
Genetic Landscape of Congenital Cataracts in a Swiss Cohort: Addressing Diagnostic Oversights in Nance–Horan Syndrome

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Subjects
2.2. Target Genes
2.3. Exome Sequencing and Data Analysis
2.4. Segregation
3. Results

{kind=link}
| Subjects | Gene | cDNA | Predicted AA Change | Ex/Int | Zygosity | Inheritance | GnomAD | ACMG | PolyPhen2 | Segregation | First Publication | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| f1: | 2-2♂ 2-3♂ | CRYBA1 | NM_005208.5: c.272_274del | p.Gly91del | ex4 | het | ad | N/A | P | N/A | Maternal (affected) | Qi et al. (2003) [38] |
| f2 | 2-1♀ 2-2♂ | MAF | NM_005360.5: c.880C>T | p.Arg294 Trp | ex1 | het | ad | N/A | P | 1.0 | Maternal (affected) | Sun et al. (2014) [39] |
| f3 | 2-1♀ | EPHA2 | NM_004431.5: c.2826-9G>A | p.? | int16 | het | ad | N/A | P | N/A | Maternal (affected) | Zhang et al. (2009) [37] |
| f4 | 3-1♀ 3-2♀ | GJA8 | NM_005267.5: c.592C>T | p.Arg198 Trp | ex2 | het | ad | N/A | P | 1.0 | Maternal (affected) | Hu et al. (2010) [40] |
| f5 | 2-3♂ | CRYGC | NM_020989.4: c.471G>A | p.Trp157Ter | ex3 | het | ad | N/A | P | N/A | De novo | Guo et al. (2012) [41] |
| f6 | 2-2♂ | MIP | NM_012064.4: c.97C>T | p.Arg33Cys | ex1 | het | ad | N/A | P | 0.982 | De novo | Gu et al. (2007) [42] |
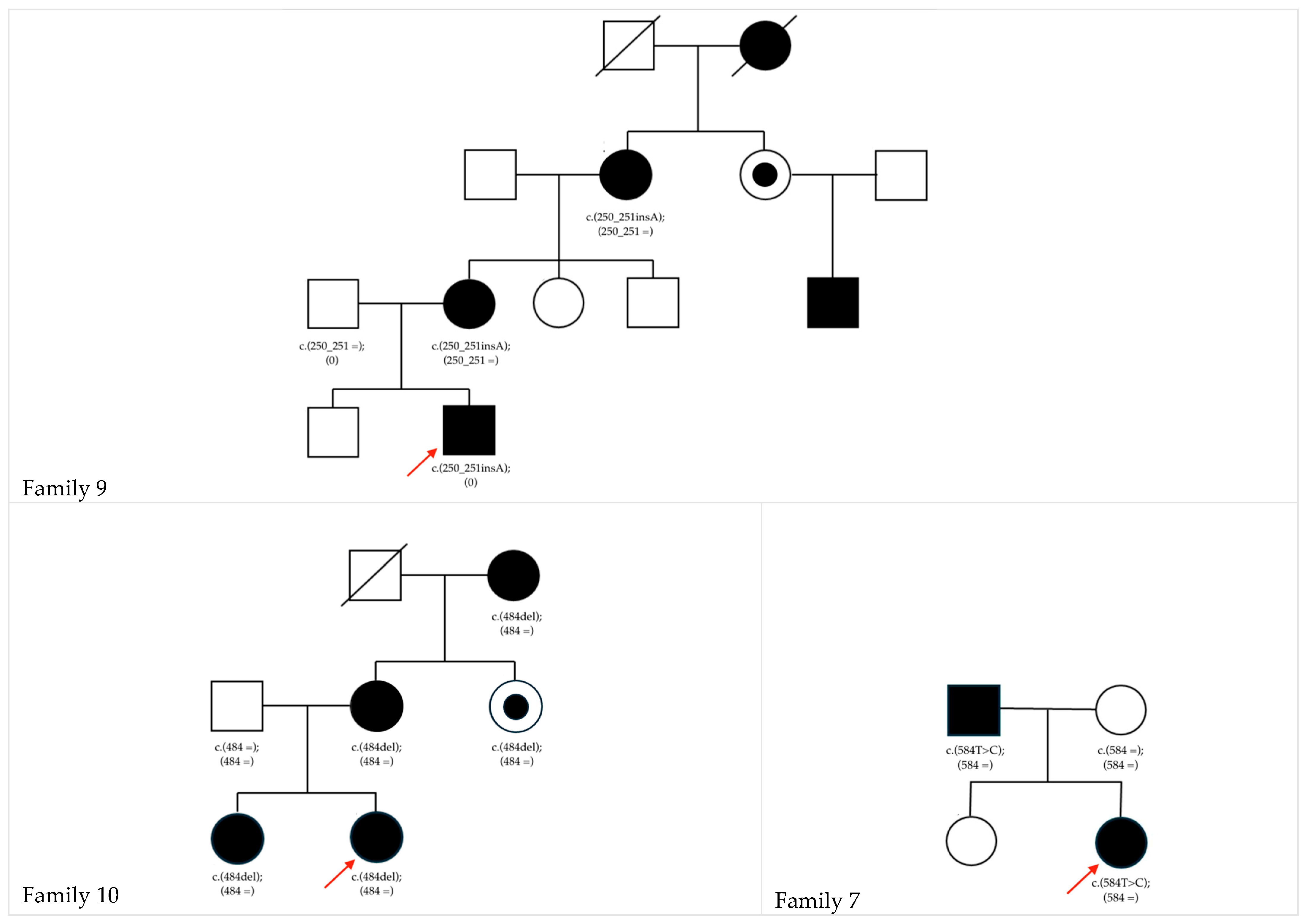
| f7 | 2-2♀ | GJA8 | NM_005267.5: c.584T>C | p.Phe195Ser | ex2 | het | ad | N/A | LP | 0.999 | Paternal (affected) | This study |
| f8 | 2-1♀ | CRYAA | NM_000394.4: c.347G>A | p.Arg116His | ex3 | het | ad | N/A | P | 1.0 | De novo | Hansen et al. (2007) [43] |
| f9 | 4-2♂ | NHS | NM_001291867.2:c.250_251insA | p.Pro84His fsTer99 | ex1 | hemi | xld | N/A | LP | N/A | Maternal (affected) | This study |
| f10 | 3-1♀ 3-2♀ | NHS | NM_001291867.2:c.484del | p.Arg162Ala fsTer34 | ex1 | het | xld | N/A | LP | N/A | Maternal (affected) | This study |
Genotype–Phenotype Comparison and Clinical Case Presentation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fan, F.; Luo, Y.; Wu, J.; Gao, C.; Liu, X.; Mei, H.; Zhou, X. The mutation spectrum in familial versus sporadic congenital cataract based on next-generation sequencing. BMC Ophthalmol. 2020, 20, 361. [Google Scholar] [CrossRef]
- Sheeladevi, S.; Lawrenson, J.G.; Fielder, A.R.; Suttle, C.M. Global prevalence of childhood cataract: A systematic review. Eye 2016, 30, 1160–1169. [Google Scholar] [CrossRef]
- Messina-Baas, O.; Cuevas-Covarrubias, S.A. Inherited Congenital Cataract: A Guide to Suspect the Genetic Etiology in the Cataract Genesis. Mol. Syndromol. 2017, 8, 58–78. [Google Scholar] [CrossRef]
