
Cerebral Edema in Traumatic Brain Injury

 ,
,  ,
,  , and
, and 
Abstract
1. Introduction
2. Historical Aspects
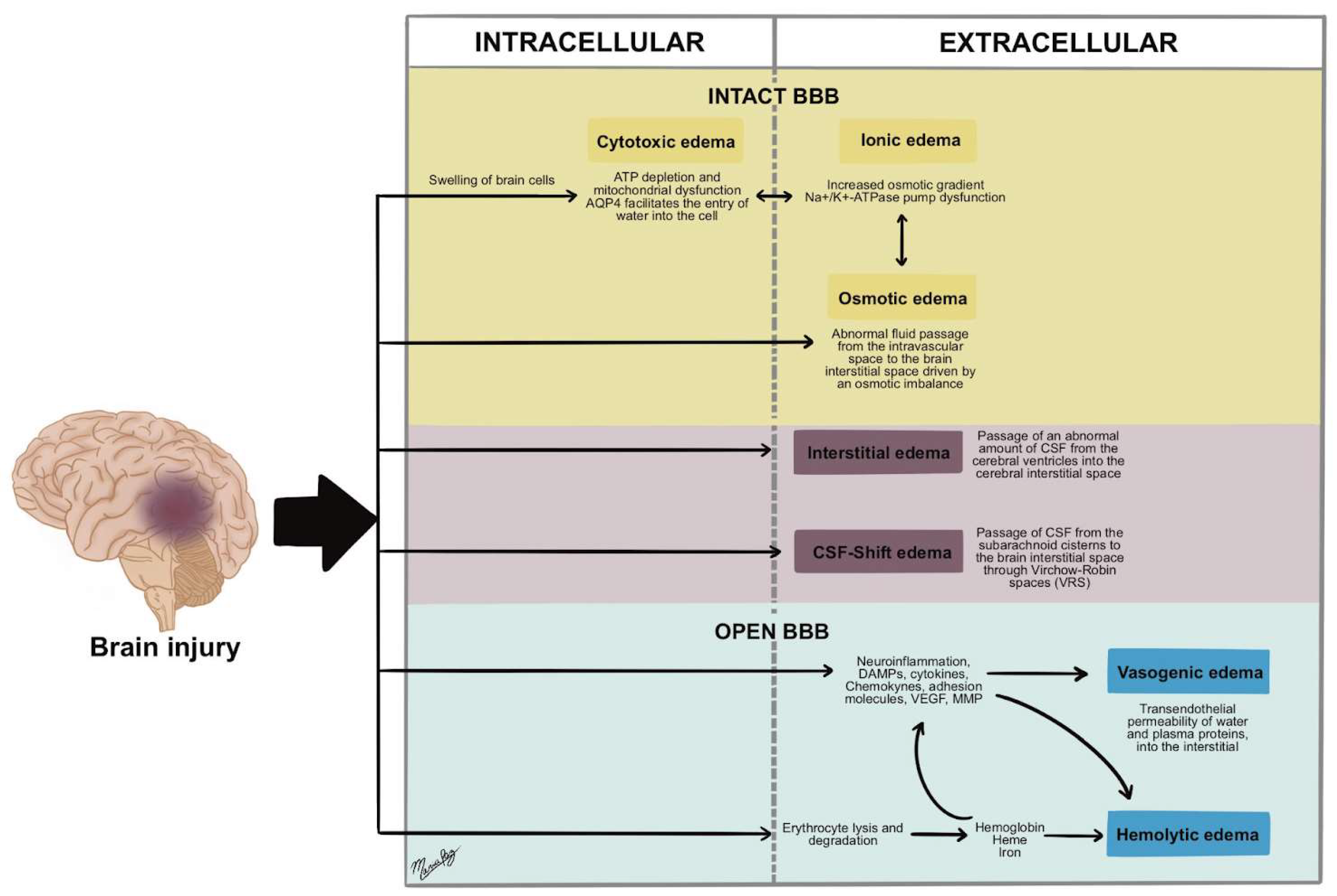
3. Types of Post-Traumatic Cerebral Edema
3.1. Cytotoxic Edema
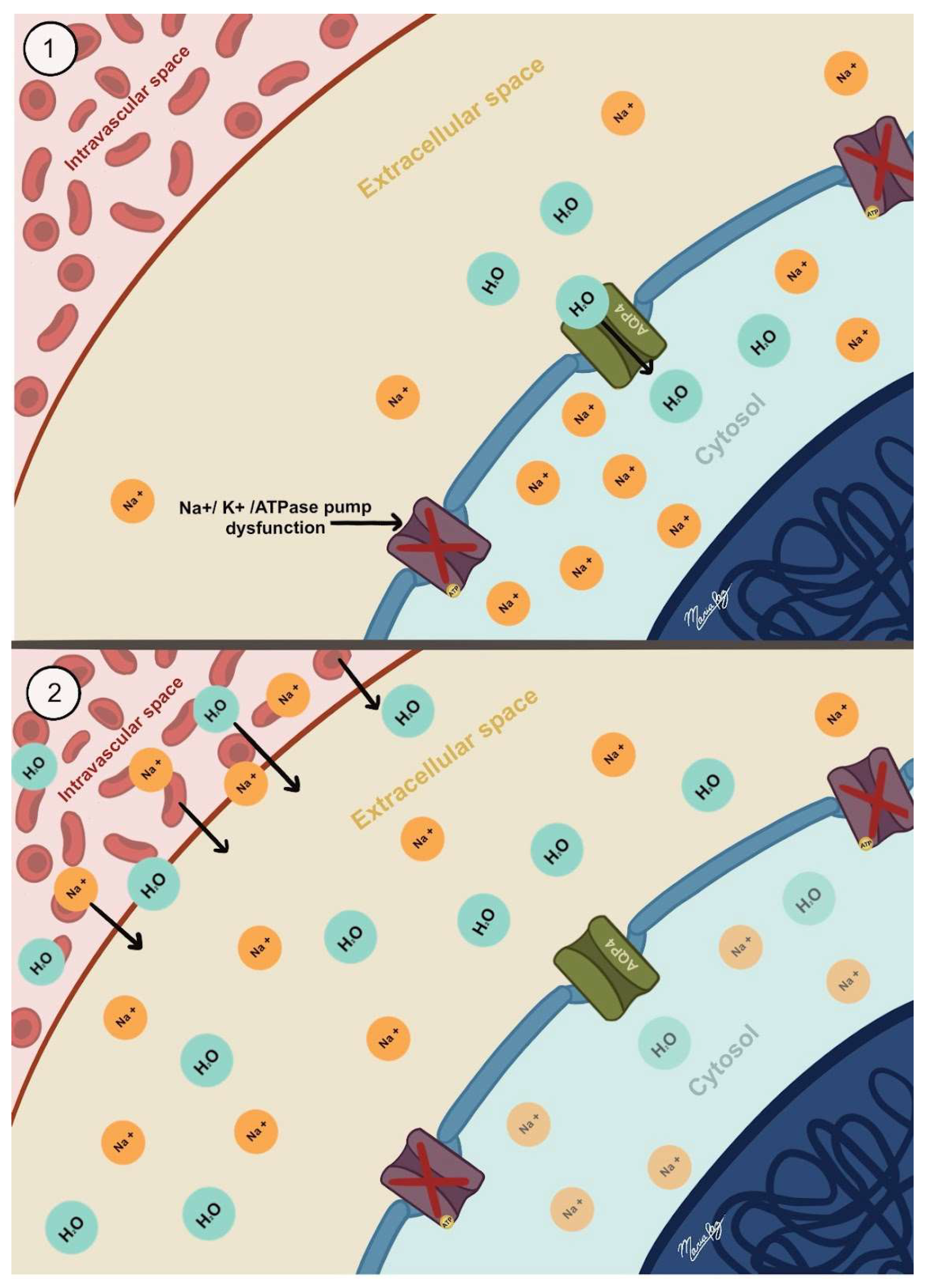
3.2. Ionic Edema
3.3. Vasogenic Edema
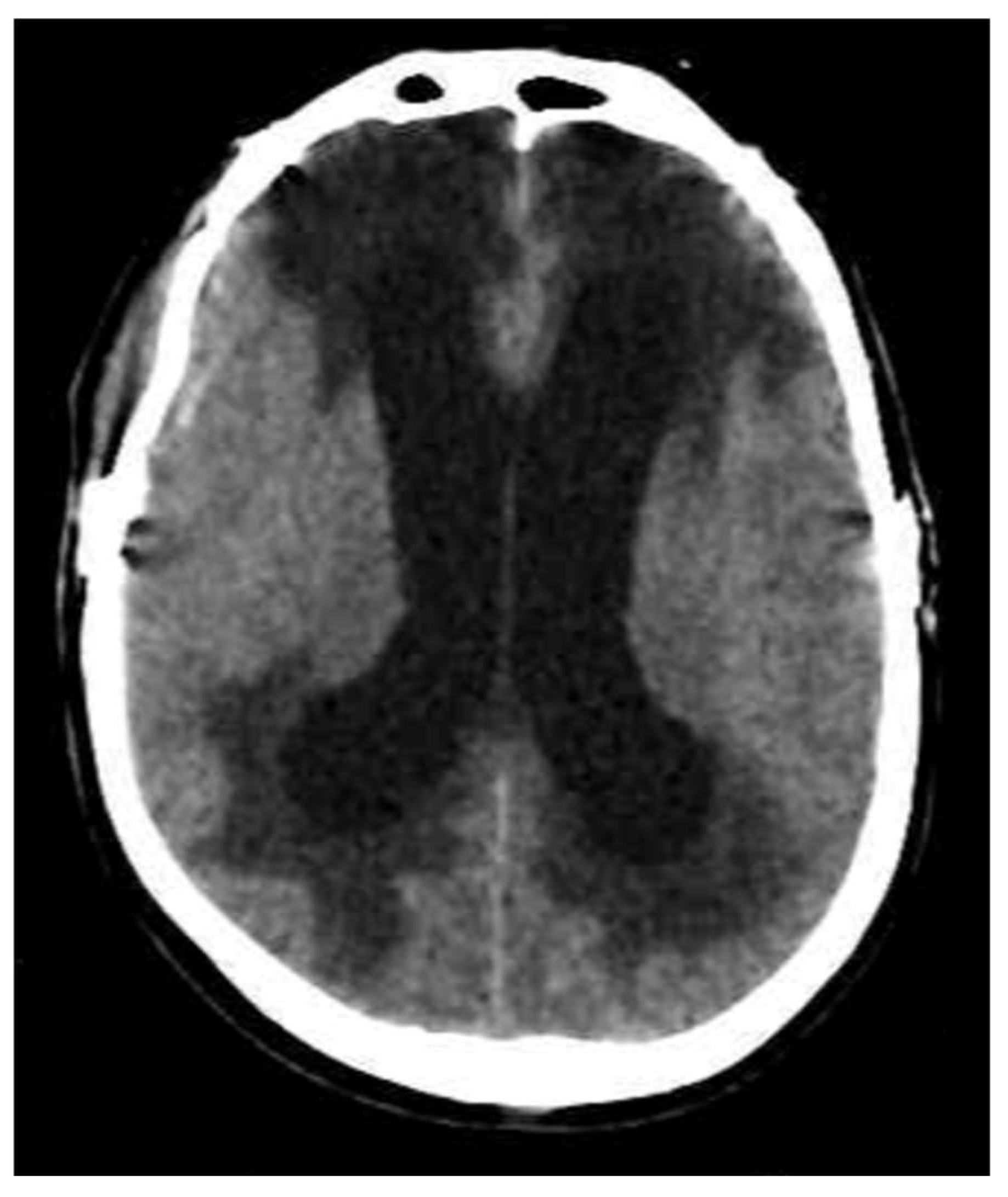
3.4. Interstitial Edema
3.5. Osmotic Edema
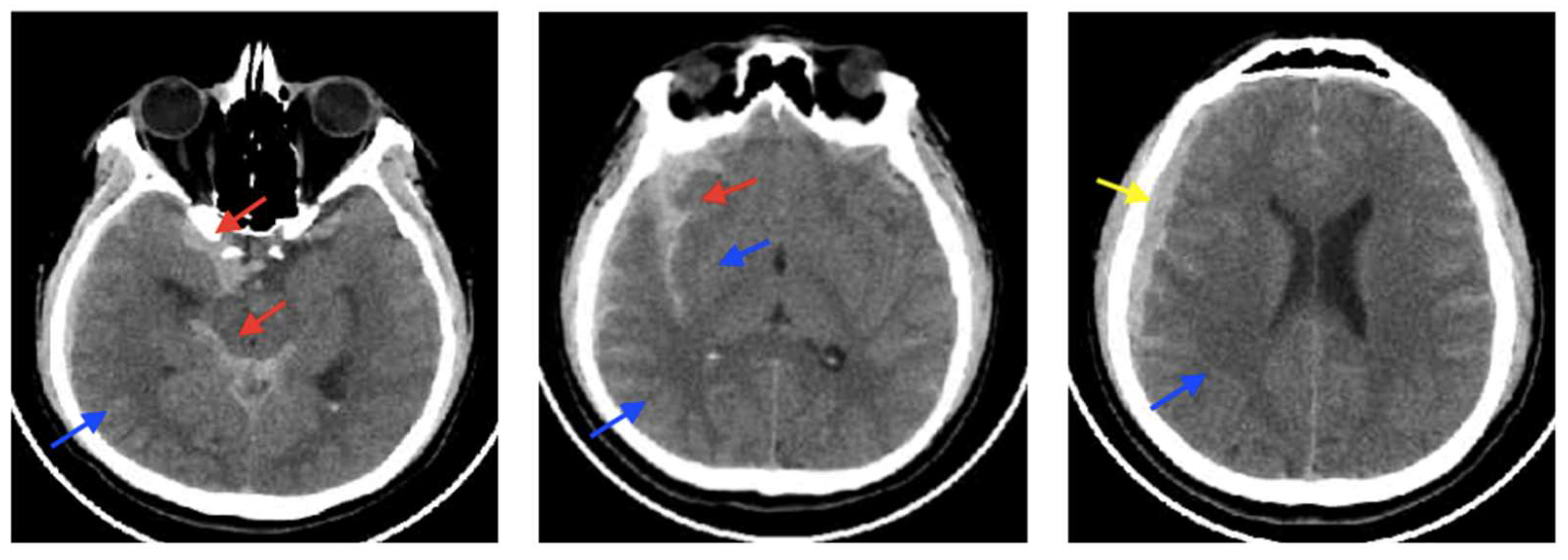
3.6. CSF-Shift Edema
3.7. Hemolytic Edema
4. Conclusions
Funding
Conflicts of Interest
References
- Iencean, S.M. Brain edema—A new classification. Med. Hypotheses 2003, 61, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.L.; Rojas, R.; Eisenberg, R.L. Cerebral Edema. Am. J. Roentgenol. 2012, 199, W258–W273. [Google Scholar] [CrossRef] [PubMed]
- Marmarou, A. A review of progress in understanding the pathophysiology and treatment of brain edema. Neurosurg. Focus 2007, 22, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Stokum, J.A.; Gerzanich, V.; Simard, J.M. Molecular pathophysiology of cerebral edema. J. Cereb. Blood Flow Metab. 2016, 36, 513–538. [Google Scholar] [CrossRef] [PubMed]
- Jha, R.M.; Kochanek, P.M.; Simard, J.M. Pathophysiology and treatment of cerebral edema in traumatic brain injury. Neuropharmacology 2019, 145, 230–246. [Google Scholar] [CrossRef] [PubMed]
- Cook, A.M.; Jones, G.M.; Hawryluk, G.W.J.; Mailloux, P.; McLaughlin, D.; Papangelou, A.; Samuel, S.; Tokumaru, S.; Venkatasubramanian, C.; Zacko, C.; et al. Guidelines for the Acute Treatment of Cerebral Edema in Neurocritical Care Patients. Neurocrit. Care 2020, 32, 647–666. [Google Scholar] [CrossRef] [PubMed]
- Michel, C.C.; Woodcock, T.E.; Curry, F.E. Understanding and extending the Starling principle. Acta Anaesthesiol. Scand. 2020, 64, 1032–1037. [Google Scholar] [CrossRef] [PubMed]
- Levick, J.R.; Michel, C.C. Microvascular fluid exchange and the revised Starling principle. Cardiovasc. Res. 2010, 87, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Igor Klatzo, M.D. Neuropathological Aspects Of Brain Edema. J. Neuropathol. Exp. Neurol. 1967, 26, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Dalby, T.; Wohl, E.; Dinsmore, M.; Unger, Z.; Chowdhury, T.; Venkatraghavan, L. Pathophysiology of Cerebral Edema—A Comprehensive Review. J. Neuroanaesth. Crit. Care 2021, 08, 163–172. [Google Scholar] [CrossRef]
