

Chlorogenic Acid and Cinnamaldehyde in Breast Cancer Cells: Predictive Examination of Pharmacokinetics and Binding Thermodynamics with the Key Mediators of PI3K/Akt Signaling


Abstract
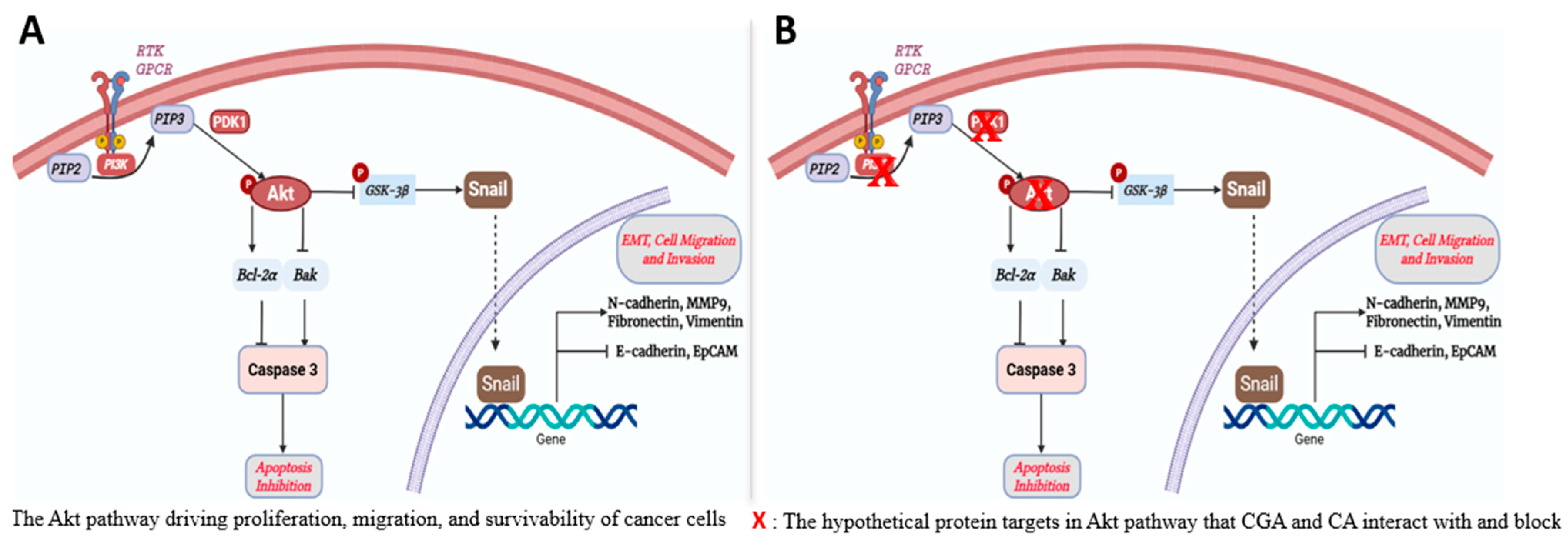
1. Introduction
2. Materials and Methods
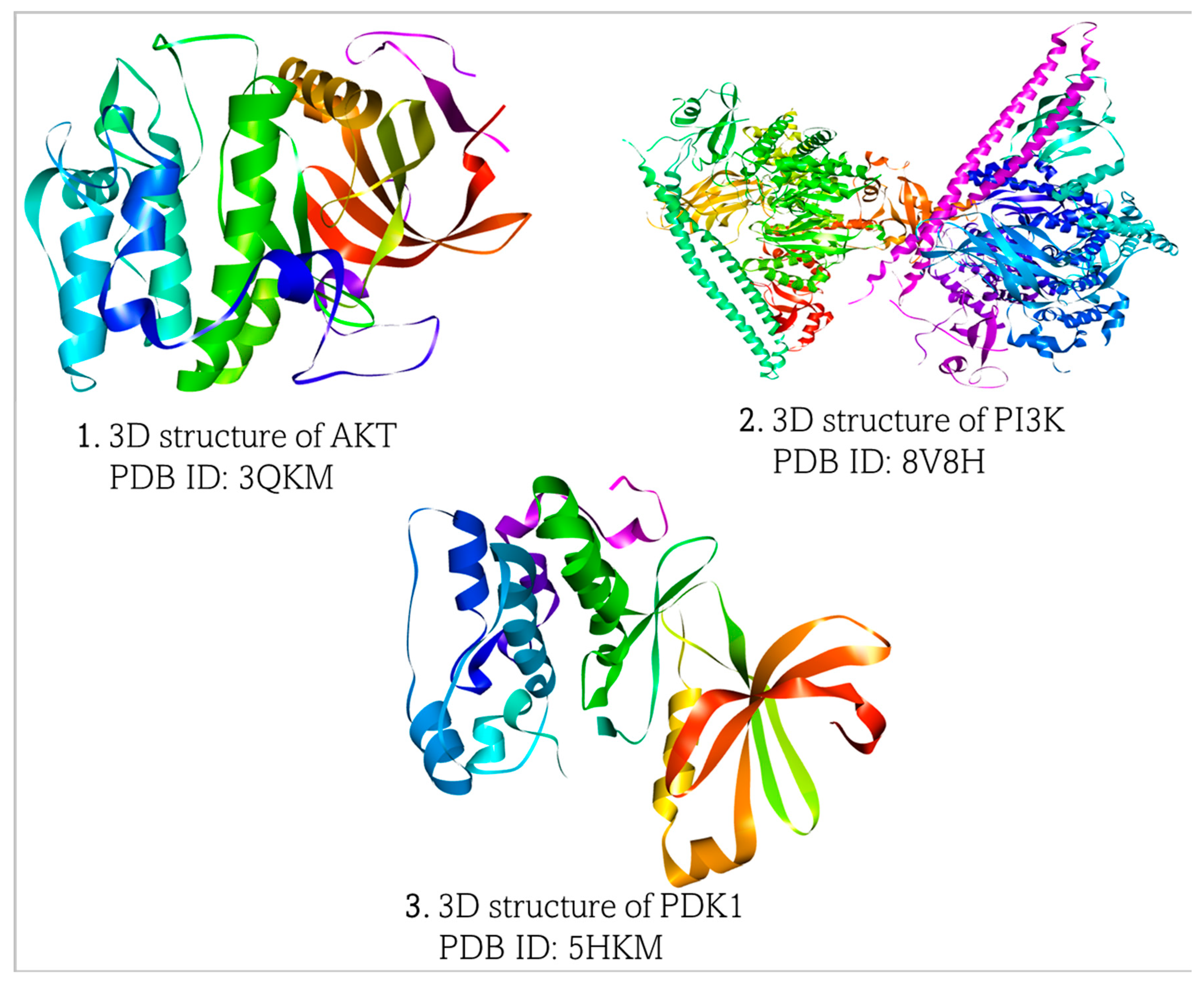
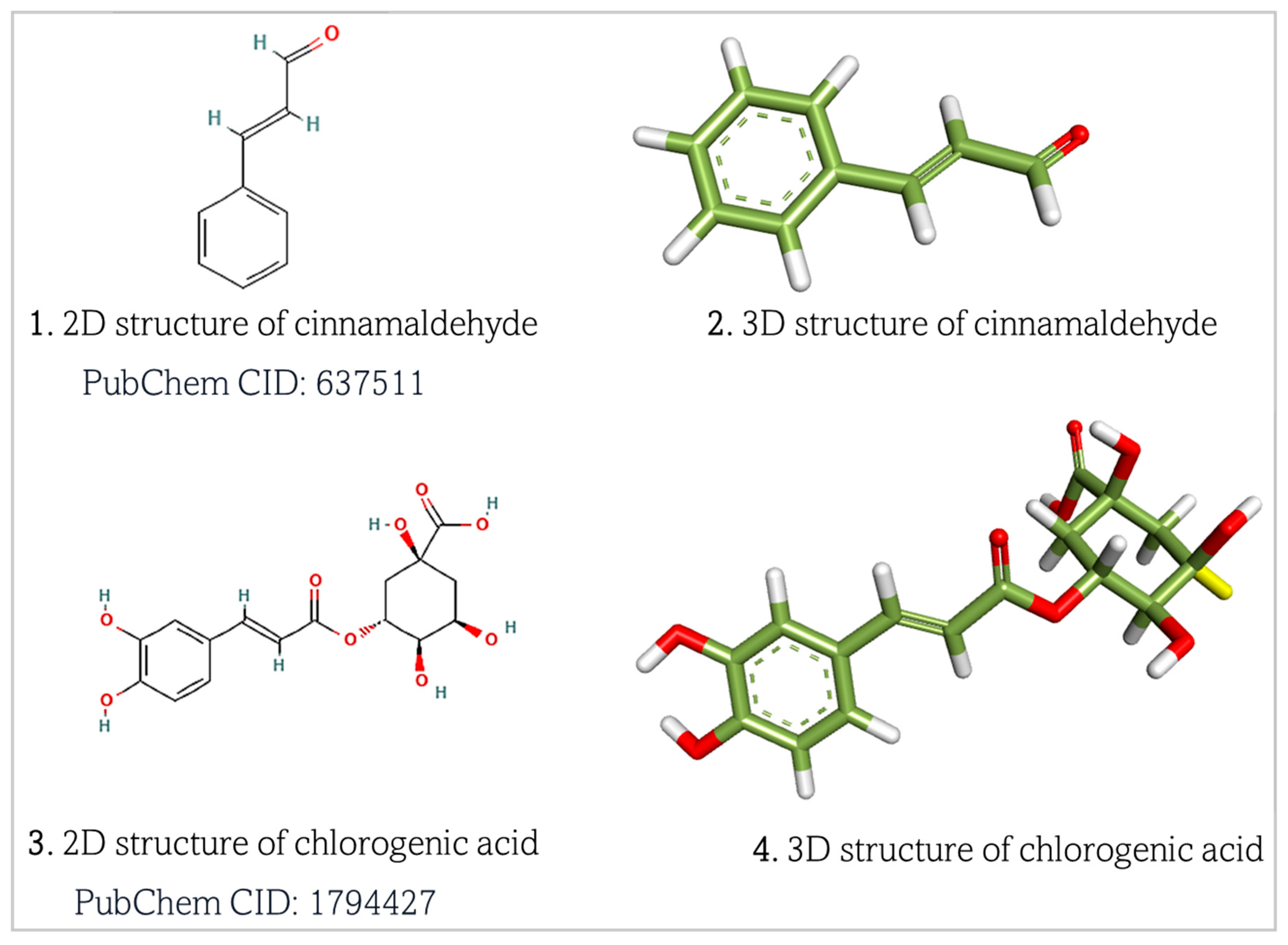
2.1. Retrieval and Preparation of Target Proteins and Ligands
2.2. Molecular Docking Approach
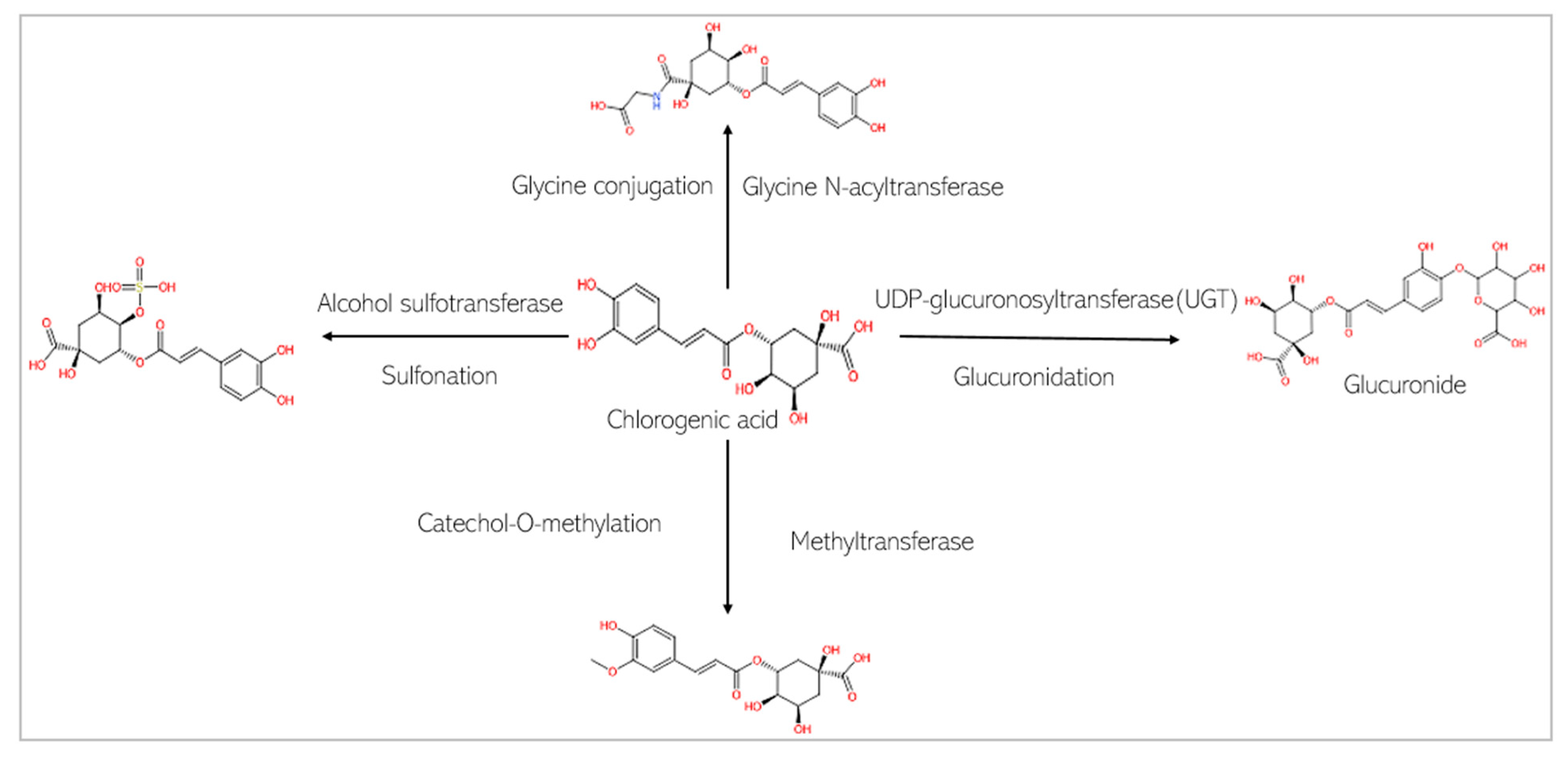
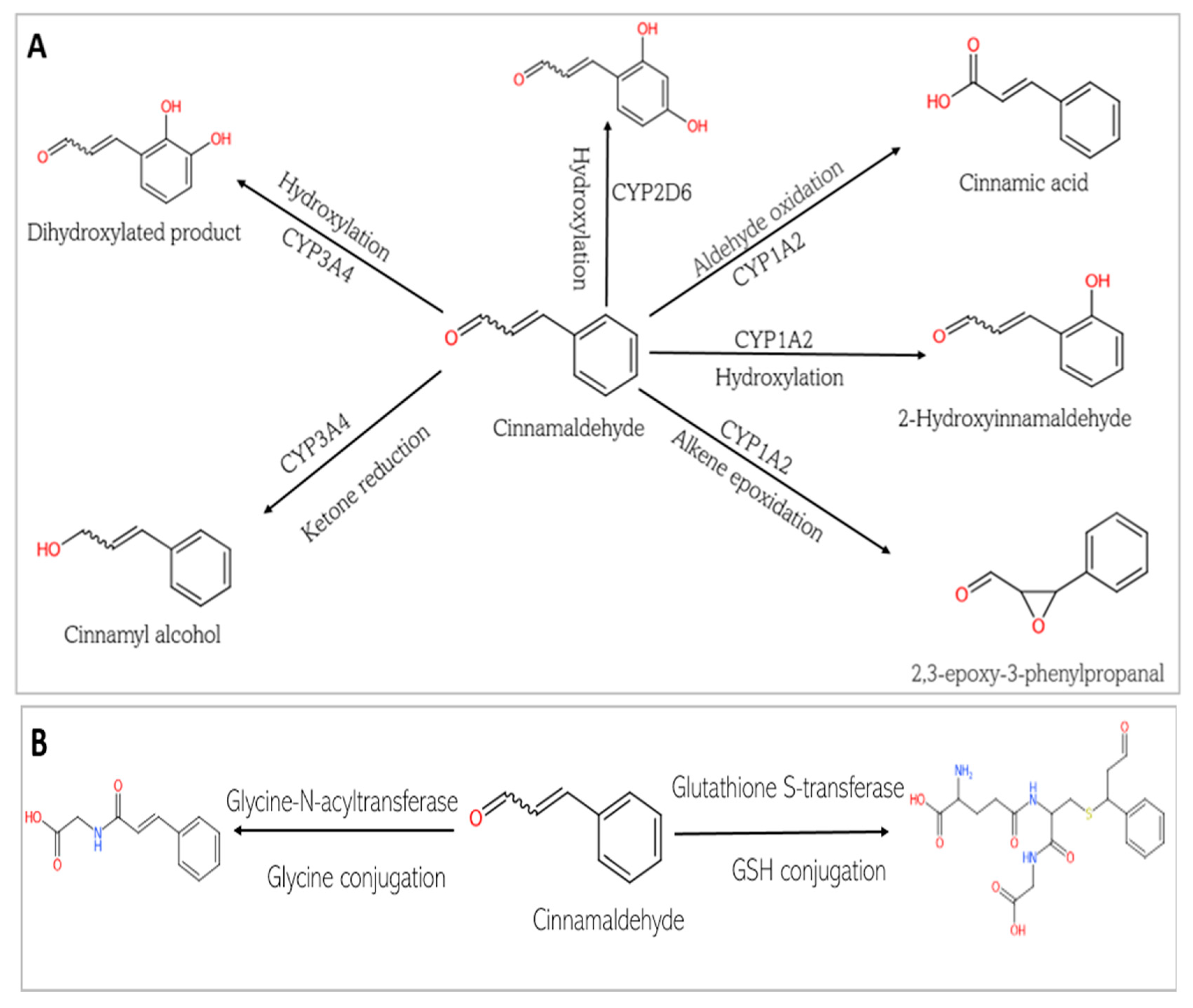
2.3. Approach to Pharmacokinetic Analysis of Chlorogenic Acid and Cinnamaldehyde
2.4. Drug-Likeness Analysis
3. Results
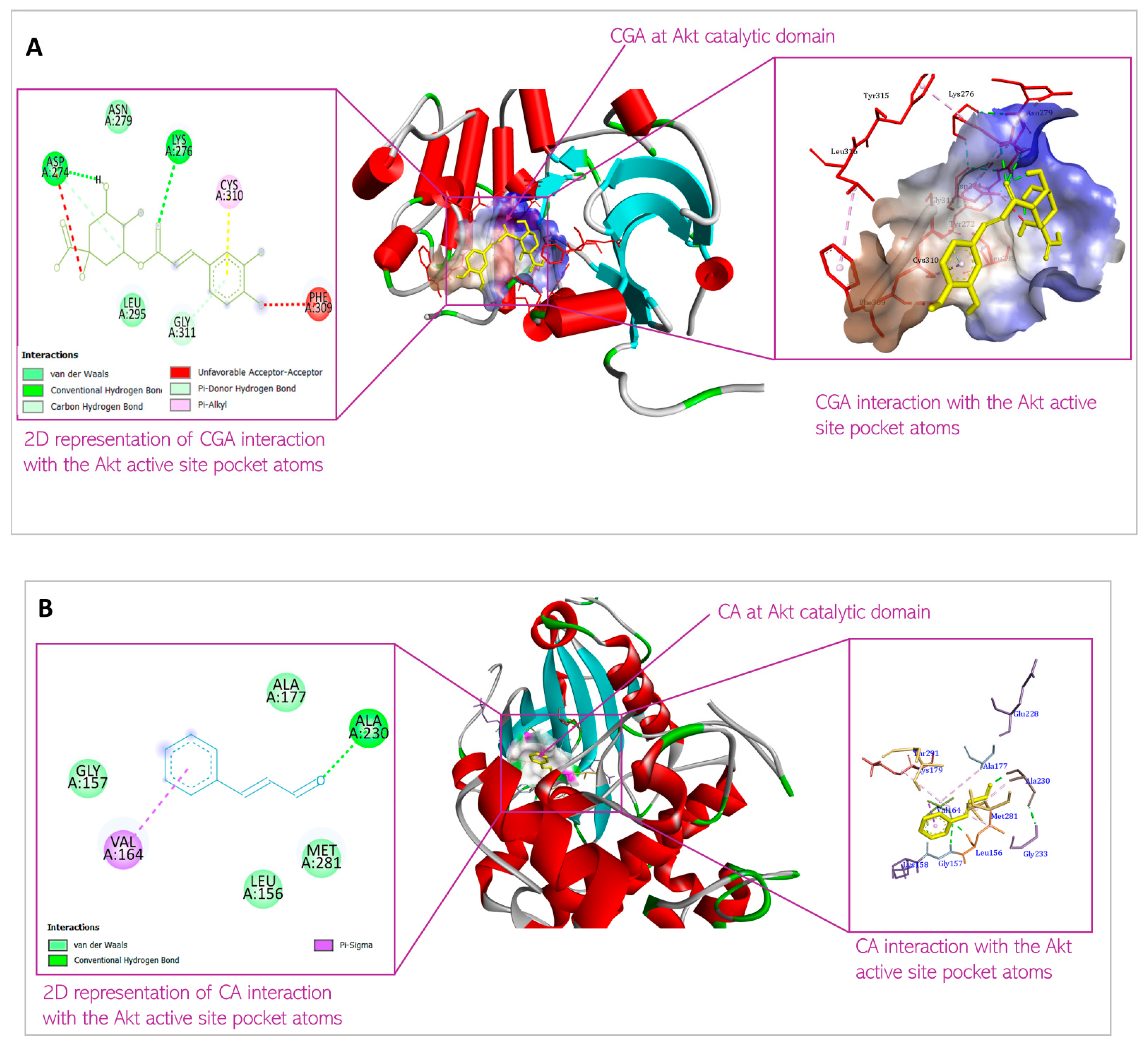
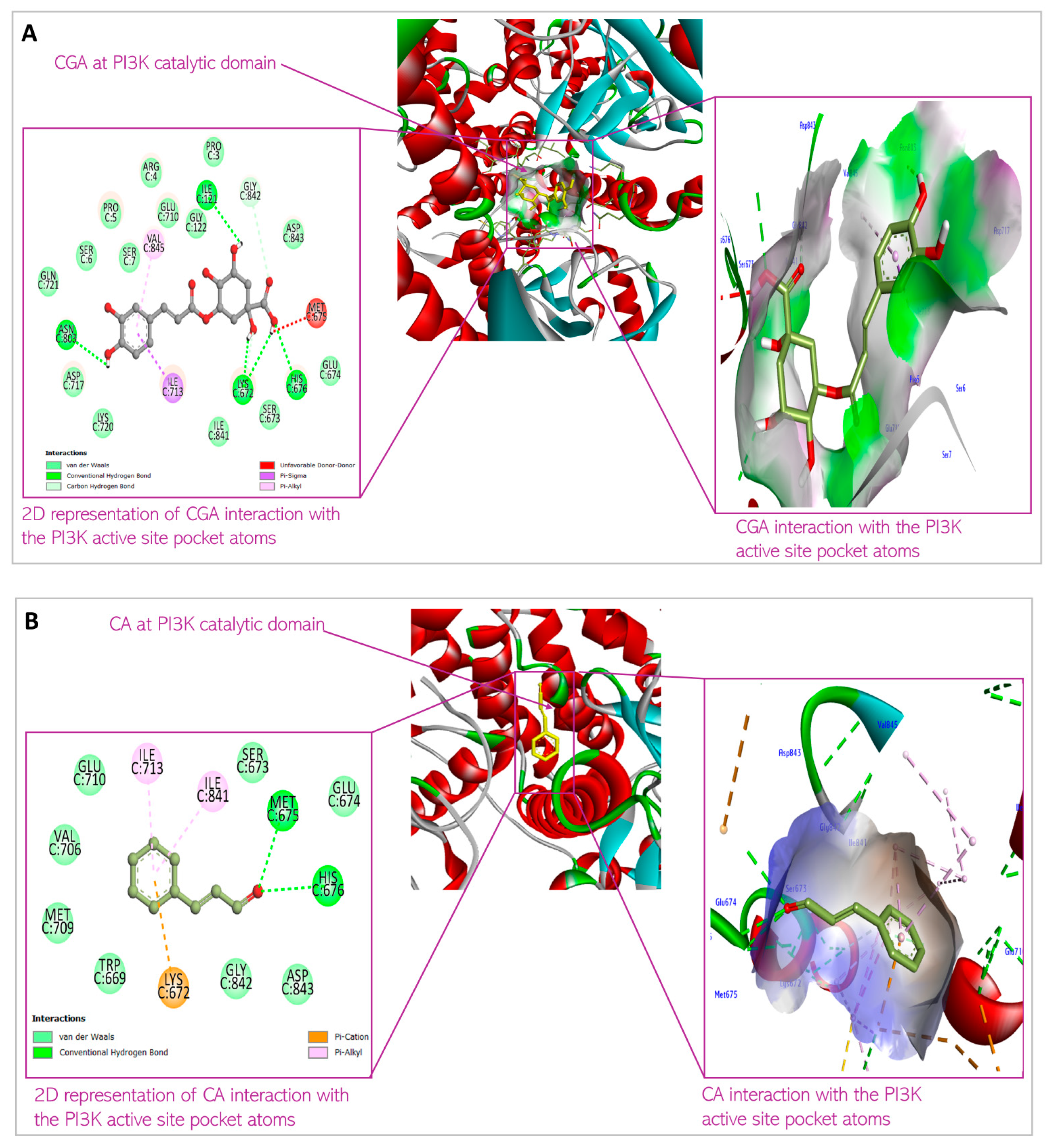
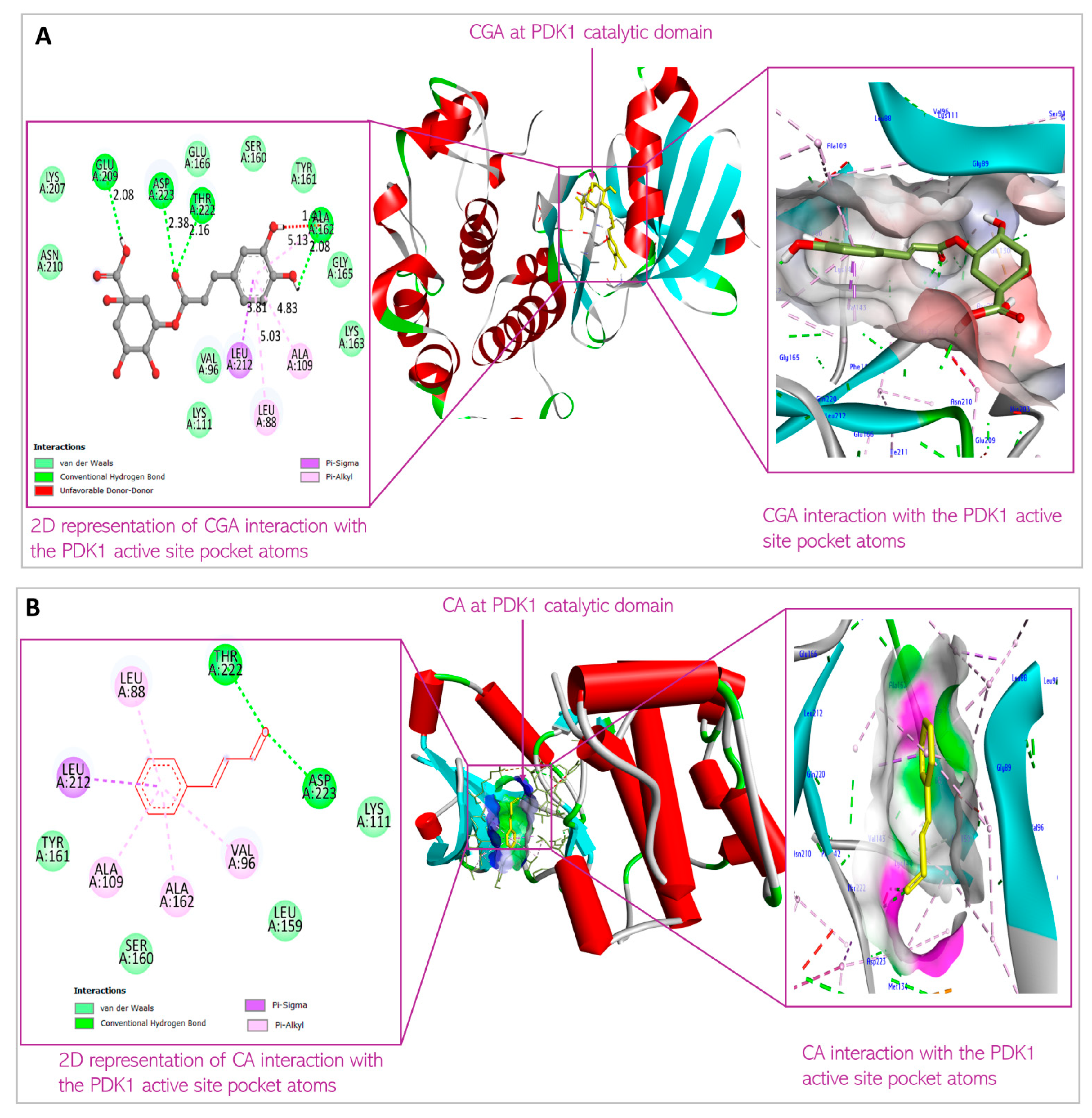
3.1. Predicted Molecular Docking Results
3.2. Predicted Pharmacokinetic Results for Chlorogenic Acid and Cinnamaldehyde
4. Discussion
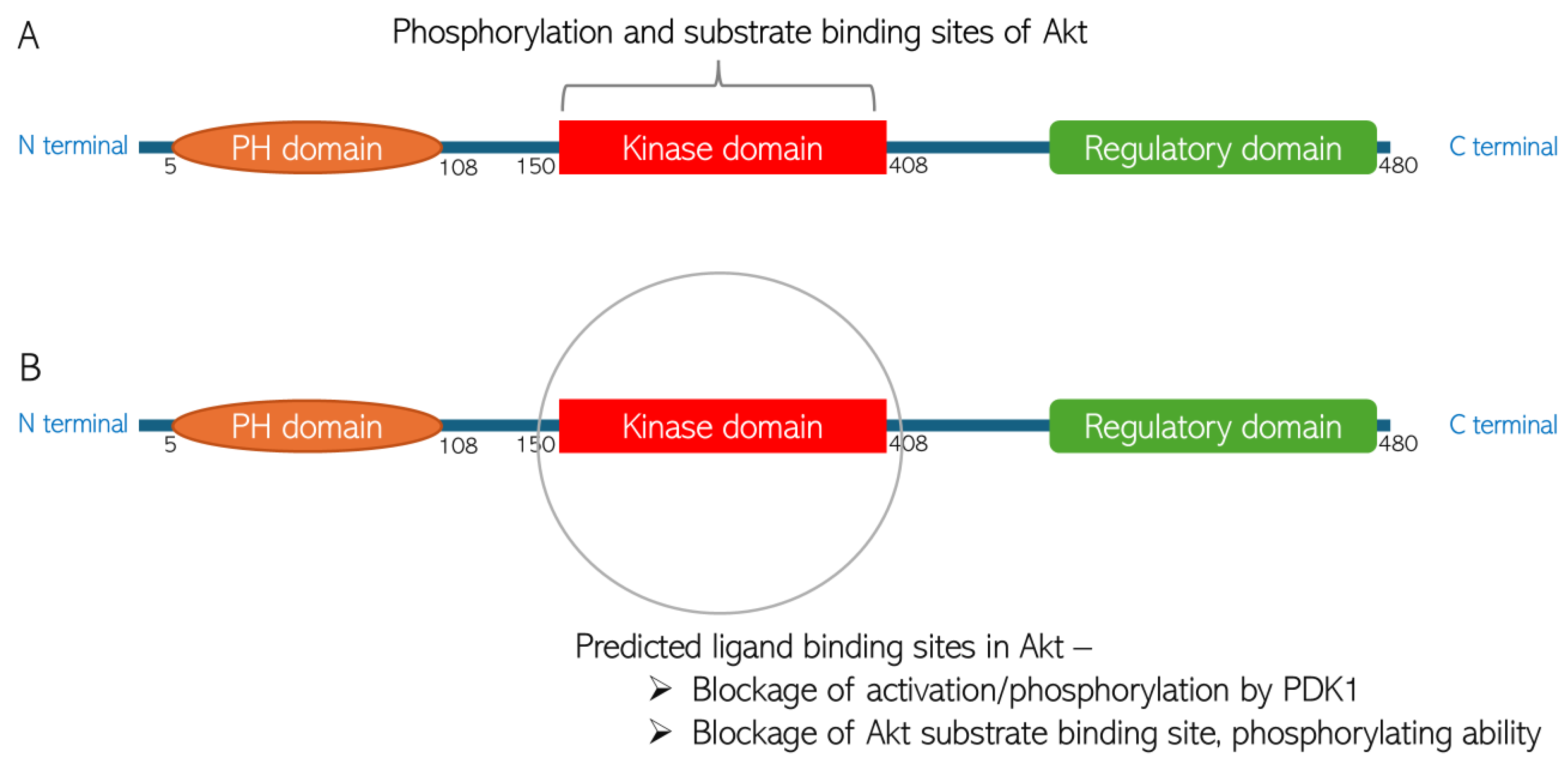
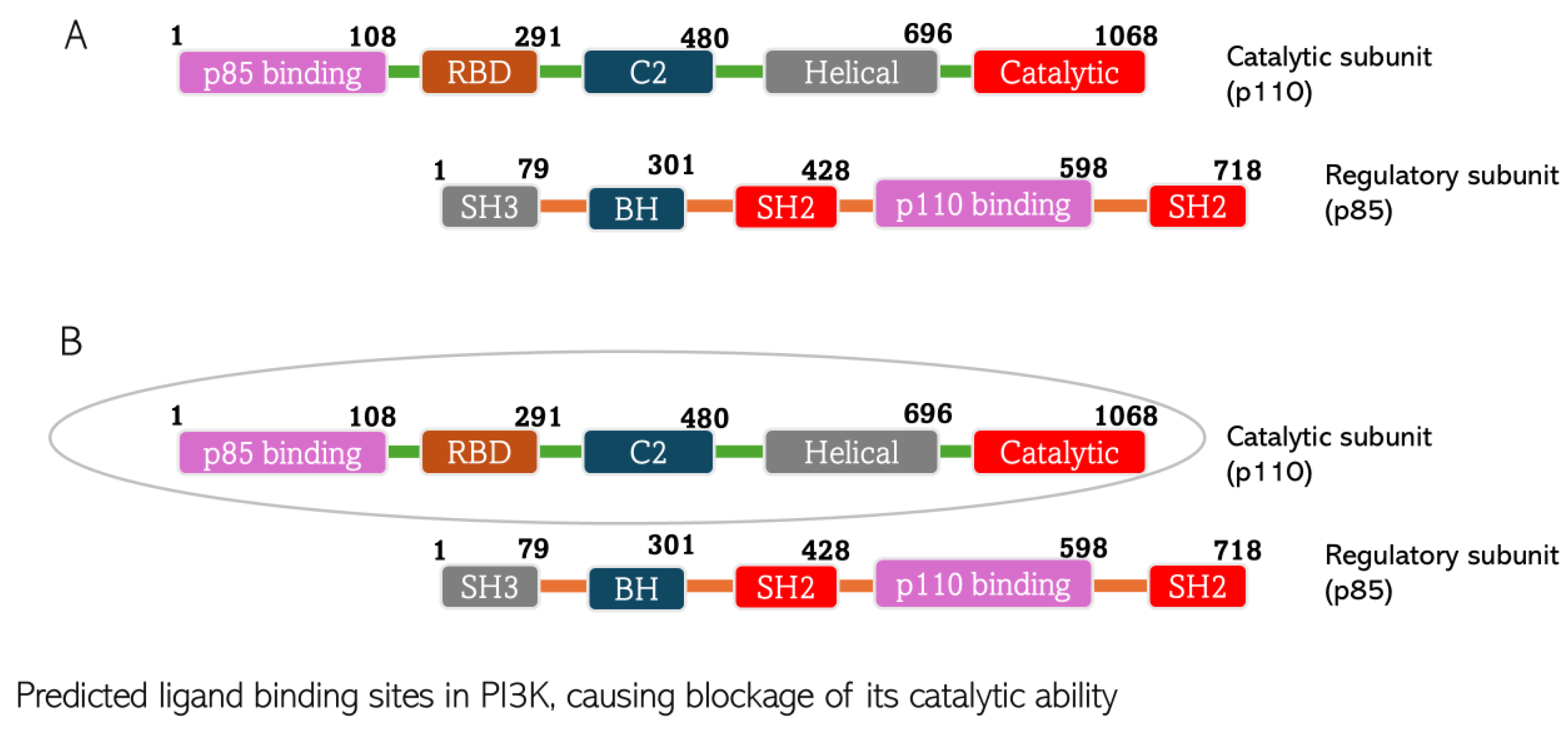
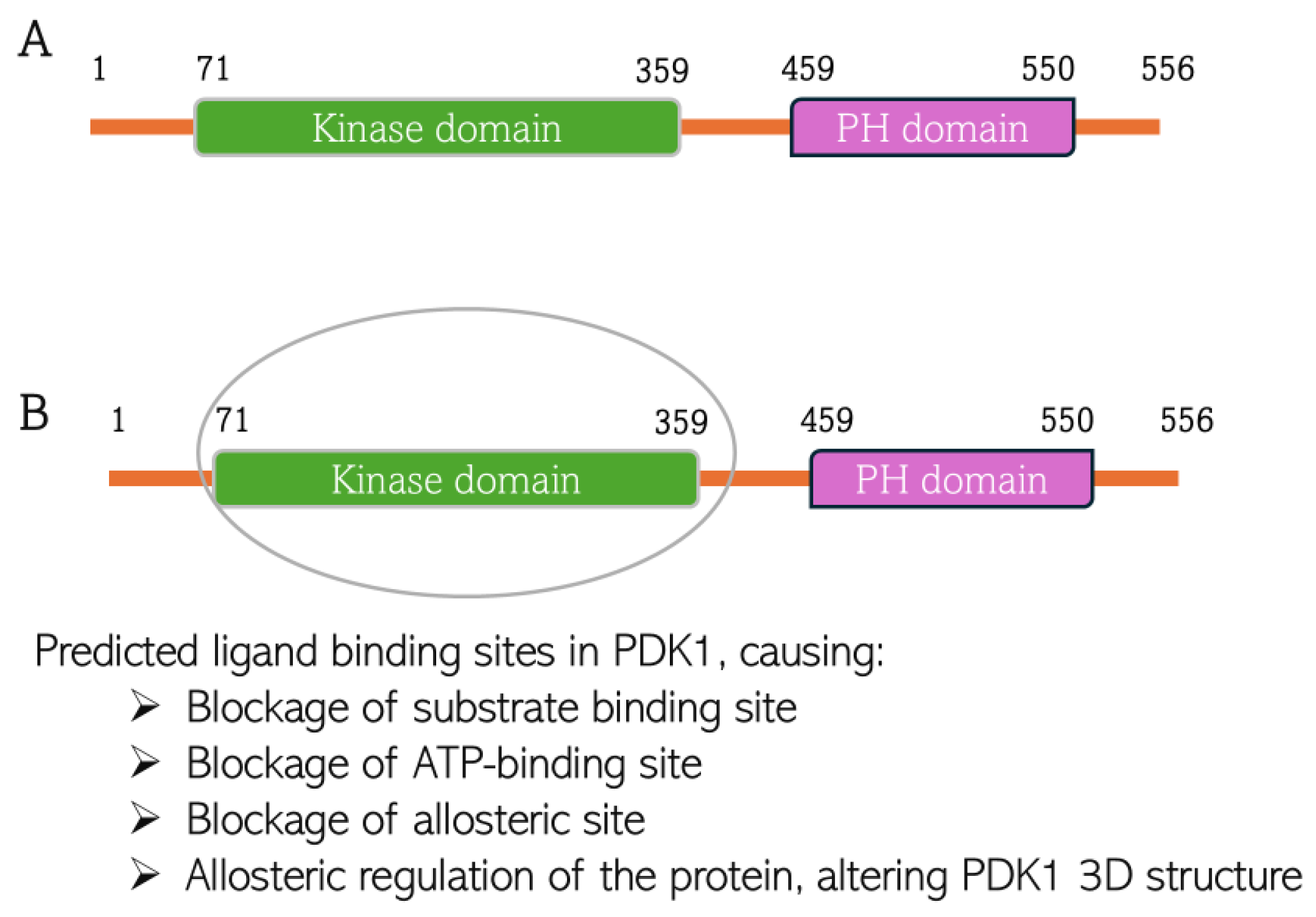
4.1. Molecular Docking
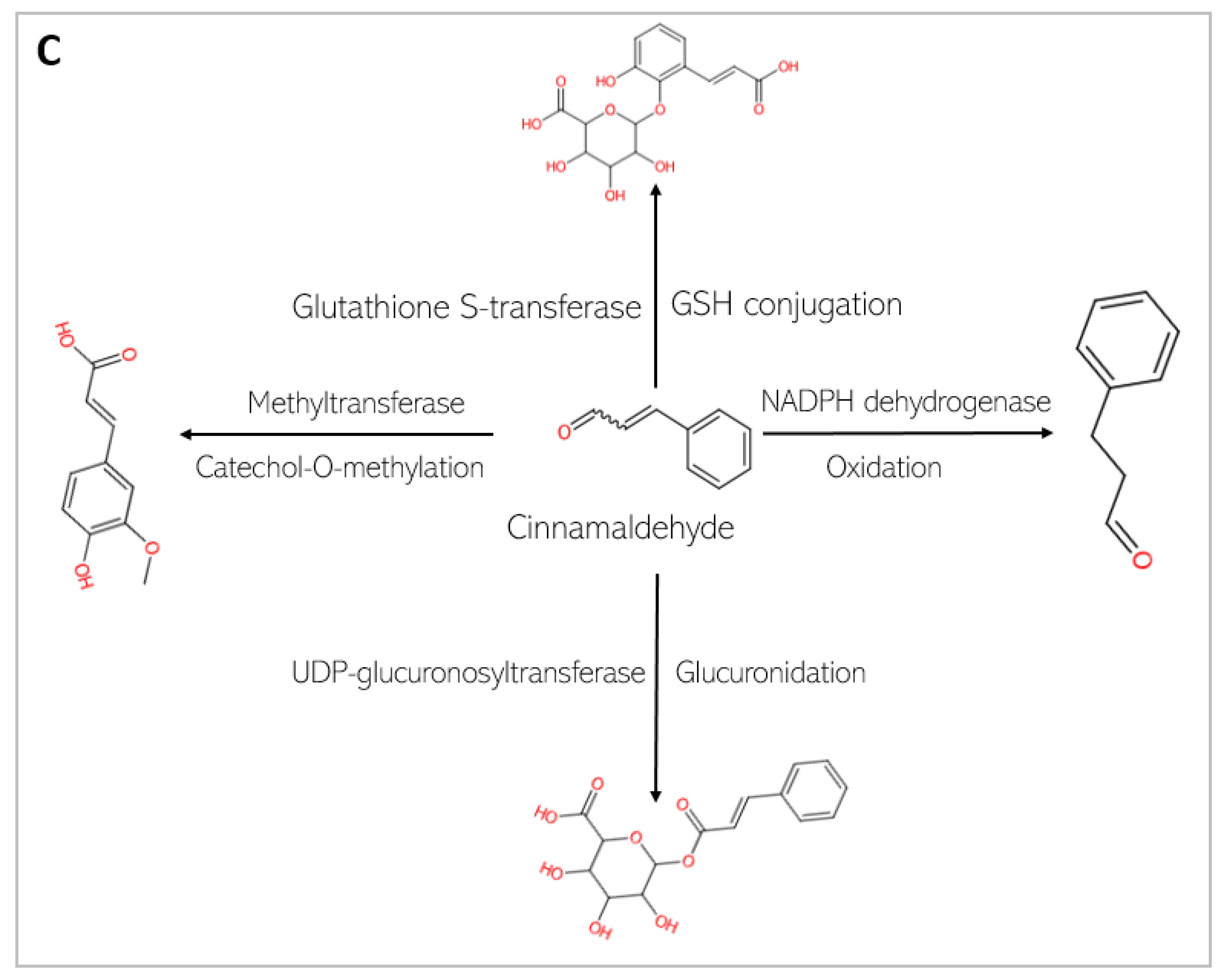
4.2. Pharmacokinetics of Chlorogenic Acid and Cinnamaldehyde
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Courtney, D.; Davey, M.G.; Moloney, B.M.; Barry, M.K.; Sweeney, K.; McLaughlin, R.P.; Malone, C.M.; Lowery, A.J.; Kerin, M.J. Breast cancer recurrence: Factors impacting occurrence and survival. Ir. J. Med. Sci. 2022, 191, 2501–2510. [Google Scholar] [CrossRef] [PubMed]
- Olayiwola, Y.; Gollahon, L. Natural Compounds and Breast Cancer: Chemo-Preventive and Therapeutic Capabilities of Chlorogenic Acid and Cinnamaldehyde. Pharmaceuticals 2024, 17, 361. [Google Scholar] [CrossRef] [PubMed]
- Burguin, A.; Diorio, C.; Durocher, F. Breast Cancer Treatments: Updates and New Challenges. J. Pers. Med. 2021, 11, 808. [Google Scholar] [CrossRef] [PubMed]
- Schuster, C.; Wolpert, N.; Moustaid-Moussa, N.; Gollahon, L.S. Combinatorial Effects of the Natural Products Arctigenin, Chlorogenic Acid, and Cinnamaldehyde Commit Oxidation Assassination on Breast Cancer Cells. Antioxidants 2022, 11, 591. [Google Scholar] [CrossRef] [PubMed]