- Jain, I.S.; Pillay, P.; Gangwar, D.N.; Dhir, S.P.; Kaul, V.K. Congenital cataract: Etiology and morphology. J. Pediatr. Ophthalmol. Strabismus 1983, 20, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.A.; Francis, P.J.; Berry, V.; Bhattacharya, S.S.; Moore, A.T. Molecular genetic basis of inherited cataract and associated phenotypes. Surv. Ophthalmol. 2004, 49, 300–315. [Google Scholar] [CrossRef] [PubMed]
- Delas, F.; Koller, S.; Feil, S.; Dacheva, I.; Gerth-Kahlert, C.; Berger, W. Novel CRYGC Mutation in Conserved Ultraviolet-Protective Tryptophan (p.Trp131Arg) Is Linked to Autosomal Dominant Congenital Cataract. Int. J. Mol. Sci. 2023, 24, 16594. [Google Scholar] [CrossRef]
- Berry, V.; Ionides, A.; Pontikos, N.; Moghul, I.; Moore, A.T.; Quinlan, R.A.; Michaelides, M. Whole exome sequencing reveals novel and recurrent disease-causing variants in lens specific gap junctional protein encoding genes causing congenital cataract. Genes 2020, 11, 512. [Google Scholar] [CrossRef]
- Shiels, A.; Hejtmancik, J.F. Molecular Genetics of Cataract. Prog. Mol. Biol. Transl. Sci. 2015, 134, 203–218. [Google Scholar] [CrossRef]
- Redwood, A.; Douzgou, S.; Waller, S.; Ramsden, S.; Roberts, A.; Bonin, H.; Lloyd, I.C.; Ashworth, J.; Black, G.C.M.; Clayton-Smith, J. Congenital cataracts in females caused by BCOR mutations; report of six further families demonstrating clinical variability and diverse genetic mechanisms. Eur. J. Med. Genet. 2020, 63, 103658. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.A.; Nussbaum, R.L.; Brewer, E.D. Lowe Syndrome; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Mickler, C.; Boden, J.; Trivedi, R.H.; Wilson, M.E. Pediatric cataract. Pediatr. Ann. 2011, 40, 83–87. [Google Scholar] [CrossRef]
- Berry, V.; Georgiou, M.; Fujinami, K.; Quinlan, R.; Moore, A.; Michaelides, M. Inherited cataracts: Molecular genetics, clinical features, disease mechanisms and novel therapeutic approaches. Br. J. Ophthalmol. 2020, 104, 1331–1337. [Google Scholar] [CrossRef]
- Wu, X.; Long, E.; Lin, H.; Liu, Y. Prevalence and epidemiological characteristics of congenital cataract: A systematic review and meta-analysis. Sci. Rep. 2016, 6, 28564. [Google Scholar] [CrossRef]
- Cassidy, L.; Taylor, D. Congenital cataract and multisystem disorders. Eye 1999, 13, 464–473. [Google Scholar] [CrossRef]
- Yi, J.; Yun, J.; Li, Z.K.; Xu, C.T.; Pan, B.R. Epidemiology and molecular genetics of congenital cataracts. Int. J. Ophthalmol. 2011, 4, 422–432. [Google Scholar] [CrossRef]