- Rosenblum, W.I. Cytotoxic Edema: Monitoring Its Magnitude and Contribution to Brain Swelling. J. Neuropathol. Exp. Neurol. 2007, 66, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Cherian, I.; Beltran, M.; Kasper, E.; Bhattarai, B.; Munokami, S.; Grasso, G. Exploring the Virchow-Robin spaces function: A unified theory of brain diseases. Surg. Neurol. Int. 2016, 7, 711. [Google Scholar] [CrossRef] [PubMed]
- Harrison, I.F.; Siow, B.; Akilo, A.B.; Evans, P.G.; Ismail, O.; Ohene, Y.; Nahavandi, P.; Thomas, D.L.; Lythgoe, M.F.; Wells, J.A. Non-invasive imaging of CSF-mediated brain clearance pathways via assessment of perivascular fluid movement with diffusion tensor MRI. eLife 2018, 7, e34028. [Google Scholar] [CrossRef] [PubMed]
- Leinonen, V.; Vanninen, R.; Rauramaa, T. Raised intracranial pressure and brain edema. In Handbook of Clinical Neurology [Internet]; Elsevier: Amsterdam, The Netherlands, 2018; pp. 25–37. Available online: https://linkinghub.elsevier.com/retrieve/pii/B9780128023952000043 (accessed on 1 November 2024).
- Winkler, E.A.; Minter, D.; Yue, J.K.; Manley, G.T. Cerebral Edema in Traumatic Brain Injury. Neurosurg. Clin. N. Am. 2016, 27, 473–488. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.K.; Kwon, M.S.; Ivanov, A.; Gerzanich, V.; Simard, J.M. The Sulfonylurea Receptor 1 (Sur1)-Transient Receptor Potential Melastatin 4 (Trpm4) Channel. J. Biol. Chem. 2013, 288, 3655–3667. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Dong, Y.; Simard, J.M. Functional Coupling between Sulfonylurea Receptor Type 1 and a Nonselective Cation Channel in Reactive Astrocytes from Adult Rat Brain. J. Neurosci. 2003, 23, 8568–8577. