
Therapeutic Targeting of Apoptosis, Autophagic Cell Death, Necroptosis, Pyroptosis, and Ferroptosis Pathways in Oral Squamous Cell Carcinoma: Molecular Mechanisms and Potential Strategies



Abstract

1. Introduction
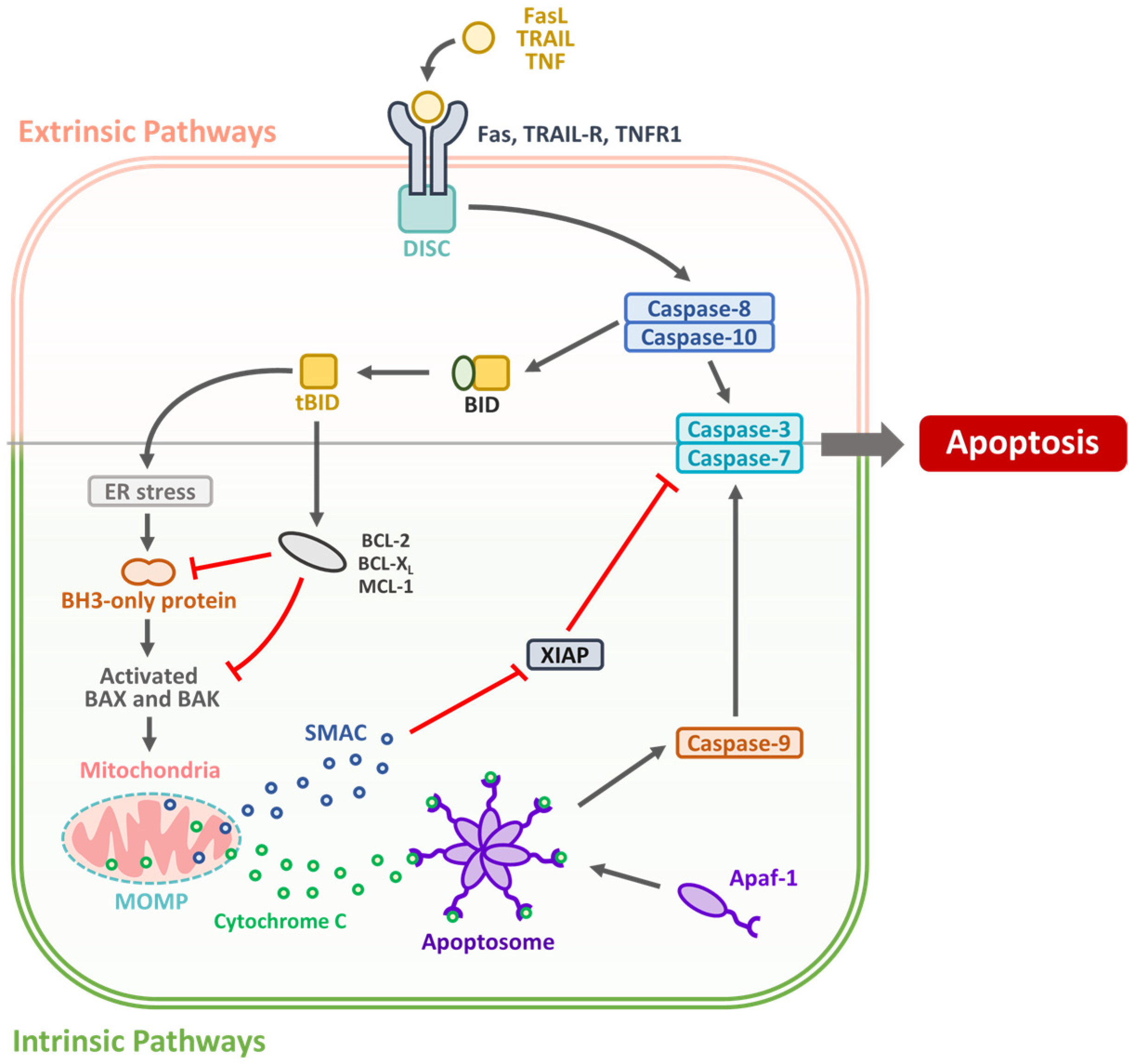
2. Targeting Apoptotic Pathways in OSCC: Mechanisms and Therapeutic Strategies
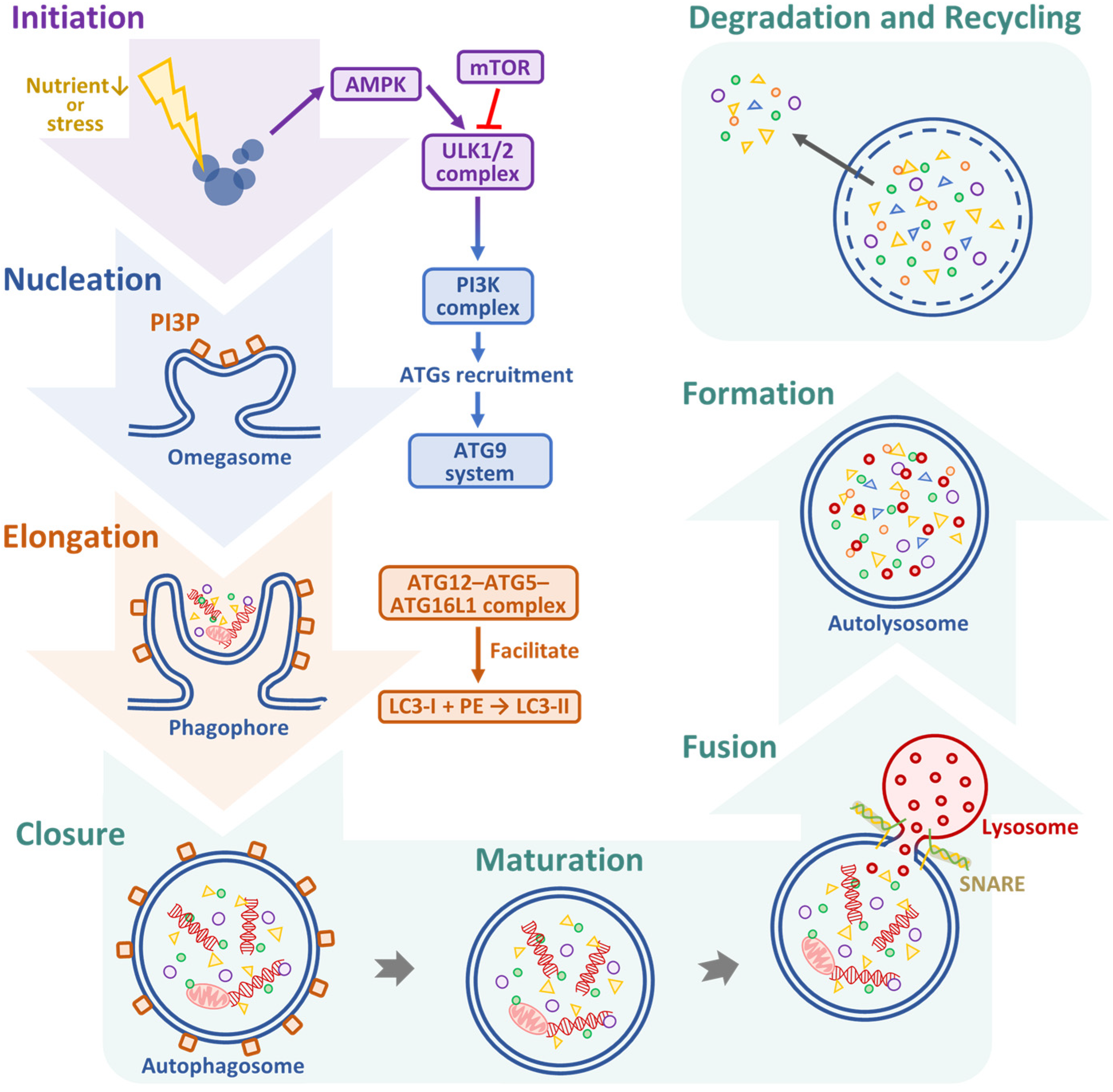
3. Therapeutic Targeting of ACD in OSCC: Dual Roles, Regulatory Pathways, and Emerging Strategies
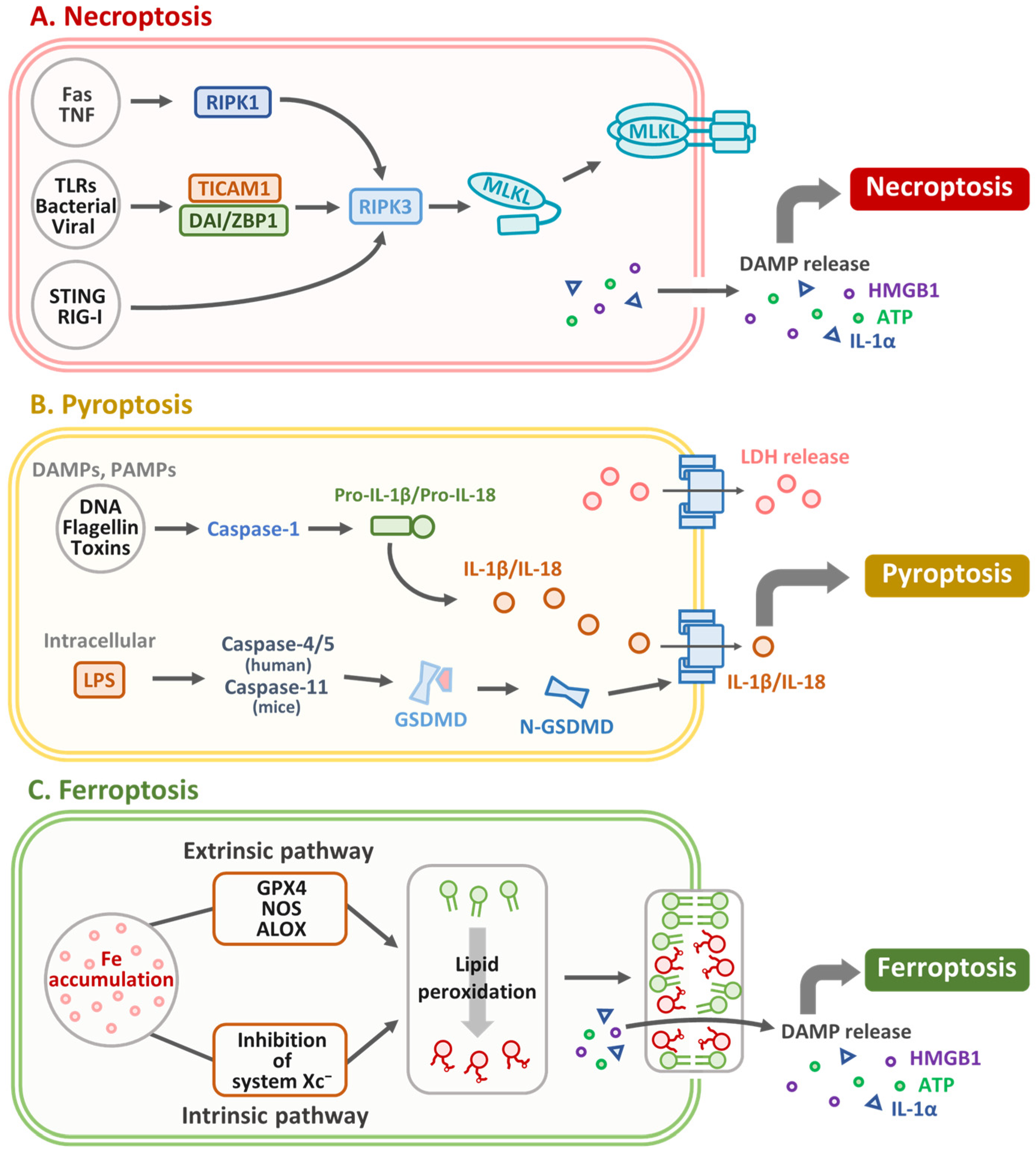
4. Necroptosis in OSCC: Mechanisms, Molecular Markers, and Therapeutic Implications
5. Targeting Pyroptosis in OSCC: Therapeutic Strategies and Prognostic Biomarkers
6. Targeting Ferroptosis in OSCC: Mechanisms, Prognostic Biomarkers, and Therapeutic Strategies
7. Regulated Cell Death Pathways as Therapeutic Targets in OSCC: Apoptosis, ACD, Necroptosis, Pyroptosis, and Ferroptosis
8. Disulfidptosis in SLC7A11-High OSCC: Mechanisms, Therapeutic Implications, and Prognostic Signatures
9. Summary
10. Limitations
11. Future Perspectives
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACD | Autophagic Cell Death |
| ACSL4 | Acyl-CoA synthetase long chain family member 4 |
| AIF | Apoptosis-inducing factor |
| AKT | Protein Kinase B |
| AMPK | AMP-activated Protein Kinase |
| Apaf-1 | Apoptotic Protease-activating Factor 1 |
| ATG | Autophagy-related |
| ATM | Ataxia-telangiectasia mutated |
| ATP | Adenosine triphosphate |
| AUC | Area under the curve |
| Bak | Bcl-2 homologous antagonist/killer |
| Bax | Bcl2-associated X protein |
| BECN1 | Beclin-1 |
| Bcl-2 | B-cell lymphoma 2 |
| Bcl-xL | B-cell lymphoma-extra large |
| BID | BH3 interacting-domain death agonist |
| BNIP3 | BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 |
| CA9 | Carbonic anhydrase 9 |
| CAP | Cancer-associated pyroptosis |
| CAR | Chimeric antigen receptor |
| Caspase | Cysteine-dependent aspartate-specific protease |
| CD5 | Cluster of differentiation 5 |
| CHK | Checkpoint kinase |
| CISD2 | CDGSH iron sulfur domain 2 |
| CK19 | Cytokeratin 19 |
| CQ | Chloroquine |
| CTLA4 | Cytotoxic T-lymphocyte-associated protein 4 |
| DAMPs | Damage-associated molecular patterns |
| DCA | Dichloroacetate |
| DDIT4 | DNA damage inducible transcript 4 |
| DISC | Death-inducing Signaling Complex |
| DRIGs | Disulfidptosis-related Immune Genes |
| DRs | Death Receptors |
| EGFR | Epidermal Growth Factor Receptor |
| EMT | Epithelial–mesenchymal Transition |
| Endo G | Endonuclease G |
| ER | Endoplasmic Reticulum |
| ERK1/2 | Extracellular signal-regulated kinase 1 and 2 |
| FADD | Fas-associated Death Domain Protein |
| Fas | Fas cell surface death receptor |
| FRGs | Ferroptosis-related Genes |
| GPX4 | Glutathione Peroxidase 4 |
| GSDM | Gasdermin |
| GSDMD | Gasdermin D |
| GSH | Glutathione |
| GTPase | Guanosine triphosphatase |
| HCQ | Hydroxychloroquine |
| HDAC | Histone Deacetylase |
| HNSCC | Head and Neck Squamous Cell Carcinoma |
| HOXA10 | Homeobox A10 |
| HOPS | Homotypic fusion and vacuolar protein sorting |
| IAP | Inhibitor of Apoptosis Protein |
| IFN | Interferon |
| IL | Interleukin |
| IL12RB2 | Interleukin-12 receptor subunit beta-2 |
| IRF1 | Interferon regulatory factor 1 |
| LAIR2 | Leukocyte-associated immunoglobulin-like receptor 2 |
| LC3 | Microtubule-associated Protein 1A/1B-light Chain 3 |
| LDH | Lactate dehydrogenase |
| lncRNAs | Long Noncoding RNAs |
| LPS | Lipopolysaccharide |
| MAP1LC3A | Microtubule-associated proteins 1A/1B light chain 3A |
| MDA | Malondialdehyde |
| MLKL | Mixed Lineage Kinase Domain-like Pseudokinase |
| MOMP | Mitochondrial Outer Membrane Permeabilization |
| mTOR | Mammalian Target of Rapamycin |
| NEC-1 | Necrostatin-1 |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NKX2-3 | NK2 Homeobox 3 |
| OSCC | Oral Squamous Cell Carcinoma |
| P-gp | P-glycoprotein |
| PARK2 | Parkin RBR E3 ubiquitin protein ligase |
| PARL | Plasma-activated Ringer’s Lactate |
| PARP1 | Poly (ADP-ribose) Polymerase 1 |
| PE | Phosphatidylethanolamine |
| PDT | Photodynamic Therapy |
| PI3K | Phosphoinositide 3-kinase |
| PPAR-α | Peroxisome proliferator activated receptor α |
| PPT1 | Palmitoyl-protein Thioesterase 1 |
| PRDX2 | Peroxiredoxin-2 |
| PRDX5 | Peroxiredoxin 5 |
| Rab7 | Ras-related protein 7 |
| RCD | Regulated Cell Death |
| RIG-I | Retinoic acid-inducible gene I |
| RIPK1/3 | Receptor-interacting Protein Kinases 1 and 3 |
| ROC | Receiver operating characteristic |
| RONS | Reactive Oxygen and Nitrogen Species |
| ROS | Reactive Oxygen Species |
| RUNX2 | RUNT-related Transcription Factor 2 |
| SL7A11 | Solute carrier family 7 member 11 |
| SMAC | Second Mitochondria-derived Activator of Caspases |
| SNARE | Soluble N-ethylmaleimide-sensitive factor activating protein receptor |
| SPD | Spermidine |
| SQSTM1 | Sequestosome 1 |
| STAT1 | Signal transducer and activator of transcription 1 |
| STING | Stimulator of interferon genes |
| TCGA | The Cancer Genome Atlas |
| TFP | Trifluoperazine |
| TICAM1 | TIR domain containing adaptor molecule 1 |
| TLRs | Toll-like receptors |
| TNF | Tumor Necrosis Factor |
| TP53 | Tumor protein p53 |
| TRAF6 | Tumor necrosis factor receptor-associated factor 6 |
| TRAIL | TNF-related Apoptosis-inducing Ligand |
| TRAIL-R | TNF-related apoptosis-inducing ligand receptor |
| TSCC | Tongue Squamous Cell Carcinoma |
| ULK1 | Unc-51-like Kinase 1 |
| VPS34 | Vacuolar protein sorting 34 |
| XIAP | X-linked IAP |