- Olayiwola, Y.; Gollahon, L.S. Chlorogenic Acid and Cinnamaldehyde in Combination Inhibit Metastatic Traits and Induce Apoptosis via Akt Downregulation in Breast Cancer Cells. Int. J. Mol. Sci. 2024, 25, 6417. [Google Scholar] [CrossRef] [PubMed]
- Gollahon, L.S.; Lee, K.; Finckbone, V.; Jeong, Y. The natural product NI-07 demonstrates effective anti-cancer properties against numerous cancer cell types. J. Solid Tumors 2013, 3, 30. [Google Scholar] [CrossRef]
- Kapinova, A.; Kubatka, P.; Liskova, A.; Baranenko, D.; Kruzliak, P.; Matta, M.; Büsselberg, D.; Malicherova, B.; Zulli, A.; Kwon, T.K. Controlling metastatic cancer: The role of phytochemicals in cell signaling. J. Cancer Res. Clin.Oncol. 2019, 145, 1087–1109. [Google Scholar] [CrossRef] [PubMed]
- Pang, S.; Jia, M.; Gao, J.; Liu, X.; Guo, W.; Zhang, H. Effects of dietary patterns combined with dietary phytochemicals on breast cancer metastasis. Life Sci. 2021, 264, 118720. [Google Scholar] [CrossRef] [PubMed]
- Majrashi, T.A.; Alshehri, S.A.; Alsayari, A.; Muhsinah, A.B.; Alrouji, M.; Alshahrani, A.M.; Shamsi, A.; Atiya, A. Insight into the biological roles and mechanisms of phytochemicals in different types of cancer: Targeting cancer therapeutics. Nutrients 2023, 15, 1704. [Google Scholar] [CrossRef] [PubMed]
- Younas, M.; Hano, C.; Giglioli-Guivarc’h, N.; Abbasi, B.H. Mechanistic evaluation of phytochemicals in breast cancer remedy: Current understanding and future perspectives. RSC Adv. 2018, 8, 29714–29744. [Google Scholar] [CrossRef] [PubMed]
- Akhtar Siddiqui, J.; Singh, A.; Chagtoo, M.; Singh, N.; Madhav Godbole, M.; Chakravarti, B. Phytochemicals for breast cancer therapy: Current status and future implications. Curr. Cancer Drug Targets 2015, 15, 116–135. [Google Scholar] [CrossRef] [PubMed]
- Issinger, O.-G.; Guerra, B. Phytochemicals in cancer and their effect on the PI3K/AKT-mediated cellular signaling. Biomed. Pharmacother. 2021, 139, 111650. [Google Scholar] [CrossRef] [PubMed]