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.M.; Chen, M.; Tarasov, K.V.; Bhatta, S.; Ivanova, S.; Melnitchenko, L.; Tsymbalyuk, N.; West, G.A.; Gerzanich, V. Newly expressed SUR1-regulated NCCa-ATP channel mediates cerebral edema after ischemic stroke. Nat. Med. 2006, 12, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, M.C.; Verkman, A.S. Aquaporin water channels in the nervous system. Nat. Rev. Neurosci. 2013, 14, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Yang, G.Y. Aquaporin-4: A Potential Therapeutic Target for Cerebral Edema. Int. J. Mol. Sci. 2016, 17, 1413. [Google Scholar] [CrossRef] [PubMed]
- Burhan, H.; Cherian, I. CSF Shift Edema-Role of “Fluid Shift” in Adopting a Microsurgical Technique for Moderate to Severe Head Injuries. Neurol. India 2021, 69, 334–335. [Google Scholar] [CrossRef] [PubMed]
- Biller, A.; Badde, S.; Heckel, A.; Guericke, P.; Bendszus, M.; Nagel, A.M.; Heiland, S.; Mairbäurl, H.; Bärtsch, P.; Schommer, K. Exposure to 16 h of normobaric hypoxia induces ionic edema in the healthy brain. Nat. Commun. 2021, 12, 5987. [Google Scholar] [CrossRef] [PubMed]
- Stephens, B.; Spessert, E.; Knaysi, G.; Cota, M.; Latour, L.; Diaz-Arrastia, R. Volumetric Measurements of Cytotoxic and Vasogenic Edema Following Traumatic Brain Injury Using Magnetic Resonance Imaging (P7.156). Neurology 2015, 84, 156. [Google Scholar] [CrossRef]
- Vlodavsky, E.; Palzur, E.; Shehadeh, M.; Soustiel, J.F. Post-traumatic cytotoxic edema is directly related to mitochondrial function. J. Cereb. Blood Flow Metab. 2017, 37, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Exner, C.; Sienel, R.I.; When, A.C.; Seker, F.B.; Boldoczki, F.M.; Guo, Y.; Duering, M.; Pasternak, O.; Plesnila, N.; et al. Characterization of Vasogenic and Cytotoxic Brain Edema Formation After Experimental Traumatic Brain Injury by Free Water Diffusion Magnetic Resonance Imaging. J. Neurotrauma. 2024, 41, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Stokum, J.A.; Gerzanich, V.; Sheth, K.N.; Kimberly, W.T.; Simard, J.M. Emerging Pharmacological Treatments for Cerebral Edema: Evidence from Clinical Studies. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 291–309. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Wang, Q.; Deng, Y.; Fang, M.; Chen, C.; Fu, Y.; Jiang, W.; Jiang, X. Hypertonic saline ameliorates cerebral edema through downregulation of aquaporin-4 expression in the astrocytes. Neuroscience 2010, 166, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Yu, X.; Liao, Z.; Zhu, N.; Huo, H.; Wang, M.; Ji, G.; She, H.; Luo, Z.; Yue, S. Hypertonic saline reduces lipopolysaccharide-induced mouse brain edema through inhibiting aquaporin 4 expression. Crit. Care 2012, 16, R186. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, H.M.; Shenton, M.E.; Pasternak, O.; Simard, J.M.; Okonkwo, D.O.; Aldrich, C.; He, F.; Jain, S.; Hayman, E.G. Magnetic Resonance Imaging Pilot Study of Intravenous Glyburide in Traumatic Brain Injury. J. Neurotrauma. 2020, 37, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Griepp, D.W.; Lee, J.; Moawad, C.M.; Davati, C.; Runnels, J.; Fiani, B. BIIB093 (intravenous glibenclamide) for the prevention of severe cerebral edema. Surg. Neurol. Int. 2021, 12, 80. [Google Scholar] [CrossRef] [PubMed]
- Khanna, A.; Walcott, B.P.; Kahle, K.T.; Simard, J.M. Effect of glibenclamide on the prevention of secondary brain injury following ischemic stroke in humans. Neurosurg. Focus 2014, 36, E11. [Google Scholar] [CrossRef] [PubMed]
- O’pHelan, K.H.; Park, D.; Efird, J.T.; Johnson, K.; Albano, M.; Beniga, J.; Green, D.M.; Chang, C.W.J. Patterns of Increased Intracranial Pressure After Severe Traumatic Brain Injury. Neurocrit. Care 2009, 10, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Kadry, H.; Noorani, B.; Cucullo, L. A blood–brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS 2020, 17, 69. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.M.; Kent, T.A.; Chen, M.; Tarasov, K.V.; Gerzanich, V. Brain oedema in focal ischaemia: Molecular pathophysiology and theoretical implications. Lancet Neurol. 2007, 6, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Nag, S.; Manias, J.L.; Stewart, D.J. Pathology and new players in the pathogenesis of brain edema. Acta Neuropathol. 2009, 118, 197–217. [Google Scholar] [CrossRef] [PubMed]