| ZBP1 | Z-DNA binding protein 1 |
References
- Badwelan, M.; Muaddi, H.; Ahmed, A.; Lee, K.T.; Tran, S.D. Oral Squamous Cell Carcinoma and Concomitant Primary Tumors, What Do We Know? A Review of the Literature. Curr. Oncol. 2023, 30, 3721–3734. [Google Scholar] [CrossRef] [PubMed]
- Irani, S. New Insights into Oral Cancer-Risk Factors and Prevention: A Review of Literature. Int. J. Prev. Med. 2020, 11, 202. [Google Scholar] [CrossRef] [PubMed]
- Thakral, A.; Lee, J.J.; Hou, T.; Hueniken, K.; Dudding, T.; Gormley, M.; Virani, S.; Olshan, A.; Diergaarde, B.; Ness, A.R.; et al. Smoking and alcohol by HPV status in head and neck cancer: A Mendelian randomization study. Nat. Commun. 2024, 15, 7835. [Google Scholar] [CrossRef] [PubMed]
- Park, W.; Wei, S.; Kim, B.S.; Kim, B.; Bae, S.J.; Chae, Y.C.; Ryu, D.; Ha, K.T. Diversity and complexity of cell death: A historical review. Exp. Mol. Med. 2023, 55, 1573–1594. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Guo, S.; Guo, J.; Du, Q.; Wu, C.; Wu, Y.; Zhang, Y. Cell death pathways: Molecular mechanisms and therapeutic targets for cancer. MedComm 2024, 5, e693. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Yang, R.; Zhang, Y.; Guo, M.; Takehiro, K.; Zhan, M.; Yang, L.; Wang, H. Molecular mechanisms and therapeutic strategies in overcoming chemotherapy resistance in cancer. Mol. Biomed. 2025, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C. Exploring cell death pathways in oral cancer: Mechanisms, therapeutic strategies, and future perspectives. Discov. Oncol. 2025, 16, 395. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, Y.; Lei, J.; Wu, Y. Autophagy in oral cancer: Promises and challenges (Review). Int. J. Mol. Med. 2024, 54, 116. [Google Scholar] [CrossRef] [PubMed]
- Siquara da Rocha, L.O.; de Morais, E.F.; de Oliveira, L.Q.R.; Barbosa, A.V.; Lambert, D.W.; Gurgel Rocha, C.A.; Coletta, R.D. Exploring beyond Common Cell Death Pathways in Oral Cancer: A Systematic Review. Biology 2024, 13, 103. [Google Scholar] [CrossRef] [PubMed]
- Vo, T.T.T.; Peng, T.Y.; Nguyen, T.H.; Bui, T.N.H.; Wang, C.S.; Lee, W.J.; Chen, Y.L.; Wu, Y.C.; Lee, I.T. The crosstalk between copper-induced oxidative stress and cuproptosis: A novel potential anticancer paradigm. Cell Commun. Signal 2024, 22, 353. [Google Scholar] [CrossRef] [PubMed]
- Adeshakin, F.O.; Adeshakin, A.O.; Afolabi, L.O.; Yan, D.; Zhang, G.; Wan, X. Mechanisms for Modulating Anoikis Resistance in Cancer and the Relevance of Metabolic Reprogramming. Front. Oncol. 2021, 11, 626577. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, D.; Kim, S.E.; Overholtzer, M. Entosis: The core mechanism and crosstalk with other cell death programs. Exp. Mol. Med. 2024, 56, 870–876. [Google Scholar] [CrossRef] [PubMed]
- Mohd Faizal, N.F.; Shai, S.; Savaliya, B.P.; Karen-Ng, L.P.; Kumari, R.; Kumar, R.; Vincent-Chong, V.K. A Narrative Review of Prognostic Gene Signatures in Oral Squamous Cell Carcinoma Using LASSO Cox Regression. Biomedicines 2025, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- Wallach, D.; Kovalenko, A. Keeping inflammation at bay. eLife 2014, 3, e02583. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, M.; Ahmad, R.; Tantry, I.Q.; Ahmad, W.; Siddiqui, S.; Alam, M.; Abbas, K.; Moinuddin; Hassan, M.I.; Habib, S.; et al. Apoptosis: A Comprehensive Overview of Signaling Pathways, Morphological Changes, and Physiological Significance and Therapeutic Implications. Cells 2024, 13, 1838. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Chakraborty, R.; Ranganathan, S. Proliferation and Apoptosis Pathways and Factors in Oral Squamous Cell Carcinoma. Int. J. Mol. Sci. 2022, 23, 1562. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.; Wei, Z.; Yang, W.; Huang, J.; Yang, Y.; Wang, J. The role of BCL-2 family proteins in regulating apoptosis and cancer therapy. Front. Oncol. 2022, 12, 985363. [Google Scholar] [CrossRef] [PubMed]
- Lindemann, A.; Takahashi, H.; Patel, A.A.; Osman, A.A.; Myers, J.N. Targeting the DNA Damage Response in OSCC with TP53 Mutations. J. Dent. Res. 2018, 97, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Hu, A.; Sun, L.; Lin, H.; Liao, Y.; Yang, H.; Mao, Y. Harnessing innate immune pathways for therapeutic advancement in cancer. Signal Transduct. Target. Ther. 2024, 9, 68. [Google Scholar] [CrossRef] [PubMed]
- Lo Muzio, L.; Farina, A.; Rubini, C.; Pezzetti, F.; Stabellini, G.; Laino, G.; Santarelli, A.; Pannone, G.; Bufo, P.; de Lillo, A.; et al. Survivin as prognostic factor in squamous cell carcinoma of the oral cavity. Cancer Lett. 2005, 225, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Khan, Z.; Khan, A.A.; Yadav, H.; Prasad, G.; Bisen, P.S. Survivin, a molecular target for therapeutic interventions in squamous cell carcinoma. Cell Mol. Biol. Lett. 2017, 22, 8. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.; Singh, B.; Schuster, G. Induction of apoptosis in oral cancer cells: Agents and mechanisms for potential therapy and prevention. Oral Oncol. 2004, 40, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Montero, J.; Haq, R. Adapted to Survive: Targeting Cancer Cells with BH3 Mimetics. Cancer Discov. 2022, 12, 1217–1232. [Google Scholar] [CrossRef] [PubMed]
- de Bakker, T.; Journe, F.; Descamps, G.; Saussez, S.; Dragan, T.; Ghanem, G.; Krayem, M.; Van Gestel, D. Restoring p53 Function in Head and Neck Squamous Cell Carcinoma to Improve Treatments. Front. Oncol. 2021, 11, 799993. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.T.; Sun, S.Y. Regulation of Cancer Metastasis by TRAIL/Death Receptor Signaling. Biomolecules 2021, 11, 499. [Google Scholar] [CrossRef] [PubMed]