- Ham, S.L.; Nasrollahi, S.; Shah, K.N.; Soltisz, A.; Paruchuri, S.; Yun, Y.H.; Luker, G.D.; Bishayee, A.; Tavana, H. Phytochemicals potently inhibit migration of metastatic breast cancer cells. Integr. Biol. 2015, 7, 792–800. [Google Scholar] [CrossRef] [PubMed]
- Israel, B.E.B.; Tilghman, S.L.; Parker-Lemieux, K.; Payton-Stewart, F. Phytochemicals: Current strategies for treating breast cancer. Oncol. Lett. 2018, 15, 7471–7478. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Aoki, R.; Terasaki, M. Potential chemo-preventive effects of dietary combination of phytochemicals against cancer development. Pharmaceuticals 2023, 16, 1591. [Google Scholar] [CrossRef] [PubMed]
- Wani, A.K.; Akhtar, N.; Mir, T.U.G.; Singh, R.; Jha, P.K.; Mallik, S.K.; Sinha, S.; Tripathi, S.K.; Jain, A.; Jha, A.; et al. Targeting Apoptotic Pathway of Cancer Cells with Phytochemicals and Plant-Based Nanomaterials. Biomolecules 2023, 13, 194. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A Target for Anticancer Therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef] [PubMed]
- Tautermann, C.S. Current and future challenges in modern drug discovery. Quantum Mech. Drug Discov. 2020, 2114, 1–17. [Google Scholar]
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and oppor-tunities in drug discovery: Miniperspective. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Hawkins, B.A.; Du, J.J.; Groundwater, P.W.; Hibbs, D.E.; Lai, F. A Guide to In Silico Drug Design. Pharmaceutics 2022, 15, 49. [Google Scholar] [CrossRef] [PubMed]
- Asiamah, I.; Obiri, S.A.; Tamekloe, W.; Armah, F.A.; Borquaye, L.S. Applications of molecular docking in natural products-based drug discovery. Sci. Afr. 2023, 20, e01593. [Google Scholar] [CrossRef]
- Terefe, E.M.; Ghosh, A. Molecular Docking, Validation, Dynamics Simulations, and Pharmacokinetic Prediction of Phytochemicals Isolated from Croton dichogamus Against the HIV-1 Reverse Transcriptase. Bioinform. Biol. Insights 2022, 16, 11779322221125605. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, B.T.; Smith, D.L.; Ram, P.T.; Lu, Y.; Mills, G.B. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat. Rev. Drug Discov. 2005, 4, 988–1004. [Google Scholar] [CrossRef] [PubMed]