- Casaca-Carreira, J.; Temel, Y.; Hescham, S.A.; Jahanshahi, A. Transependymal Cerebrospinal Fluid Flow: Opportunity for Drug Delivery? Mol. Neurobiol. 2018, 55, 2780–2788. [Google Scholar] [CrossRef] [PubMed]
- Durward, Q.J.; Maestro, R.F.D.; Amacher, A.L.; Farrar, J.K. The influence of systemic arterial pressure and intracranial pressure on the development of cerebral vasogenic edema. J. Neurosurg. 1983, 59, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, M.C.; Manley, G.T.; Krishna, S.; Verkman, A.S. Aquaporin-4 facilitates reabsorption of excess fluid in vasogenic brain edema. FASEB J. 2004, 18, 1291–1293. [Google Scholar] [CrossRef] [PubMed]
- Cherian, I.; Beltran, M.; Landi, A.; Alafaci, C.; Torregrossa, F.; Grasso, G. Introducing the concept of “CSF-shift edema” in traumatic brain injury. J. Neurosci. Res. 2018, 96, 744–752. [Google Scholar] [CrossRef] [PubMed]
- Cherian, I.; Bernardo, A.; Grasso, G. Cisternostomy for Traumatic Brain Injury: Pathophysiologic Mechanisms and Surgical Technical Notes. World Neurosurg. 2016, 89, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Cherian, I.; Burhan, H.; Dashevskiy, G.; Motta, S.J.H.; Parthiban, J.; Wang, Y.; Tong, H.; Torregrossa, F.; Grasso, G. Cisternostomy: A Timely Intervention in Moderate to Severe Traumatic Brain Injuries: Rationale, Indications, and Prospects. World Neurosurg. 2019, 131, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Al Masri, M.; Corell, A.; Michaëlsson, I.; Jakola, A.S.; Skoglund, T. The glymphatic system for neurosurgeons: A scoping review. Neurosurg. Rev. 2024, 47, 61. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Nedergaard, M. Is There a Cerebral Lymphatic System? Stroke 2013, 44 (Suppl. 1), S93–S95. [Google Scholar] [CrossRef] [PubMed]