- Kansal, V.; Kinney, B.L.C.; Uppada, S.; Saba, N.F.; Stokes, W.A.; Buchwald, Z.S.; Schmitt, N.C. The expanding role of IAP antagonists for the treatment of head and neck cancer. Cancer Med. 2023, 12, 13958–13965. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.P.N.; Pinto, B.; Monteiro, L.; Silva, P.M.A.; Bousbaa, H. Combination Therapy as a Promising Way to Fight Oral Cancer. Pharmaceutics 2023, 15, 1653. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Tao, L.; Qiu, J.; Xu, J.; Yang, X.; Zhang, Y.; Tian, X.; Guan, X.; Cen, X.; Zhao, Y. Tumor biomarkers for diagnosis, prognosis and targeted therapy. Signal Transduct. Target. Ther. 2024, 9, 132. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Virgilio, L.; Silva-Lucero, M.D.; Flores-Morelos, D.S.; Gallardo-Nieto, J.; Lopez-Toledo, G.; Abarca-Fernandez, A.M.; Zacapala-Gomez, A.E.; Luna-Munoz, J.; Montiel-Sosa, F.; Soto-Rojas, L.O.; et al. Autophagy: A Key Regulator of Homeostasis and Disease: An Overview of Molecular Mechanisms and Modulators. Cells 2022, 11, 2262. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.; O’Sullivan, J. The two faces of autophagy in oral squamous cell carcinoma. Arch. Oral Biol. 2022, 134, 105321. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Xue, J.; Ji, X. Radiosensitizing effects of Withaferin A in gastric cancer cells via autophagy Inhibition and mitochondrial disruption. Sci. Rep. 2025, 15, 19695. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Cáceres, M.P.; Munoz, L.; Pradenas, J.M.; Pena, F.; Lagos, P.; Aceiton, P.; Owen, G.I.; Morselli, E.; Criollo, A.; Ravasio, A.; et al. Mechanobiology of Autophagy: The Unexplored Side of Cancer. Front. Oncol. 2021, 11, 632956. [Google Scholar] [CrossRef] [PubMed]
- Melia, T.J.; Lystad, A.H.; Simonsen, A. Autophagosome biogenesis: From membrane growth to closure. J. Cell Biol. 2020, 219, 2085. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Lei, X.; Zhao, G.; Guo, R.; Cui, N. mTOR in programmed cell death and its therapeutic implications. Cytokine Growth Factor. Rev. 2023, 71–72, 66–81. [Google Scholar] [CrossRef] [PubMed]
- Pareek, G.; Kundu, M. Physiological functions of ULK1/2. J. Mol. Biol. 2024, 436, 168472. [Google Scholar] [CrossRef] [PubMed]
- Noda, T.; Matsunaga, K.; Taguchi-Atarashi, N.; Yoshimori, T. Regulation of membrane biogenesis in autophagy via PI3P dynamics. Semin. Cell Dev. Biol. 2010, 21, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Tian, K.; Ran, Y.; Zhou, H.; Zhou, L.; Ding, Y.; Tang, X. Beclin-1: A therapeutic target at the intersection of autophagy, immunotherapy, and cancer treatment. Front. Immunol. 2024, 15, 1506426. [Google Scholar] [CrossRef] [PubMed]
- Iriondo, M.N.; Etxaniz, A.; Varela, Y.R.; Ballesteros, U.; Lázaro, M.; Valle, M.; Fracchiolla, D.; Martens, S.; Montes, L.R.; Goñi, F.M.; et al. Effect of ATG12-ATG5-ATG16L1 autophagy E3-like complex on the ability of LC3/GABARAP proteins to induce vesicle tethering and fusion. Cell Mol. Life Sci. 2023, 80, 56. [Google Scholar] [CrossRef] [PubMed]
- Ueno, T.; Komatsu, M. Monitoring Autophagy Flux and Activity: Principles and Applications. Bioessays 2020, 42, e2000122. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Cao, X.; Zhu, Q. p62/SQSTM1 in cancer: Phenomena, mechanisms, and regulation in DNA damage repair. Cancer Metastasis Rev. 2025, 44, 33. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Zhou, C.; Que, H.; Wang, Y.; Rong, Y. The fate of autophagosomal membrane components. Autophagy 2023, 19, 370–371. [Google Scholar] [CrossRef] [PubMed]
- Schleinitz, A.; Pöttgen, L.A.; Keren-Kaplan, T.; Pu, J.; Saftig, P.; Bonifacino, J.S.; Haas, A.; Jeschke, A. Consecutive functions of small GTPases guide HOPS-mediated tethering of late endosomes and lysosomes. Cell Rep. 2023, 42, 111969. [Google Scholar] [CrossRef]
- Jarocki, M.; Turek, K.; Saczko, J.; Tarek, M.; Kulbacka, J. Lipids associated with autophagy: Mechanisms and therapeutic targets. Cell Death Discov. 2024, 10, 460. [Google Scholar] [CrossRef] [PubMed]
- Jalali, P.; Shahmoradi, A.; Samii, A.; Mazloomnejad, R.; Hatamnejad, M.R.; Saeed, A.; Namdar, A.; Salehi, Z. The role of autophagy in cancer: From molecular mechanism to therapeutic window. Front. Immunol. 2025, 16, 1528230. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Serrano, A.J.; Sanchez-Maldonado, J.M.; Gonzalez-Olmedo, C.; Carretero-Fernandez, M.; Diaz-Beltran, L.; Gutierrez-Bautista, J.F.; Garcia-Verdejo, F.J.; Galvez-Montosa, F.; Lopez-Lopez, J.A.; Garcia-Martin, P.; et al. Crosstalk Between Autophagy and Oxidative Stress in Hematological Malignancies: Mechanisms, Implications, and Therapeutic Potential. Antioxidants 2025, 14, 264. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Li, Y.; Ge, H.; Wu, Y.; Zhang, Y.; Guo, S.; Zhang, P.; Cheng, J.; Wang, Y. Identification of an autophagy-related prognostic signature in head and neck squamous cell carcinoma. J. Oral Pathol. Med. 2021, 50, 1040–1049. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wu, W.; Xu, M.; Mi, J.; Xu, L.; Wang, R. Development and Validation of an Autophagy-Related Signature for Head and Neck Squamous Cell Carcinoma. Biomed. Res. Int. 2021, 2021, 1028158. [Google Scholar] [CrossRef] [PubMed]