- Gómez-Laguna, L.; Martínez-Herrera, A.; del Pilar Reyes-de la Rosa, A.; García-Delgado, C.; Nieto-Martínez, K.; Fernández-Ramírez, F.; Valderrama-Atayupanqui, T.Y.; Morales-Jiménez, A.B.; Villa-Morales, J.; Kofman, S.; et al. Nance–Horan syndrome in females due to a balanced X;1 translocation that disrupts the NHS gene: Familial case report and review of the literature. Ophthalmic Genet. 2018, 39, 56–62. [Google Scholar] [CrossRef]
- Yu, X.; Zhao, Y.; Yang, Z.; Chen, X.; Kang, G. Genetic research on Nance-Horan syndrome caused by a novel mutation in the NHS gene. Gene 2024, 906, 148223. [Google Scholar] [CrossRef] [PubMed]
- Zin, O.A.; Neves, L.M.; Motta, F.L.; Junior, D.C.; Cunha, D.P.; Agonigi, B.N.S.; Malacarne, J.; Rodrigues, A.P.S.; Rodrigues, G.D.; Tinoco, M.L.C.; et al. Genotype–Phenotype Correlations of Nance–Horan Syndrome in Male and Female Carriers of a Novel Variant. Genes 2025, 16, 91. [Google Scholar] [CrossRef]
- Walpole, I.R.; Hockey, A.; Nicoll, A. The Nance-Horan syndrome supported by reports that affected subjects have long. Clin. Genet. 1990, 27, 632–634. [Google Scholar]
- Hernández, V.; Pascual-Camps, I.; Aparisi, M.J.; Martínez-Matilla, M.; Martínez, F.; Cerón, J.A.; Pedrola, L. Great clinical variability of Nance Horan syndrome due to deleterious NHS mutations in two unrelated Spanish families. Ophthalmic Genet. 2019, 40, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Burdon, K.P.; McKay, J.D.; Sale, M.M.; Russell-Eggitt, I.M.; Mackey, D.A.; Wirth, M.G.; Elder, J.E.; Nicoll, A.; Clarke, M.P.; FitzGerald, L.M.; et al. Mutations in a Novel Gene, NHS, Cause the Pleiotropic Effects of Nance-Horan Syndrome, Including Severe Congenital Cataract, Dental Anomalies, and Mental Retardation. Am. J. Hum. Genet. 2003, 73, 1120–1130. [Google Scholar] [CrossRef]
- Shiels, A. Through the Cat-Map Gateway: A Brief History of Cataract Genetics. Genes 2024, 15, 785. [Google Scholar] [CrossRef]
- Shiels, A.; Bennett, T.M.; Hejtmancik, J.F. Cat-Map: Putting cataract on the map. Mol. Vis. 2010, 16, 2007–2015. [Google Scholar]
- Prabu, R.V.; Priyambada, P.; Ranjini, H.; Wasnik, R.B. A case report of children of the same family presenting with congenital cataract- as part of a rare genetic disorder-Sengers Syndrome. Indian J. Ophthalmol. 2020, 68, 2567–2569. [Google Scholar] [CrossRef]