- Møllgård, K.; Beinlich, F.R.M.; Kusk, P.; Miyakoshi, L.M.; Delle, C.; Plá, V.; Hauglund, N.L.; Esmail, T.; Rasmussen, M.K.; Gomolka, R.S.; et al. A mesothelium divides the subarachnoid space into functional compartments. Science 2023, 379, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Bodmer, D.; Vaughan, K.A.; Zacharia, B.E.; Hickman, Z.L.; Connolly, E.S. The Molecular Mechanisms that Promote Edema After Intracerebral Hemorrhage. Transl. Stroke Res. 2012, 3, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, S.; Chang, J.; Wei, J.; Feng, M.; Wang, R. Perihematomal Edema After Intracerebral Hemorrhage: An Update on Pathogenesis, Risk Factors, and Therapeutic Advances. Front. Immunol. 2021, 12, 740632. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Keep, R.F.; Hoff, J.T.; Xi, G. Brain Injury After Intracerebral Hemorrhage: The Role of Thrombin and Iron. Stroke 2007, 38, 759–762. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.R.; Colon, G.P.; Betz, A.L.; Keep, R.F.; Kim, S.; Hoff, J.T. Edema from intracerebral hemorrhage: The role of thrombin. J. Neurosurg. 1996, 84, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Urday, S.; Kimberly, W.T.; Beslow, L.A.; Vortmeyer, A.O.; Selim, M.H.; Rosand, J.; Simard, J.M.; Sheth, K.N. Targeting secondary injury in intracerebral haemorrhage—Perihaematomal oedema. Nat. Rev. Neurol. 2015, 11, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Ducruet, A.F.; Zacharia, B.E.; Hickman, Z.L.; Grobelny, B.T.; Yeh, M.L.; Sosunov, S.A.; Connolly, E.S. The complement cascade as a therapeutic target in intracerebral hemorrhage. Exp. Neurol. 2009, 219, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.P.; Xi, G.; Keep, R.F.; Hua, Y.; Nemoianu, A.; Hoff, J.T. Brain edema after experimental intracerebral hemorrhage: Role of hemoglobin degradation products. J. Neurosurg. 2002, 96, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Holste, K.G.; Hua, Y.; Keep, R.F.; Xi, G. Brain edema formation and therapy after intracerebral hemorrhage. Neurobiol. Dis. 2023, 176, 105948. [Google Scholar] [CrossRef] [PubMed]
