e83. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Fang, Y.H.; Luo, Y.Y.; Lou, J.G.; Chen, J. Congenital diarrhea disorders caused by diacylglycerol acyl transferase 1 gene mutation. Chin. J. Pediatr. 2020, 58, 1018–1020. [Google Scholar]
- Li, S.; Zhao, S.; Sinson, J.C.; Bajic, A.; Rosenfeld, J.A.; Neeley, M.B.; Pena, M.; Worley, K.C.; Burrage, L.C.; Weisz-Hubshman, M.; et al. The clinical utility and diagnostic implementation of human subject cell transdifferentiation followed by RNA sequencing. Am. J. Hum. Genet. 2024, 111, 841–862. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Patient No. | Variants | Phenotype | Onset Age | Reference |
|---|---|---|---|---|
| 1–2 | c.895-1G>A; | vomiting, PLE, malnutrition, watery diarrhea, hypertriglyceridemia | N/A | Ye et al., 2019 [7] |
| 3 | c.1249-6T>G | early-onset PLE, CDD, malnutrition, hypoalbuminemia, lymphopenia, edema | N/A | Ye et al., 2019 [7] |
| 4 | c.895-1G>A;c.751+1G>C | vomiting, PLE, malnutrition, watery diarrhea, hypertriglyceridemia | 8 months | Xu et al., 2020 [6] |
| 5–6 | c.314T>C (p.Leu105Pro) | early-onset vomiting and/or diarrhea, hypoalbuminemia, PLE | 19 months/ 26 months | van Rijn et al., 2018 [8]; Gluchowski et al., 2017 [9] |
| 7 | c.1202G>A (p.W401*) | failure to thrive, vomiting, diarrhea, hypoalbuminemia, hypogammaglobulinemia, edema | 5 months | van Rijn et al., 2018 [8] |
| 8 | c.573_574delinsCCCATCCCCCCTCGCCCATCT, p.Val192Profs*99) | failure to thrive, vomiting, diarrhea, hypoalbuminemia, hypogammaglobulinemia, edema | 11 months | van Rijn et al., 2018 [8] |
| 9 | c.937-1G>A | failure to thrive, vomiting, diarrhea, hypoalbuminemia, hypogammaglobulinemia | 8 years | van Rijn et al., 2018 [8] |
| 10 | c.953insG (p.Ile319Hisfs*33) | failure to thrive, vomiting, bloody and watery diarrhea, hypoalbuminemia, hypogammaglobulinemia | 2 years | van Rijn et al., 2018 [8] |
| 11 | c.629_631delCCT (p.Ser210del) | severe vomiting, diarrhea | 20 months | van Rijn et al., 2018 [8] |
| 12 | c.751+2T>C | vomiting, CDD, PLE, malnutrition, electrolyte abnormalities, TPN dependence | 7 weeks | Schlegel et al., 2018 [10] |
| 13 | c.884T>C (p.Leu295Pro) | vomiting, CDD, PLE, malnutrition | 2 months | Stephen et al., 2016 [11] |
| 14 | c.629_631delCCT (p.Ser210del) | vomiting, diarrhea, severe malnutrition, hypotonia | 2 weeks | Gupta et al., 2020 [12] |
| 15 | c.676+1G>A | vomiting, diarrhea, vitamin D deficiency | N/A | Gupta et al., 2020 [12] |
| 16 | c1013_1015delTCT (p.Phe338del) | severe diarrhea, PLE, fat-soluble vitamin deficiency, secondary hyperparathyroidism | 2 months | Ratchford et al., 2017 [13] |
| c.1260c>G (p.Ser420Arg) | ||||
| 17 | c.1310A>G (p.Gln437Arg) | vomiting, growth failure, vitamin D deficiency | N/A | Gupta et al., 2020 [12] |
| c.981+1G>T | ||||
| 18 | c.1311+1G>A | vomiting, growth failure, hypogammaglobulinemia, TPN dependence | N/A | Gupta et al., 2020 [12] |
| c.1462delG (p.Ala488Profs*226) | ||||
| 19 | c.676+1G>T | vomiting, diarrhea, malnutrition | 1 month | Chen et al., 2020 [14] |
| c.367_368delCT | ||||
| Our case | c.620A>G, p.Lys207Arg | vomiting, watery diarrhea, hypotonia, severe malnutrition, TPN dependence | 4 months | |
| c.1249-6T>G |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, J.; Ma, J.; Wang, L.; Deng, Z.; Yao, R. Combined Genetic and Transcriptional Study Unveils the Role of DGAT1 Gene Mutations in Congenital Diarrhea. Biomedicines 2025, 13, 1897. https://doi.org/10.3390/biomedicines13081897
Zeng J, Ma J, Wang L, Deng Z, Yao R. Combined Genetic and Transcriptional Study Unveils the Role of DGAT1 Gene Mutations in Congenital Diarrhea. Biomedicines. 2025; 13(8):1897. https://doi.org/10.3390/biomedicines13081897
Chicago/Turabian StyleZeng, Jingqing, Jing Ma, Lan Wang, Zhaohui Deng, and Ruen Yao. 2025. "Combined Genetic and Transcriptional Study Unveils the Role of DGAT1 Gene Mutations in Congenital Diarrhea" Biomedicines 13, no. 8: 1897. https://doi.org/10.3390/biomedicines13081897
APA StyleZeng, J., Ma, J., Wang, L., Deng, Z., & Yao, R. (2025). Combined Genetic and Transcriptional Study Unveils the Role of DGAT1 Gene Mutations in Congenital Diarrhea. Biomedicines, 13(8), 1897. https://doi.org/10.3390/biomedicines13081897