- Haghighi, A.; Haack, T.B.; Atiq, M.; Mottaghi, H.; Haghighi-Kakhki, H.; Bashir, R.A.; Ahting, U.; Feichtinger, R.G.; Mayr, J.A.; Rötig, A.; et al. Sengers syndrome: Six novel AGK mutations in seven new families and review of the phenotypic and mutational spectrum of 29 patients. Orphanet J. Rare Dis. 2014, 9, 119. [Google Scholar] [CrossRef]
- Li, W.; Liu, J.; Galvin, J.A. Epigenetics and Common Ophthalmic Diseases. Yale J. Biol. Med. 2016, 89, 597–600. [Google Scholar] [PubMed]
- Wang, Y.; Guan, H. The Role of DNA Methylation in Lens Development and Cataract Formation. Cell. Mol. Neurobiol. 2017, 37, 979–984. [Google Scholar] [CrossRef] [PubMed]
- Bari, K.J.; Sharma, S.; Chary, K.V.R. Structure of G57W mutant of human γS-crystallin and its involvement in cataract formation. J. Struct. Biol. 2019, 205, 72–78. [Google Scholar] [CrossRef]
- Li, J.; Chen, X.; Yan, Y.; Yao, K. Molecular genetics of congenital cataracts. Exp. Eye Res. 2020, 191, 107872. [Google Scholar] [CrossRef] [PubMed]
- Rechsteiner, D.; Issler, L.; Koller, S.; Lang, E.; Bähr, L.; Feil, S.; Rüegger, C.M.; Kottke, R.; Toelle, S.P.; Zweifel, N.; et al. Genetic Analysis in a Swiss Cohort of Bilateral Congenital Cataract. JAMA Ophthalmol. 2021, 139, 691–700. [Google Scholar] [CrossRef]
- Rabbani, B.; Tekin, M.; Mahdieh, N. The promise of whole-exome sequencing in medical genetics. J. Hum. Genet. 2014, 59, 5–15. [Google Scholar] [CrossRef]
- Bamshad, M.J.; Ng, S.B.; Bigham, A.W.; Tabor, H.K.; Emond, M.J.; Nickerson, D.A.; Shendure, J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 2011, 12, 745–755. [Google Scholar] [CrossRef]
- Jackson, D.; Malka, S.; Harding, P.; Palma, J.; Dunbar, H.; Moosajee, M. Molecular diagnostic challenges for non-retinal developmental eye disorders in the United Kingdom. Am. J. Med. Genet. Part C Semin. Med. Genet. 2020, 184, 578–589. [Google Scholar] [CrossRef]
- Association, W.M. World Medical Association Declaration of Helsinki: Ethical Principles for Medical Research Involving Human Subjects. JAMA 2013, 310, 2191–2194. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Haug, P.; Koller, S.; Maggi, J.; Lang, E.; Feil, S.; Wlodarczyk, A.; Bähr, L.; Steindl, K.; Rohrbach, M.; Gerth-Kahlert, C.; et al. Whole exome sequencing in coloboma/microphthalmia: Identification of novel and recurrent variants in seven genes. Genes 2021, 12, 65. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Hua, R.; Xiao, W.; Burdon, K.P.; Bhattacharya, S.S.; Craig, J.E.; Shang, D.; Zhao, X.; Mackey, D.A.; Moore, A.T.; et al. Mutations of the EPHA2 receptor tyrosine kinase gene cause autosomal dominant congenital cataract. Hum. Mutat. 