- Vande Vyvere, T.; Pisică, D.; Wilms, G.; Claes, L.; Van Dyck, P.; Snoeckx, A.; van den Hauwe, L.; Pullens, P.; Verheyden, J.; Wintermark, M.; et al. Imaging Findings in Acute Traumatic Brain Injury: A National Institute of Neurological Disorders and Stroke Common Data Element-Based Pictorial Review and Analysis of Over 4000 Admission Brain Computed Tomography Scans from the Collaborative European NeuroTrauma Effectiveness Research in Traumatic Brain Injury (CENTER-TBI) Study. J. Neurotrauma 2024, 41, 2248–2297. [Google Scholar] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Edema | BBB Integrity | Tissue Compartment | Composition | Mechanism | Proposed Treatment and Starting Time |
|---|---|---|---|---|---|
| Cytotoxic | Preserved | Intracellular | Water and ions | Intracellular water accumulation due to ionic imbalance from dysfunctional pumps causes fluid shift from the interstitial to the intracellular compartment. | Hypertonic saline in the first week after the injury |
| Ionic | Preserved | Interstitial | Water and ions | Fluid buildup in the interstitial compartment occurs from ionic imbalance due to cytotoxic edema, where ions accumulate inside cells. This creates an osmotic gradient that shifts fluid from the intravascular to the extracellular space across a functional BBB. | Hypertonic saline, generally starting before day 5–7. |
| Vasogenic | Compromised | Interstitial | Plasma ultrafiltrate | Fluid accumulation in the interstitial compartment caused by a dysfunctional BBB permeable to macromolecules and proteins drives a fluid shift from the intravascular to the extracellular space. | Corticosteroids, starting on the second week after injury. |
| Interstitial | Preserved | Interstitial | CSF | Abnormal fluid accumulation in the extracellular space surrounding the cerebral ventricles is caused by an increased pressure within the ventricular system that damages the ependymal lining and generates CSF passage from the intraventricular space to the periventricular extracellular space. | EVD/Ventricular shunt/Endoscopic third ventriculostomy, when identification of the imaging findings. |
| Osmotic | Preserved | Interstitial | Water and ions | An osmotic imbalance between the intravascular and extracellular spaces leads to abnormal fluid accumulation in the interstitial compartments, occurring when solute concentrations differ between the brain parenchyma and blood plasma across an intact BBB. | Achieving water homeostasis and avoiding misuse of hyperosmolar therapy, especially in the second week after injury. |