- Pena-Oyarzun, D.; Reyes, M.; Hernandez-Caceres, M.P.; Kretschmar, C.; Morselli, E.; Ramirez-Sarmiento, C.A.; Lavandero, S.; Torres, V.A.; Criollo, A. Role of Autophagy in the Microenvironment of Oral Squamous Cell Carcinoma. Front. Oncol. 2020, 10, 602661. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi-Dehlaghi, F.; Mohammadi, P.; Valipour, E.; Pournaghi, P.; Kiani, S.; Mansouri, K. Autophagy: A challengeable paradox in cancer treatment. Cancer Med. 2023, 12, 11542–11569. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Aziz, Y.S.; Leck, L.Y.W.; Jansson, P.J.; Sahni, S. Emerging Role of Autophagy in the Development and Progression of Oral Squamous Cell Carcinoma. Cancers 2021, 13, 6152. [Google Scholar] [CrossRef] [PubMed]
- Benbelkacem, M.; Moulai, N.; Chader, H.; Ouahioune, W.; Bourouba, M. Dichloroacetate and chloroquine in combination with arsenite suppress ROS-induced oral squamous cell carcinoma (OSCC) development and improve BALB/c mice survival. Free Radic. Biol. Med. 2025, 227, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.Y.; Ahn, S.G.; Yoon, J.H. Apicidin, a histone deaceylase inhibitor, induces both apoptosis and autophagy in human oral squamous carcinoma cells. Oral Oncol. 2011, 47, 1032–1038. [Google Scholar] [CrossRef] [PubMed]
- Coeli-Lacchini, F.B.; da Silva, G.; Belentani, M.; Alves, J.S.F.; Ushida, T.R.; Lunardelli, G.T.; Garcia, C.B.; Silva, T.A.; Lopes, N.P.; Leopoldino, A.M. Spermidine Suppresses Oral Carcinogenesis through Autophagy Induction, DNA Damage Repair, and Oxidative Stress Reduction. Am. J. Pathol. 2023, 193, 2172–2181. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Lee, C.Y.; Lu, C.C.; Tsai, F.J.; Hsu, Y.M.; Tsao, J.W.; Juan, Y.N.; Chiu, H.Y.; Yang, J.S.; Wang, C.C. Resveratrol-induced autophagy and apoptosis in cisplatin-resistant human oral cancer CAR cells: A key role of AMPK and Akt/mTOR signaling. Int. J. Oncol. 2017, 50, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Han, E.J.; Choi, E.Y.; Jeon, S.J.; Lee, S.W.; Moon, J.M.; Jung, S.H.; Jung, J.Y. Piperine Induces Apoptosis and Autophagy in HSC-3 Human Oral Cancer Cells by Regulating PI3K Signaling Pathway. Int. J. Mol. Sci. 2023, 24, 13949. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.H.; Wu, W.S.; Lin, L.C.; Liu, C.S.; Ho, S.Y.; Wang, B.J.; Huang, B.M.; Yeh, Y.L.; Chiu, H.W.; Yang, W.L.; et al. Bortezomib enhances radiosensitivity in oral cancer through inducing autophagy-mediated TRAF6 oncoprotein degradation. J. Exp. Clin. Cancer Res. 2018, 37, 91. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Wu, B.W. Recognition of necroptosis: From molecular mechanisms to detection methods. Biomed. Pharmacother. 2024, 178, 117196. [Google Scholar] [CrossRef] [PubMed]
- Ye, K.; Chen, Z.; Xu, Y. The double-edged functions of necroptosis. Cell Death Dis. 2023, 14, 163. [Google Scholar] [CrossRef] [PubMed]
- Dhuriya, Y.K.; Sharma, D. Necroptosis: A regulated inflammatory mode of cell death. J. Neuroinflammation 2018, 15, 199. [Google Scholar] [CrossRef] [PubMed]
- Krishnasamy, N.; Ramadoss, R.; Panneer Selvam, S.; Sundar, S.; Hemashree, K. Necroptosis could serve as an indicator of therapeutic resistance in viral induced well-differentiated oral squamous cell carcinoma? Oral Oncol. Rep. 2024, 10, 100378. [Google Scholar] [CrossRef]
- Huang, K.; Gu, X.; Xu, H.; Li, H.; Shi, M.; Wei, D.; Wang, S.; Li, Y.; Liu, B.; Li, Y. Prognostic Value of Necroptosis-Related Genes Signature in Oral Squamous Cell Carcinoma. Cancers 2023, 15, 4539. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, R.P.; Pandiar, D.; Ramani, P.; Jayaraman, S. Necroptosis in human cancers with special emphasis on oral squamous cell carcinoma. J. Stomatol. Oral Maxillofac. Surg. 2023, 124, 101565. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, R.P.; Pandiar, D.; Dilipan, E.; Inbarajan, A.; Ramani, P.; Jayaraman, S. Role of PDGFRA-Associated miR-140 in Modulating Necroptotic Proteins in Oral Squamous Cell Carcinoma: A Molecular Docking Study. J. Int. Oral Health 2025, 17, 73–81. [Google Scholar] [CrossRef]
- Saravanan, L.; Mahale, A.; Gota, V.; Khandelia, P.; Kulkarni, O.P. Necrostatin-1 attenuates oral squamous cell carcinoma by modulating tumour immune response in mice. Fundam. Clin. Pharmacol. 2025, 39, e70008. [Google Scholar] [CrossRef] [PubMed]
- Shao, K.Y.; Luo, S.D.; Huang, E.Y.; Chang, T.M.; Botcha, L.; Sehar, M.; Liu, J.F.; Chuang, P.K. Acetylshikonin induces cell necroptosis via mediating mitochondrial function and oxidative stress-regulated signaling in human Oral Cancer cells. Bioorg. Chem. 2025, 159, 108396. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Xie, F.; Zhou, X.; Wu, Y.; Yan, H.; Liu, T.; Huang, J.; Wang, F.; Zhou, F.; Zhang, L. Role of pyroptosis in inflammation and cancer. Cell Mol. Immunol. 2022, 19, 971–992. [Google Scholar] [CrossRef] [PubMed]
- Kong, Q.; Zhang, Z. Cancer-associated pyroptosis: A new license to kill tumor. Front. Immunol. 2023, 14, 1082165. [Google Scholar] [CrossRef] [PubMed]
- Chia, W.T.; Chen, K.Y.; Yang, C.Y.; Hsieh, C.C.; Tsao, C.H.; Lin, C.K.; Peng, B.; Ho, S.L.; Chen, Y.L.; Chang, S.C.; et al. Okanin Inhibits Cell Growth and Induces Apoptosis and Pyroptosis in Oral Cancer. Cancers 2024, 16, 3195. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Mo, R.; Yang, M.; Xie, H.; Ma, F.; Ding, Z.; Wu, S.; Lam, J.W.Y.; Du, J.; Zhang, J.; et al. Mitochondria-Targeting AIEgens as Pyroptosis Inducers for Boosting Type-I Photodynamic Therapy of Tongue Squamous Cell Carcinoma. ACS Nano 2024, 18, 26140–26152. [Google Scholar] [CrossRef] [PubMed]