- Shariati, M.; Meric-Bernstam, F. Targeting AKT for cancer therapy. Expert Opin. Investig. Drugs 2019, 28, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Paplomata, E.; O’Regan, R. The PI3K/AKT/mTOR pathway in breast cancer: Targets, trials and biomarkers. Ther. Adv. Med. Oncol. 2014, 6, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.U.; Ahemad, N.; Chuah, L.-H.; Naidu, R.; Htar, T.T. Illustrated step by step protocol to perform molecular docking: Human estrogen receptor complex with 4-hydroxytamoxifen as a case study. Prog. Drug Discov. Biomed. Sci. 2020, 3, a0000054. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2. 0: New docking methods expanded force field, and python bindings. J. Chem. Information. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Chirumbolo, S.; Bjørklund, G.; Lysiuk, R.; Vella, A.; Lenchyk, L.; Upyr, T. Targeting Cancer with Phytochemicals via Their Fine Tuning of the Cell Survival Signaling Pathways. Int. J. Mol. Sci. 2018, 19, 3568. [Google Scholar] [CrossRef] [PubMed]
- Reichel, A.; Lienau, P. Pharmacokinetics in Drug Discovery: An Exposure-Centred Approach to Optimising and Predicting Drug Efficacy and Safety. In New Approaches to Drug Discovery; Handbook of Experimental, Pharmacology; Nielsch, U., Fuhrmann, U., Jaroch, S., Eds.; Springer: New York, NY, USA, 2015; Volume 232, pp. 235–260. [Google Scholar]
- Hassan, M.; Sallam, H.; Hassan, Z. The role of pharmacokinetics and pharmaco-dynamics in early drug development with reference to the cyclin-dependent kinase (Cdk) inhibitor-roscovitine. Sultan Qaboos Univ. Med. J. 2011, 11, 165. [Google Scholar] [PubMed]
- Kim, D.H.; Kim, J.Y.; Yu, B.P.; Chung, H.Y. The activation of NF-κB through Akt-induced FOXO1 phosphorylation during aging and its modulation by calorie re-striction. Biogerontology 2008, 9, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Majewska, E.; Szeliga, M. AKT/GSK3β signaling in glioblastoma. Neurochem. Res. 2017, 42, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Y.; Wang, L.; Chen, B.; Zhu, M.; Ma, C.; Mu, C.; Tao, A.; Li, S.; Luo, L. Cinnamaldehyde suppressed EGF-induced EMT process and inhibits ovarian cancer progression through PI3K/AKT pathway. Front. Pharmacol. 