2009, 30, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Jia, H.; Huang, S.; Lin, H.; Gu, J.; Su, H.; Zhang, T.; Gao, Y.; Qu, L.; Li, D.; et al. A deletion mutation in the betaA1/A3 crystallin gene (CRYBA1/A3) is associated with autosomal dominant congenital nuclear cataract in a Chinese family. Hum. Genet. 2004, 114, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Xiao, X.; Li, S.; Guo, X.; Zhang, Q. Exome sequencing of 18 Chinese families with congenital cataracts: A new sight of the NHS gene. PLoS ONE 2014, 9, e100455. [Google Scholar] [CrossRef]
- Hu, S.; Wang, B.; Zhou, Z.; Zhou, G.; Wang, J.; Ma, X.; Qi, Y. A novel mutation in GJA8 causing congenital cataract-microcornea syndrome in a Chinese pedigree. Mol. Vis. 2010, 16, 1585–1592. [Google Scholar]
- Guo, Y.; Su, D.; Li, Q.; Yang, Z.; Ma, Z.; Ma, X.; Zhu, S. A nonsense mutation of CRYGC associated with autosomal dominant congenital nuclear cataracts and microcornea in a Chinese pedigree. Mol. Vis. 2012, 18, 1874–1880. [Google Scholar]
- Gu, F.; Zhai, H.; Li, D.; Zhao, L.; Li, C.; Huang, S.; Ma, X. A novel mutation in major intrinsic protein of the lens gene (MIP) underlies autosomal dominant cataract in a Chinese family. Mol. Vis. 2007, 13, 1651–1666. [Google Scholar] [PubMed]
- Hansen, L.; Yao, W.; Eiberg, H.; Kjaer, K.W.; Baggesen, K.; Hejtmancik, J.F.; Rosenberg, T. Genetic heterogeneity in microcornea-cataract: Five novel mutations in CRYAA, CRYGD, and GJA8. Investig. Ophthalmol. Vis. Sci. 2007, 48, 3937–3944. [Google Scholar] [CrossRef]
- Bennett, R.L.; French, K.S.; Resta, R.G.; Austin, J. Practice resource-focused revision: Standardized pedigree nomenclature update centered on sex and gender inclusivity: A practice resource of the National Society of Genetic Counselors. J. Genet. Couns. 2022, 31, 1238–1248. [Google Scholar] [CrossRef]
- Rossen, J.L.; Bohnsack, B.L.; Zhang, K.X.; Ing, A.; Drackley, A.; Castelluccio, V.; Ralay-ranaivo, H. Evaluation of Genetic Testing in a Cohort of Diverse Pediatric Patients in the United States with Congenital Cataracts. Genes 2023, 14, 608. [Google Scholar] [CrossRef]
- Ling, C.; Sui, R.; Yao, F.; Wu, Z.; Zhang, X.; Zhang, S. Whole exome sequencing identified a novel truncation mutation in the NHS gene associated with Nance-Horan syndrome. BMC Med. Genet. 2019, 20, 14. [Google Scholar] [CrossRef]
- Toutain, A.; Dessay, B.; Ronce, N.; Ferrante, M.I.; Tranchemontagne, J.; Newbury-Ecob, R.; Wallgren-Pettersson, C.; Burn, J.; Kaplan, J.; Rossi, A.; et al. Refinement of the NHS locus on chromosome Xp22.13 and analysis of five candidate genes. Eur. J. Hum. Genet. 2002, 10, 516–520. [Google Scholar] [CrossRef] [PubMed]