- Zi, M.; Xingyu, C.; Yang, C.; Xiaodong, S.; Shixian, L.; Shicheng, W. Improved antitumor immunity of chemotherapy in OSCC treatment by Gasdermin-E mediated pyroptosis. Apoptosis 2023, 28, 348–361. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wu, Z.-Z.; Zhu, S.-W.; Wan, S.-C.; Zhang, M.-J.; Zhang, B.-X.; Yang, Q.-C.; Xiao, Y.; Li, H.; Mao, L.; et al. CTLA-4 blockade induces tumor pyroptosis via CD8+ T cells in head and neck squamous cell carcinoma. Mol. Ther. 2023, 31, 2154–2168. [Google Scholar] [CrossRef] [PubMed]
- Xin, Y.; Zhang, J.; Jiang, Q.; Qiu, J. Construction of prognostic signature of patients with oral squamous cell carcinoma based on pyroptosis-related long non-coding RNAs. Front. Surg. 2022, 9, 935765. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Olzmann, J.A. The cell biology of ferroptosis. Nat. Rev. Mol. Cell Biol. 2024, 25, 424–442. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zhu, S.; Zhao, R.; Liu, W.; Jin, L.; Ren, X.; He, H. Targeting ferroptosis as a potential strategy to overcome the resistance of cisplatin in oral squamous cell carcinoma. Front. Pharmacol. 2024, 15, 1402514. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Wu, Y.; Ying, Y.; Ding, Y. Current knowledge of ferroptosis in the pathogenesis and prognosis of oral squamous cell carcinoma. Cell Signal 2024, 119, 111176. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-H.; Chiu, V.; Huang, C.-Y.; Tzeng, I.-S.; Hsieh, P.-C.; Kuo, C.-Y.J.C.T.i.N.R. Promotion of Ferroptosis in Oral Cancer Cell Lines by Chrysophanol. Curr. Top. Nutraceutical Res. 2020, 18, 273. [Google Scholar] [CrossRef]
- Hsu, P.C.; Hsu, C.C.; Hsia, Y.J.; Kuo, C.Y. Chrysophanol Suppresses Cell Growth via mTOR/PPAR-alpha Regulation and ROS Accumulation in Cultured Human Tongue Squamous Carcinoma SAS Cells. Curr. Issues Mol. Biol. 2022, 44, 1528–1538. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Yang, M.; Nakamura, K.; Tanaka, H.; Hori, M.; Nishio, M.; Suzuki, A.; Hibi, H.; Toyokuni, S. Ferroptosis induced by plasma-activated Ringer’s lactate solution prevents oral cancer progression. Oral Dis. 2024, 30, 3912–3924. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, C.; Li, X.; Tao, Z.; Zhu, W.; Su, Y.; Choi, W.S. Melatonin and erastin emerge synergistic anti-tumor effects on oral squamous cell carcinoma by inducing apoptosis, ferroptosis, and inhibiting autophagy through promoting ROS. Cell Mol. Biol. Lett. 2023, 28, 36. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.C.; Chang, P.C.; Lin, Y.T.; Huang, P.T.; Chen, J.Y.; Lin, C.S.; Wu, B.N.; Chang, H.M.; Wu, W.J.; Chang, C.I.; et al. Repurposing of the Antipsychotic Trifluoperazine Induces SLC7A11/GPX4- Mediated Ferroptosis of Oral Cancer via the ROS/Autophagy Pathway. Int. J. Biol. Sci. 2024, 20, 6090–6113. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Zhong, Y.; Yuan, F.; Zhang, L.; Cai, Y.; Liao, L. A ferroptosis-related prognostic model with excellent clinical performance based on the exploration of the mechanism of oral squamous cell carcinoma progression. Sci. Rep. 2023, 13, 1461. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Hu, S.; Tang, Y.; Yang, D.; Chen, Q. PPT1 Promotes Growth and Inhibits Ferroptosis of Oral Squamous Cell Carcinoma Cells. Curr. Cancer Drug Targets 2024, 24, 1047–1060. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Lan, T.; Zheng, D.L.; Ding, L.C.; Lu, Y.G. CDH4 inhibits ferroptosis in oral squamous cell carcinoma cells. BMC Oral Health 2023, 23, 329. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Jia, R.; Guo, J. RUNX2 isoform II protects cancer cells from ferroptosis and apoptosis by promoting PRDX2 expression in oral squamous cell carcinoma. eLife 2025, 13, RP99122. [Google Scholar] [CrossRef] [PubMed]
- Rao, Y.; Li, J.; Shi, L.; Chen, X.; Hu, Y.; Mao, Y.; Zhang, X.; Liu, X. Silencing CK19 regulates ferroptosis by affecting the expression of GPX4 and ACSL4 in oral squamous cell carcinoma in vivo and in vitro. Sci. Rep. 2024, 14, 15968. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.W.; Liu, C.L.; Li, X.M.; Shang, Y. Quercetin induces ferroptosis by inactivating mTOR/S6KP70 pathway in oral squamous cell carcinoma. Toxicol. Mech. Methods 2024, 34, 669–675. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Fu, J.; Zhao, Y.; Xu, J.; Chen, C.; Zheng, L.; Wang, B. Comprehensive Analysis of the Significance of Ferroptosis-Related Genes in the Prognosis and Immunotherapy of Oral Squamous Cell Carcinoma. Bioinform. Biol. Insights 2022, 16, 11779322221115548. [Google Scholar] [CrossRef] [PubMed]
- Melo, G.; Silva, C.A.B.; Hague, A.; Parkinson, E.K.; Rivero, E.R.C. Anticancer effects of putative and validated BH3-mimetic drugs in head and neck squamous cell carcinomas: An overview of current knowledge. Oral Oncol. 2022, 132, 105979. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; Mikelis, C.M.; Zhang, Y.; Gutkind, J.S.; Zhang, B. TRAIL induces apoptosis in oral squamous carcinoma cells--a crosstalk with oncogenic Ras regulated cell surface expression of death receptor 5. Oncotarget 2013, 4, 206–217. [Google Scholar] [CrossRef] [PubMed]
- Vogler, M.; Braun, Y.; Smith, V.M.; Westhoff, M.A.; Pereira, R.S.; Pieper, N.M.; Anders, M.; Callens, M.; Vervliet, T.; Abbas, M.; et al. The BCL2 family: From apoptosis mechanisms to new advances in targeted therapy. Signal Transduct. Target. Ther. 2025, 10, 91. [Google Scholar] [CrossRef] [PubMed]