2022, 13, 779608. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, T.M.; Leal, J.F.M.; Seger, R.; Taya, Y.; Oren, M. Crosstalk between Akt, p53 and Mdm2: Possible implications for the regulation of apoptosis. Oncogene 2002, 21, 1299–1303. [Google Scholar] [CrossRef] [PubMed]
- Asati, V.; Mahapatra, D.K.; Bharti, S.K. PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways inhibitors as anticancer agents: Structural and pharmacological perspectives. Eur. J. Med. Chem. 2016, 109, 314–341. [Google Scholar] [CrossRef] [PubMed]
- Hasan, G.M.; Hassan, M.I.; Sohal, S.S.; Shamsi, A.; Alam, M. Therapeutic Target-ing of Regulated Signaling Pathways of Non-Small Cell Lung Carcinoma. ACS Omega 2023, 8, 26685–26698. [Google Scholar] [CrossRef] [PubMed]
- Ren, D.; Qin, G.; Zhao, J.; Sun, Y.; Zhang, B.; Li, D.; Wang, B.; Jin, X.; Wu, H. Metformin activates the STING/IRF3/IFN-β pathway by inhibiting AKT phosphorylation in pancreatic cancer. Am. J. Cancer Res. 2020, 10, 2851–2864. [Google Scholar] [PubMed]
- Shen, Z.; Xue, D.; Wang, K.; Zhang, F.; Shi, J.; Jia, B.; Yang, D.; Zhang, Q.; Zhang, S.; Jiang, H.; et al. Metformin exerts an antitumor effect by inhibiting bladder cancer cell migration and growth, and promoting apoptosis through the PI3K/AKT/mTOR pathway. BMC Urol. 2022, 22, 79. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.; Lux, A.; O’Callaghan, F. The journey of metformin from glycaemic control to mTOR inhibition and the suppression of tumour growth. Br. J. Clin. Pharmacol. 2019, 85, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Thent, Z.C.; Zaidun, N.H.; Azmi, M.F.; Senin, M.I.; Haslan, H.; Salehuddin, R. Is Metformin a Therapeutic Paradigm for Colorectal Cancer: Insight into the Molecular Pathway? Curr. Drug Targets 2017, 18, 734–750. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Cao, F.; Qiu, S.; Jiang, W.; Tao, L.; Zhu, Y. Metformin Promotes Differentiation and Attenuates H2O2-Induced Oxidative Damage of Osteoblasts via the PI3K/AKT/Nrf2/HO-1 Pathway. Front. Pharmacol. 