- Chograni, M.; Rejeb, I.; Jemaa, L.B.; Chabouni, M.; Bouhamed, H.C. The first missense mutation of NHS gene in a Tunisian family with clinical features of NHS syndrome including cardiac anomaly. Eur. J. Hum. Genet. 2011, 19, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Coccia, M.; Brooks, S.P.; Webb, T.R.; Christodoulou, K.; Wozniak, I.O.; Murday, V.; Balicki, M.; Yee, H.A.; Wangensteen, T.; Riise, R.; et al. X-linked cataract and Nance-Horan syndrome are allelic disorders. Hum. Mol. Genet. 2009, 18, 2643–2655. [Google Scholar] [CrossRef]
- Li, H.; Yang, L.; Sun, Z.; Yuan, Z.; Wu, S.; Sui, R. A novel small deletion in the NHS gene associated with Nance-Horan syndrome. Sci. Rep. 2018, 8, 2398. [Google Scholar] [CrossRef]
- Accogli, A.; Traverso, M.; Madia, F.; Bellini, T.; Vari, M.S.; Pinto, F.; Capra, V. A novel Xp22.13 microdeletion in Nance-Horan syndrome. Birth Defects Res. 2017, 109, 866–868. [Google Scholar] [CrossRef]
- Shoshany, N.; Avni, I.; Morad, Y.; Weiner, C.; Einan-Lifshitz, A.; Pras, E. NHS Gene Mutations in Ashkenazi Jewish Families with Nance–Horan Syndrome. Curr. Eye Res. 2017, 42, 1240–1244. [Google Scholar] [CrossRef]
- Merepa, S.S.; Reis, L.M.; Damián, A.; Bardakjian, T.; Schneider, A.; Trujillo-tiebas, M.J.; Ayuso, C.; Galarza, L.C.; Villaverde, R.S.; Ortiz-cabrera, N.V.; et al. GJA8 -associated developmental eye disorders: A new multicentre study highlights mutational hotspots and genotype-phenotype correlations. Eur. J. Hum. Genet. 2025, 33, 860–869. [Google Scholar] [CrossRef]
- Ma, A.S.; Grigg, J.R.; Ho, G.; Prokudin, I.; Farnsworth, E.; Holman, K.; Cheng, A.; Billson, F.A.; Martin, F.; Fraser, C.; et al. Sporadic and Familial Congenital Cataracts: Mutational Spectrum and New Diagnoses Using Next-Generation Sequencing. Hum. Mutat. 2016, 37, 371–384. [Google Scholar] [CrossRef]
- Wen, H.; Li, Q.; Mei, S.; Cai, J.; Huang, X.; Zhao, J. A novel frameshift mutation in the NHS gene causes Nance-Horan syndrome in a Chinese family. Gene 2024, 907, 148268. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Leng, Y.; Han, S.; Yan, L.; Lu, C.; Luo, Y.; Zhang, X.; Cao, L. Clinical and genetic characteristics of Chinese patients with familial or sporadic pediatric cataract. Orphanet J. Rare Dis. 2018, 13, 94. [Google Scholar] [CrossRef]