- Mortezagholi, B.; Nasiri, K.; Movahed, E.; Dadgar, E.; Nejati, S.T.; Hassani, P.; Esfahaniani, M.; Rafieyan, S. MiR-34 by targeting p53 induces apoptosis and DNA damage in paclitaxel-resistant human oral squamous carcinoma cells. Chem. Biol. Drug Des. 2023, 102, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.L.; Wu, X.; Yin, D.; Jia, X.H.; Chen, X.; Gu, Z.Y.; Zhu, X.M. Autophagy inhibitors for cancer therapy: Small molecules and nanomedicines. Pharmacol. Ther. 2023, 249, 108485. [Google Scholar] [CrossRef] [PubMed]
- Semlali, A.; Papadakos, S.; Contant, C.; Zouaoui, I.; Rouabhia, M. Rapamycin inhibits oral cancer cell growth by promoting oxidative stress and suppressing ERK1/2, NF-kappaB and beta-catenin pathways. Front. Oncol. 2022, 12, 873447. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Tang, Z. Prognostic model revealing pyroptosis-related signatures in oral squamous cell carcinoma based on bioinformatics analysis. Sci. Rep. 2024, 14, 6149. [Google Scholar] [CrossRef] [PubMed]
- Zeng, D.; Wang, X.; Zhang, S.; Zheng, A.; Huang, Q.; Cao, L. Pyroptosis-related gene-based prognostic signature for predicting the overall survival of oral squamous cell carcinoma patients. Front. Surg. 2022, 9, 903271. [Google Scholar] [CrossRef] [PubMed]
- Ru, Q.; Li, Y.; Chen, L.; Wu, Y.; Min, J.; Wang, F. Iron homeostasis and ferroptosis in human diseases: Mechanisms and therapeutic prospects. Signal Transduct. Target. Ther. 2024, 9, 271. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, A.; Battaglia, A.M.; Sacco, A.; Petriaggi, L.; Giorgio, E.; Barone, S.; Biamonte, F.; Giudice, A. Ferroptosis and oral squamous cell carcinoma: Connecting the dots to move forward. Front. Oral Health 2024, 5, 1461022. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.; Gao, L.; Makitie, A.A.; Florek, E.; Czarnywojtek, A.; Saba, N.F.; Ferlito, A. Iron, Ferroptosis, and Head and Neck Cancer. Int. J. Mol. Sci. 2023, 24, 15127. [Google Scholar] [CrossRef] [PubMed]
- Zheng, T.; Liu, Q.; Xing, F.; Zeng, C.; Wang, W. Disulfidptosis: A new form of programmed cell death. J. Exp. Clin. Cancer Res. 2023, 42, 137. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Nie, L.; Zhang, Y.; Yan, Y.; Wang, C.; Colic, M.; Olszewski, K.; Horbath, A.; Chen, X.; Lei, G.; et al. Actin cytoskeleton vulnerability to disulfide stress mediates disulfidptosis. Nat. Cell Biol. 2023, 25, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhuang, L.; Gan, B. Disulfidptosis: Disulfide stress-induced cell death. Trends Cell Biol. 2024, 34, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Niu, X.; Zhou, M.; Li, W. Development and validation of a novel disulfidptosis-related lncRNAs signature in patients with HPV-negative oral squamous cell carcinoma. Sci. Rep. 2024, 14, 14436. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liu, B.; Deng, S.Z.; Xiong, T.; Dai, L.; Cheng, B. Disulfidptosis-related immune patterns predict prognosis and characterize the tumor microenvironment in oral squamous cell carcinoma. BMC Oral Health 2025, 25, 180. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Wang, X.; Zhou, Y.; Wang, X.; Yu, Y. Autophagy, ferroptosis, pyroptosis, and necroptosis in tumor immunotherapy. Signal Transduct. Target. Ther. 2022, 7, 196. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Xu, J.; Zhang, B.; Liu, J.; Liang, C.; Hua, J.; Meng, Q.; Yu, X.; Shi, S. Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J. Hematol. Oncol. 2020, 13, 110. [Google Scholar] [CrossRef] [PubMed]
- Sant’Angelo, D.; Descamps, G.; Lecomte, V.; Stanicki, D.; Penninckx, S.; Dragan, T.; Van Gestel, D.; Laurent, S.; Journe, F. Therapeutic Approaches with Iron Oxide Nanoparticles to Induce Ferroptosis and Overcome Radioresistance in Cancers. Pharmaceuticals 2025, 18, 325. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapeutic Strategy | Target | Cancer Cell Type/Model | Key Findings | References |
|---|---|---|---|---|
| IAP overexpression (e.g., XIAP and survivin) | IAP family proteins | OSCC tissues and cell lines (e.g., SCC-9 and CAL-27) | Overexpression linked to resistance to apoptosis, poor prognosis | [20,21] |
| BH3 mimetics (e.g., ABT-199/Venetoclax) | Bcl-2 family proteins | OSCC cell lines (e.g., HSC-3 and SCC-25) | Induced intrinsic apoptosis, enhanced sensitivity to chemotherapy | [23] |
| p53 reactivation (e.g., PRIMA-1 and APR-246) | Mutant p53 | OSCC models with p53 mutation | Restored p53 function, induced apoptosis | [24] |
| Death receptor agonists (e.g., TRAIL) | TRAIL receptors (DR4/DR5) | OSCC xenograft models, CAL-27, SCC-15 | Triggered extrinsic apoptosis, though variable efficacy due to resistance | [25] |
| SMAC mimetics (e.g., LCL161 and BV6) | IAP antagonism | OSCC cell lines and preclinical mouse models | Sensitized cells to apoptosis, especially when combined with TRAIL or chemo | [26] |
| Combination therapies (e.g., with ICIs) | Multiple (apoptosis + immune checkpoint) | Syngeneic or humanized OSCC mouse models | Enhanced tumor regression and immune activation | [27,28] |
| Cancer Type | Experimental Model | Autophagy Marker(s) | Findings | References |
|---|---|---|---|---|
| OSCC | Human tissue samples and cell lines (e.g., SCC-9 and HSC-3) | Beclin-1, LC3 | Loss of Beclin-1 and reduced LC3 expression correlated with tumor progression and poor prognosis, suggesting a tumor-suppressive role of autophagy. | [19] |
| HNSCC | Genomic and transcriptomic analysis of patient cohorts | NKX2-3, FADD | Identified as autophagy-related genes linked to poor prognosis and potential therapeutic targeting. | [46] |