2022, 13, 829830. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zhou, Y.; Liu, Y.; Ping, J.; Shou, Q.; Chen, F.; Ruo, R. Metformin improves hepatic IRS2/PI3K/Akt signaling in insulin-resistant rats of NASH and cirrhosis. J. Endocrinol. 2016, 229, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Chatterjee, A.; Kogan, D.; Patel, D.; Foster, D.A. 5-Aminoimidazole-4-carboxamide-1-β-4-ribofuranoside (AICAR) enhances the efficacy of rapamycin in human cancer cells. Cell Cycle 2015, 14, 3331–3339. [Google Scholar] [CrossRef] [PubMed]
- Smyth, L.M.; Tamura, K.; Oliveira, M.; Ciruelos, E.M.; Mayer, I.A.; Sablin, M.P.; Biganzoli, L.; Ambrose, H.J.; Ashton, J.; Barnicle, A.; et al. Capivasertib, an AKT Kinase Inhibitor, as Monotherapy or in Combination with Fulvestrant in Patients with AKT1 (E17K)-Mutant, ER-Positive Metastatic Breast Cancer. Clin. Cancer Res. 2020, 26, 3947–3957. [Google Scholar] [CrossRef] [PubMed]
- Gerweck, L.E.; Kozin, S.V.; Stocks, S.J. The pH partition theory predicts the accu-mulation and toxicity of doxorubicin in normal and low-pH-adapted cells. Br. J. Cancer 1999, 79, 838–842. [Google Scholar] [CrossRef] [PubMed]
- Powell, D.; Pinchuk, I.; Saada, J.; Chen, X.; Mifflin, R. Mesenchymal cells of the in-testinal lamina propria. Annu. Rev. Physiol. 2011, 73, 213–237. [Google Scholar] [CrossRef] [PubMed]
- Eloranta, J.J.; Hiller, C.; Jüttner, M.; Kullak-Ublick, G.A. The SLCO1A2 gene, en-coding human organic anion-transporting polypeptide 1A2, is transactivated by the vitamin D receptor. Mol. Pharmacol. 2012, 82, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Kinzi, J.; Grube, M.; Meyer zu Schwabedissen, H.E. OATP2B1–The underrated member of the organic anion transporting polypeptide family of drug transporters? Biochem. Pharmacol. 2021, 188, 114534. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, J.; Ariyoshi, N.; Sakakibara, M.; Nakanishi, T.; Okubo, Y.; Shiina, N.; Fujisaki, K.; Nagashima, T.; Nakatani, Y.; Tamai, I.; et al. Organic anion transporting polypeptide 2B1 expression correlates with uptake of estrone-3-sulfate and cell prolif-eration in estrogen receptor-positive breast cancer cells. Drug Metab. Pharmacokinet. 2015, 30, 133–141. [Google Scholar] [CrossRef] [PubMed]