- Huang, Y.; Ma, L.; Zhang, Z.; Nie, S.; Zhou, Y.; Zhang, J.; Wang, C.; Fang, X.; Quan, Y.; He, T.; et al. Nance–Horan syndrome pedigree due to a novel microdeletion and skewed X chromosome inactivation. Mol. Genet. Genom. Med. 2023, 11, e2100. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.; Gertsen, B.G.; Schroeder, A.L.; Fong, C.T.; Iqbal, M.A.; Zhang, B. Allelic and dosage effects of NHS in X-linked cataract and Nance–Horan syndrome: A family study and literature review. Mol. Cytogenet. 2021, 14, 48. [Google Scholar] [CrossRef]
- Żylicz, J.J.; Bousard, A.; Žumer, K.; Dossin, F.; Mohammad, E.; da Rocha, S.T.; Schwalb, B.; Syx, L.; Dingli, F.; Loew, D.; et al. The Implication of Early Chromatin Changes in X Chromosome Inactivation. Cell 2019, 176, 182–197. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Yu, H.; Wang, W.; Gong, X.; Yao, K. A novel GJA8 mutation (p.V44A) causing autosomal dominant congenital cataract. PLoS ONE 2014, 9, e115406. [Google Scholar] [CrossRef]
- Jones, J.L.; Burdon, K.P. Evaluating gap junction variants for a role in pediatric cataract: An overview of the genetic landscape and clinical classification of variants in the GJA3 and GJA8 genes. Expert Rev. Ophthalmol. 2023, 18, 71–95. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delas, F.; Gloggnitzer, J.; Maspoli, A.; Kurmann, L.; Frueh, B.E.; Dacheva, I.; Hildebrand, D.; Berger, W.; Gerth-Kahlert, C. Genetic Landscape of Congenital Cataracts in a Swiss Cohort: Addressing Diagnostic Oversights in Nance–Horan Syndrome. Biomedicines 2025, 13, 1883. https://doi.org/10.3390/biomedicines13081883
Delas F, Gloggnitzer J, Maspoli A, Kurmann L, Frueh BE, Dacheva I, Hildebrand D, Berger W, Gerth-Kahlert C. Genetic Landscape of Congenital Cataracts in a Swiss Cohort: Addressing Diagnostic Oversights in Nance–Horan Syndrome. Biomedicines. 2025; 13(8):1883. https://doi.org/10.3390/biomedicines13081883
Chicago/Turabian StyleDelas, Flora, Jiradet Gloggnitzer, Alessandro Maspoli, Lisa Kurmann, Beatrice E. Frueh, Ivanka Dacheva, Darius Hildebrand, Wolfgang Berger, and Christina Gerth-Kahlert. 2025. "Genetic Landscape of Congenital Cataracts in a Swiss Cohort: Addressing Diagnostic Oversights in Nance–Horan Syndrome" Biomedicines 13, no. 8: 1883. https://doi.org/10.3390/biomedicines13081883
APA StyleDelas, F., Gloggnitzer, J., Maspoli, A., Kurmann, L., Frueh, B. E., Dacheva, I., Hildebrand, D., Berger, W., & Gerth-Kahlert, C. (2025). Genetic Landscape of Congenital Cataracts in a Swiss Cohort: Addressing Diagnostic Oversights in Nance–Horan Syndrome. Biomedicines, 13(8), 1883. https://doi.org/10.3390/biomedicines13081883