| HNSCC | Bioinformatics and IHC validation | PARK2, EGFR | Involved in autophagy regulation; PARK2 acts as a tumor suppressor, while EGFR is implicated in autophagy-mediated survival signaling. | [47] |
| OSCC | In vitro cell culture under stress (hypoxia, nutrient deprivation) | LC3-II, p62 | Autophagy confers survival advantage under metabolic stress and may mediate resistance to therapy. | [48] |
| OSCC and other solid tumors | Preclinical studies using autophagy modulators | ROS, LC3, p62 | ROS both trigger and result from autophagy. Pathways such as PI3K/AKT/mTOR and AMPK regulate this loop, offering therapeutic targets. | [45,49] |
| Therapeutic Agent(s) | Cancer Cell Type | Key Mechanisms Targeted | Experimental Model | References |
|---|---|---|---|---|
| NaAsO2 + CQ + DCA | OSCC cells | Oxidative stress, ACD, glycolysis | In vivo (Xenograft mouse model) | [51] |
| Apicidin | OSCC cells | Apoptosis and ACD | In vitro | [52] |
| SPD | OSCC cells | ACD induction, reduced DNA damage and oxidative stress | In vivo (4NQO-induced mouse oral carcinogenesis model) | [53] |
| Resveratrol | Cisplatin-resistant oral cancer CAR cells | Mitochondria-dependent apoptosis and ACD via AMPK activation | In vitro | [54] |
| Piperine | Oral cancer cells | Apoptosis and autophagy via PI3K/AKT/mTOR inhibition | In vitro and in vivo (Xenograft mouse model) | [55] |
| Bortezomib | OSCC cells | Radiosensitization via autophagic degradation of TRAF6 | In vitro and in vivo (Xenograft mouse model) | [56] |
| Model Type | Cell/Cancer Type | Key Findings | References |
|---|---|---|---|
| In vitro and Ex vivo | OSCC cell lines and patient data | miR-140 modulates PDGFRA–RIPK3/MLKL signaling; expression correlates with patient prognosis. | [64] |
| Ex vivo | OSCC patient samples | RIPK1, RIPK3, and MLKL expression correlates with disease severity; necroptosis implicated in OSCC progression. | [65] |
| In vivo | 4-NQO-induced mouse oral carcinogenesis model | Increased expression of necroptosis markers (e.g., RIPK1, RIPK3, and MLKL) correlates with tumor severity; NEC-1 treatment reduces tumor burden and improves histopathology. | [65] |
| In vivo | OSCC xenograft mouse model | NEC-1 reduces tumor growth and reshapes the immune microenvironment (↓M2 macrophages, ↓myeloid-derived suppressor cells, ↑M1 macrophages). | [65] |
| In vitro | Human OSCC cell lines | Acetylshikonin induces necroptosis by activating RIPK1, RIPK3, and MLKL via mitochondrial and oxidative stress-regulated pathways. | [66] |
| Cancer Cell Type/Model | Experimental Model | Mechanism/Outcome | Reference |
|---|---|---|---|
| OSCC cell lines: SAS, SCC25, HSC3, OEC-M1 | In vitro; in vivo (SAS xenograft mouse model) | Okanin induced dose-dependent cytotoxicity via G2/M arrest, apoptosis, and pyroptosis (↑CASP1, GSDMC/D/E, caspase-3/7); reduced tumor volume in vivo. | [69] |
| TSCC cells | In vitro (Light-irradiated Th-M); in vivo (Tumor-bearing mice) | Th-M induced mitochondrial dysfunction, leading to caspase-3 and GSDME–mediated pyroptosis; effective tumor suppression with favorable biosafety. | [70] |
| OSCC cells (unspecified lines) | In vitro; preclinical immunotherapy setting | Decitabine restored GSDME expression, enhancing pyroptosis and chemotherapy response; improved immune microenvironment and ICB response. | [71] |
| HNSCC tumor samples (n = 57), normal tissues (n = 41); HNSCC cell line: 4MOSC1 | Ex vivo (Human tissue); in vivo (4MOSC1 orthotopic mouse model) | GSDMB/D/E upregulated in tumors; CTLA-4 blockade activated GSDMD/E via IFN-γ/TNF-α signaling (STAT1/IRF1 axis); effect CD8+ T cell–dependent. | [72] |
| OSCC (TCGA dataset) | Bioinformatic analysis (RNA-seq) | Identified 8 pyroptosis-related lncRNAs predictive of survival (AUC = 0.716); lncRNAs modulated immune infiltration and OSCC microenvironment. | [73] |
| Therapeutic Strategy | Modulator and Molecular Axis | Mechanism of Action | Key Targets | Outcomes in OSCC | References |
|---|---|---|---|---|---|
| Agent | Chrysophanol | Activates ferroptosis, modulates mTOR/PPAR-α signaling, ROS accumulation | mTOR, PPAR-α, ROS | Reduces tumor progression via iron-dependent cell death | [78,79] |
| PARL | Induces lipid peroxidation, glutathione depletion, GPX4 inactivation | RONS, GPX4, lipid ROS | Increased ferroptosis, reduced cancer cell viability | [80] | |
| Melatonin + Erastin | Enhances ROS-mediated apoptosis and ferroptosis | ROS, lipid peroxides | Promotes ferroptosis with minimal side effects | [81] | |
| TFP | Increases lipid ROS, inhibits SLC7A11/GPX4 axis, activates autophagy | SLC7A11, GPX4, lipid ROS | Induces ferroptosis, reduces mitochondrial function, poor prognosis in OSCC | [82] | |
| Gene/Pathway | Ferroptosis-Related Gene Signature | Predicts overall survival, correlates with TP53, immune response | CDGSH iron sulfur domain 2 (CISD2), DNA damage inducible transcript 4 (DDIT4), carbonic anhydrase 9 (CA9), arachidonate 15-lipoxygenase (ALOX15), ATG5, BECN1, BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3), peroxiredoxin 5 (PRDX5), microtubule-associated proteins 1A/1B light chain 3A (MAP1LC3A) | High predictive accuracy for OS in OSCC, associated with tumor progression | [83] |
| Palmitoyl-protein thioesterase 1 (PPT1) | Suppresses ferroptosis, promotes OSCC proliferation | PPT1 | Enhances tumor growth, poor prognosis | [84] | |