- van de Steeg, E.; van Esch, A.; Wagenaar, E.; Kenworthy, K.E.; Schinkel, A.H. In-fluence of human OATP1B1, OATP1B3, and OATP1A2 on the pharmacokinetics of methotrexate and paclitaxel in humanized transgenic mice. Clin. Cancer Res. 2013, 19, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Benet, L.Z.; Kroetz, D.L.; Sheiner, L.B. Pharmacokinetics: The dynamics of drug absorption, distribution, metabolism, and elimination. In Goodman and Gilman’s: The Pharmacolgical Basis of Therapeutics, 9th ed.; Hardman, J.G., Limbird, L.E., Molinoff, P.B., Ruddon, R.W., Gilman, A.G., Eds.; Mcgraw-Hill: New York, NY, USA, 1996; pp. 3–28. [Google Scholar]
- Cunningham, L. The anatomy of the arteries and veins of the breast. J. Surg. Oncol. 1977, 9, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Sachani, P.; Dhande, R.; Parihar, P.; Kasat, P.R.; Bedi, G.N.; Pradeep, U.; Kothari, P.; Mapari, S.A. Enhancing the Understanding of Breast Vascularity Through Insights from Dynamic Contrast-Enhanced Magnetic Resonance Imaging: A Comprehensive Review. Cureus 2024, 16, 70226. [Google Scholar] [CrossRef] [PubMed]
- Gautheron, J.; Jéru, I. The Multifaceted Role of Epoxide Hydrolases in Human Health and Disease. Int. J. Mol. Sci. 2020, 22, 13. [Google Scholar] [CrossRef] [PubMed]
- Garza, A.Z.; Park, S.B.; Kocz, R. Drug Elimination. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025; Available online: https://www.ncbi.nlm.nih.gov/books/NBK547662/ (accessed on 15 April 2025).
- Horde, G.W.; Gupta, V. Drug Clearance. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025; Available online: https://www.ncbi.nlm.nih.gov/books/NBK557758/ (accessed on 15 April 2025).
- Hallare, J.; Gerriets, V. Elimination Half-Life of Drugs. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025; Available online: https://www.ncbi.nlm.nih.gov/books/NBK554498/ (accessed on 15 April 2025).