| CSF-Shift | Preserved | Interstitial | CSF/Water and ions | Fluid accumulation in the interstitial space occurs when CSF moves from the subarachnoid cisterns to the brain’s interstitial space through VRS due to increased pressure in the brain cisterns from conditions like subarachnoid hemorrhage. | Cisternostomy as solely early intervention or as coadjuvant therapy when performing a craniotomy or craniectomy for hematoma drainage or brain edema control. |
| Hemolytic | Compromised | Interstitial | Plasma ultrafiltrate | Fluid accumulation in the interstitial compartment caused by a combination of BBB disruption, inflammatory response, and osmotic gradient formed by degrading products of the heme group after intracranial hemorrhages. | Evacuation of the intracranial hemorrhage that is associated with the surrounding hemolytic process (either primary or residual bleeding) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cardona-Collazos, S.; Gonzalez, W.D.; Pabon-Tsukamoto, P.; Gao, G.-Y.; Younsi, A.; Paiva, W.S.; Rubiano, A.M. Cerebral Edema in Traumatic Brain Injury. Biomedicines 2025, 13, 1728. https://doi.org/10.3390/biomedicines13071728
Cardona-Collazos S, Gonzalez WD, Pabon-Tsukamoto P, Gao G-Y, Younsi A, Paiva WS, Rubiano AM. Cerebral Edema in Traumatic Brain Injury. Biomedicines. 2025; 13(7):1728. https://doi.org/10.3390/biomedicines13071728
Chicago/Turabian StyleCardona-Collazos, Santiago, Wendy D. Gonzalez, Pamela Pabon-Tsukamoto, Guo-Yi Gao, Alexander Younsi, Wellingson S. Paiva, and Andres M. Rubiano. 2025. "Cerebral Edema in Traumatic Brain Injury" Biomedicines 13, no. 7: 1728. https://doi.org/10.3390/biomedicines13071728
APA StyleCardona-Collazos, S., Gonzalez, W. D., Pabon-Tsukamoto, P., Gao, G.-Y., Younsi, A., Paiva, W. S., & Rubiano, A. M. (2025). Cerebral Edema in Traumatic Brain Injury. Biomedicines, 13(7), 1728. https://doi.org/10.3390/biomedicines13071728