| Cadherin-4 (CDH4) | Reduces ferroptosis sensitivity, promotes proliferation and invasion | CDH4, epithelial–mesenchymal transition (EMT)-related genes | Correlation with poor survival, increased EMT and ferroptosis resistance | [85] | |
| Homeobox A10 (HOXA10)/RUNT-related transcription factor 2 (RUNX2) isoform II/peroxiredoxin-2 (PRDX2) Axis | Inhibits ferroptosis and apoptosis, promotes tumorigenesis | HOXA10, RUNX2 isoform II, PRDX2 | Ferroptosis resistance, tumor progression | [86] | |
| Cytokeratin 19 (CK19)/GPX4/acyl-CoA synthetase long chain family member 4 (ACSL4) Axis | Regulates ferroptosis, increases ROS and lipid peroxidation | GPX4, ACSL4, malondialdehyde (MDA), Fe2+ | Activation of ferroptosis pathway, increased ROS accumulation | [87] | |
| mTOR/S6KP70 Pathway | Induces ferroptosis via ROS accumulation | mTOR, S6KP70 | Quercetin-mediated ferroptosis induction, tumor suppression | [88] |
| Cell Death Pathway | Therapeutic Agent/Strategy | Mechanism of Action | Key Targets | Outcomes in OSCC | References |
|---|---|---|---|---|---|
| Apoptosis | p53 Reactivators (APR-246) | Restores p53 function, induces apoptosis | p53, ATM/ATR, CHK1/CHK2 | Increased chemosensitivity, enhanced apoptosis | [24] |
| BH3 Mimetics (Venetoclax) | Inhibits anti-apoptotic Bcl-2 proteins, restores apoptotic signaling | Bcl-2, Bax/Bak | Enhanced apoptosis, reduced tumor growth | [90,92] | |
| Death Receptor Agonists (TRAIL) | Activates extrinsic apoptotic pathway | TRAIL-R, Fas, TNFR1 | Selective apoptosis induction in OSCC cells | [91] | |
| miR-34 Restoration | Reduces P-gp expression, enhances DNA damage and apoptosis | P-gp, p53/ATM/ATR pathway | Reverses paclitaxel resistance, promotes apoptosis | [93] | |
| Autophagy | CQ, HCQ | Inhibits autophagosome–lysosome fusion | LC3, BECN1, p62 | Prevents autophagy-mediated survival under therapeutic stress | [94] |
| ACD | Rapamycin | Induces ACD, oxidative stress, inhibits signaling pathways | ERK1/2, NF-κB, β-catenin | Suppresses OSCC growth, promotes cell death | [95] |
| Necroptosis | Small Molecules (RIPK1, RIPK3, MLKL Inhibitors) | Induces necroptosis via death receptor pathways | RIPK1, RIPK3, MLKL | Promotes necroptosis, reduces tumor viability | [63] |
| Pyroptosis | CTLA-4 Blockade | Induces gasdermin-mediated pyroptosis via immune activation | CTLA-4, GSDMs | Enhances immune-mediated tumor cell death | [96] |
| Pyroptosis-related lncRNAs | Prognostic signature for risk stratification | CTLA4, CD5, IL12RB2 | Predicts patient survival, modulates immune response | [97] | |
| Ferroptosis | Erastin, RSL3 | Inhibits GPX4, promotes lipid peroxidation | GPX4, lipid ROS | Induces ferroptosis, reduces OSCC cell viability | [99,100] |
| Gene or Protein or lncRNA | Cell Death Pathway | Therapeutic Agent/Modulator | Experimental Model | References |
|---|---|---|---|---|
| Bcl-2/Bax/Bcl-xL | Apoptosis | BH3 mimetics (ABT-199/Venetoclax) | OSCC cell lines | [23,92] |
| p53 | Apoptosis | APR-246, PRIMA-1 | OSCC cell lines, clinical data | [24] |
| TRAIL-R/Fas/TNFR1 | Apoptosis | TRAIL receptor agonists | OSCC cell lines | [25] |
| NKX2-3, FADD, PARK2 | Autophagy | - | OSCC patient data | [46] |
| BECN1, LC3, p62 | ACD | Chloroquine, Rapamycin | OSCC cell lines, mouse model | [48,95] |
| RIPK1/RIPK3/MLKL | Necroptosis | Necrostatin-1 (NEC-1) | 4NQO-induced OSCC mouse model | [65] |
| Acetylshikonin | Necroptosis | Natural compound | Human OSCC cell lines | [66] |
| CASP1, GSDMD, GSDME | Pyroptosis | Okanin, Decitabine, Th-M | SAS xenograft, OSCC cell lines | [69,70,71] |
| Pyroptosis-related lncRNAs | Pyroptosis | - | TCGA OSCC datasets | [73] |
| GPX4, SLC7A11 | Ferroptosis | Erastin, RSL3, Trifluoperazine (TFP) | OSCC cell lines, patient tissues | [78,79,80,81,82] |
| HOXA10/RUNX2/PRDX2 | Ferroptosis | - | OSCC cell lines, patient tissue analysis | [86] |
| CK19, ACSL4 | Ferroptosis | - | OSCC cell lines | [87] |
| Quercetin | Ferroptosis | Natural flavonoid | OSCC cell lines | [88] |
| SLC7A11 | Disulfidptosis | Glucose transporter inhibitors | SLC7A11-high OSCC models | [102] |
| Disulfidptosis-related lncRNAs | Disulfidptosis | - | HPV-negative OSCC datasets, cell lines | [104] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, P.-C.; Tsai, C.-C.; Lin, Y.-H.; Kuo, C.-Y. Therapeutic Targeting of Apoptosis, Autophagic Cell Death, Necroptosis, Pyroptosis, and Ferroptosis Pathways in Oral Squamous Cell Carcinoma: Molecular Mechanisms and Potential Strategies. Biomedicines 2025, 13, 1745. https://doi.org/10.3390/biomedicines13071745
Hsu P-C, Tsai C-C, Lin Y-H, Kuo C-Y. Therapeutic Targeting of Apoptosis, Autophagic Cell Death, Necroptosis, Pyroptosis, and Ferroptosis Pathways in Oral Squamous Cell Carcinoma: Molecular Mechanisms and Potential Strategies. Biomedicines. 2025; 13(7):1745. https://doi.org/10.3390/biomedicines13071745
Chicago/Turabian StyleHsu, Po-Chih, Chung-Che Tsai, Ya-Hsuan Lin, and Chan-Yen Kuo. 2025. "Therapeutic Targeting of Apoptosis, Autophagic Cell Death, Necroptosis, Pyroptosis, and Ferroptosis Pathways in Oral Squamous Cell Carcinoma: Molecular Mechanisms and Potential Strategies" Biomedicines 13, no. 7: 1745. https://doi.org/10.3390/biomedicines13071745
APA StyleHsu, P.-C., Tsai, C.-C., Lin, Y.-H., & Kuo, C.-Y. (2025). Therapeutic Targeting of Apoptosis, Autophagic Cell Death, Necroptosis, Pyroptosis, and Ferroptosis Pathways in Oral Squamous Cell Carcinoma: Molecular Mechanisms and Potential Strategies. Biomedicines, 13(7), 1745. https://doi.org/10.3390/biomedicines13071745