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, H.; Tian, L.; Li, Q.; Luo, J.; Zhang, Y. Analysis of the physicochemical properties of acaricides based on Lipinski’s rule of five. J. Comput. Biol. 2020, 27, 1397–1406. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.E. What has polar surface area ever done for drug discovery? Future Med. Chem. 2011, 3, 469–484. [Google Scholar] [CrossRef] [PubMed]
- Ivanović, V.; Rančić, M.; Arsić, B.; Pavlović, A. Lipinski’s rule of five, famous ex-tensions and famous exceptions. Pop. Sci. Artic. 2020, 3, 171–177. [Google Scholar]
- Wang, Y.; Michael, S.; Huang, R.; Zhao, J.; Recabo, K.; Bougie, D.; Shu, Q.; Shinn, P.; Sun, H. Retro Drug Design: From Target Properties to Molecular Structures. bioRxiv 2021, 5, 442656. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Akt Binding | Parameters | |
|---|---|---|---|
| 1 | Chlorogenic acid | XYZ coordinates Binding energy Interacting amino acids | 25.140333; 6.181083; 8.167583 −8.2 kcal/mol Asp 274; Lys 276; Asn 279; Leu 295; Phe 309; Cys 310; Gly 311 |
| 2 | Cinnamaldehyde | XYZ coordinates Binding energy Interacting amino acids | 27.003800; 6.254100; 9.233900 −5.4 kcal/mol Leu 156; Gly 157; Val 164; Ala 177; Ala 230; Met 281 |
| 3 | Spirocyclic sulfonamides inhibitor (co-crystallized ligand) | XYZ coordinates | 28.185902; 2.786537; 11.33597 |
| 4 | Capivasertib (the first FDA-approved Akt inhibitor for treatment of breast cancer) The binding energies of FDA-approved drugs ranged from −5.6 to −6.9 kcal/mol [22] | XYZ coordinates Binding energy | 30.194838; 2.264703; 12.38245 −8.5 kcal/mol |
| Ligand | PI3K Binding | Parameters | |
|---|---|---|---|
| 1 | Chlorogenic acid | XYZ coordinates Binding energy Interacting amino acids | 58.764226; −8.98519; 81.31416 −8.5 kcal/mol Ser7; Ile121; Lys672; Ser673; Met 675; His676; Ile713; Asn803; Val845 |
| 2 | Cinnamaldehyde | XYZ coordinates Binding energy Interacting amino acids | 57.478300; −7.86420; 83.815100 −6.2 kcal/mol Trp669; Lys672; Glu674; His676; Val706; Met709; Ile 713; Ile841; Gly842; Asp 843 |
| 3 | Pyridopyrimidinones inhibitors (co-crystallized ligand) | XYZ coordinates | 54.042978; 4.928485; 87.43672 |
| Ligand | PDK1 Binding | Parameters | |
|---|---|---|---|
| 1 | Chlorogenic acid | XYZ coordinates Binding energy Interacting amino acids | 37.643194; −27.85712; −11.5306 −8.2 kcal/mol Leu88; Val96; Ala109; Lys111; Ser160; Ala 162; Glu209; Leu212; Thr222; Asp223 |
| 2 | Cinnamaldehyde | XYZ coordinates Binding energy Interacting amino acids | 34.57870; −28.64820; −12.19540 −5.8 kcal/mol Leu88; Val96; Ala109; Lys111; Leu159; Ser160; Ala162; Leu212; Thr222 |
| 3 | 7-azaindoles inhibitor (co-crystallized ligand) | XYZ coordinates | 34.78813; −27.57596; −12.39844 |
| S/N | Parameters | Predicted | Value | Recommendation | Implication |
|---|---|---|---|---|---|
| CGA | CA | ||||
| 1 | Number of rotatable bonds | 5 | 2 | ≤10, per Veber’s rule | Contributes to molecule conformational flexibility to permeate cell membrane and interact with receptors |
| 2 | Molecular weight (g/mol) | 354.31 | 132.16 | ≤500 g/mol | Small molecules cross cell membrane and have better oral bioavailability |
| 3 | Caco-2 permeability (LogPapp) | −6.49 | −4.767 | ≥0.9 | Low intestinal wall permeability |
| 4 | P-glycoprotein pump substrate | No | No | Not removed by the efflux pump in the GIT | |
| 5 | BBB permeability | No | Yes | ||
| 6 | Octanol/water partition coefficient (Lipophilicity, Log Po/w) | −0.39 | 1.97 | 0–5 | Lower value is suggestive of hydrophilicity or lipophobicity of the compound, which therefore becomes membrane impermeable |
| S/N | Parameters | Prediction | Reference | Implication | |
|---|---|---|---|---|---|
| CGA | CA | ||||
| 1 | PPB (%) | 64.8 | 94.9 | <90 | High plasma protein binding suggests drugs remain unavailable to tissue to elicit therapeutic effects |
| 2 | Volume distribution(l/kg) | 0.976 | 0.384 | 0.4–20 | Low volume reaching extravascular tissue based on blood tissue perfusion |
| S/N | Parameters | Prediction | Reference | Implication | |
|---|---|---|---|---|---|
| CGA | CA | ||||
| 1 | CLplasma (mL/min/kg) | 11.086 | 3.34 | 5–15 mL/min/kg Moderate clearance plasma rate | Varied bioavailability in the systemic circulation and plasma removal rate; this may impact volume distribution and half-life |
| 2 | T1/2 (hr) | 1.395 | 2.758 | 1–4 h short half-life | Its effect wears off quickly, and 50% is removed quickly from the body. |
| S/N | Parameters | Prediction | |
|---|---|---|---|
| CGA | CA | ||
| 1 | Lethal Dose 50 (LD50) | 5000 mg/kg | 1850 mg/kg |
| 2 | Hepatotoxicity | Inactive | Inactive |
| 3 | Neurotoxicity | Inactive | Active |
| 4 | Cardiotoxicity | Inactive | Active |
| 5 | Cytotoxicity | Inactive | Inactive |
| 6 | Mutagenicity | Inactive | Active |
| 7 | Nephrotoxicity | Fairly active | Inactive |
| 8 | Pulmonary | Fairly active | Inactive |
| 9 | Immunotoxicity | Active | Active |
| S/N | Parameters | Prediction | Optimal Value | Implication | |
|---|---|---|---|---|---|
| CGA | CA | ||||
| 1 | Number of H-bond acceptors | 9 | 1 | ≤10 | High number increases polarity and H-bonding with targets but impacts membrane penetration due to hydrophilicity conferment for CGA, but insufficient H-bond forming group in CA, making CA more lipophilic and membrane-penetrating. Undesired H-bonding in CGA, with groups such as P-gp, caused by H-bond donors. |
| 2 | Number of H-bond donors | 6 | 0 | ≤5 | |
| 3 | Topological polar surface area (TPSA)(Å2) | 164.75 | 17.07 | 0–140 Å2 | The more surface area covered by polar atoms in a drug, the poorer the drug cell permeability and effectiveness to reach target tissues. 60–80 Å2 required to penetrate mammary tissues. |
| 4 | Number of rotatable sigma bonds | 5 | 2 | ≤10 | Moderate amount confers appropriate degree of flexibility to the drug, which is important for drug conformational change to cross cell membrane and interact with target at its active site. |
| 5 | Number of heavy atoms | 25 | 10 | <36 | Appropriate number indicative of good binding affinity (target selectivity) and ligand efficiency (ligand binding energy) |
| 6 | Number of aromatic heavy atoms | 6 | 6 | 2–4 | Higher increases drug hydrophobicity, which may enhance the ligand binding affinity but may reduce target selectivity via pi orbital stacking with aromatic amino acid side chains of undesired proteins |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olayiwola, Y.; Gollahon, L. Chlorogenic Acid and Cinnamaldehyde in Breast Cancer Cells: Predictive Examination of Pharmacokinetics and Binding Thermodynamics with the Key Mediators of PI3K/Akt Signaling. Biomedicines 2025, 13, 1810. https://doi.org/10.3390/biomedicines13081810
Olayiwola Y, Gollahon L. Chlorogenic Acid and Cinnamaldehyde in Breast Cancer Cells: Predictive Examination of Pharmacokinetics and Binding Thermodynamics with the Key Mediators of PI3K/Akt Signaling. Biomedicines. 2025; 13(8):1810. https://doi.org/10.3390/biomedicines13081810
Chicago/Turabian StyleOlayiwola, Yusuff, and Lauren Gollahon. 2025. "Chlorogenic Acid and Cinnamaldehyde in Breast Cancer Cells: Predictive Examination of Pharmacokinetics and Binding Thermodynamics with the Key Mediators of PI3K/Akt Signaling" Biomedicines 13, no. 8: 1810. https://doi.org/10.3390/biomedicines13081810
APA StyleOlayiwola, Y., & Gollahon, L. (2025). Chlorogenic Acid and Cinnamaldehyde in Breast Cancer Cells: Predictive Examination of Pharmacokinetics and Binding Thermodynamics with the Key Mediators of PI3K/Akt Signaling. Biomedicines, 13(8), 1810. https://doi.org/10.3390/biomedicines13081810